Radical Dendrimers Based on Biocompatible Oligoethylene Glycol Dendrimers as Contrast Agents for MRI

 , , , , , , and
, , , , , , and
Abstract
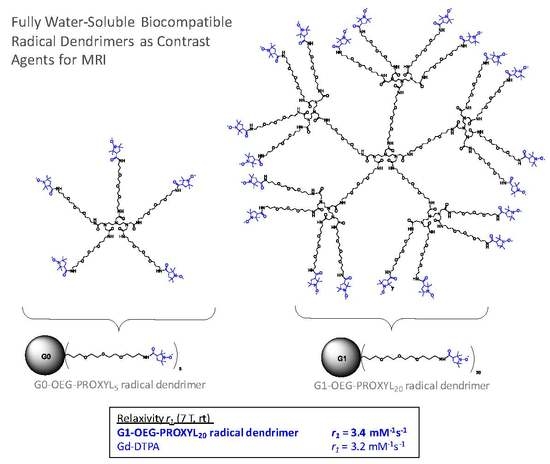
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
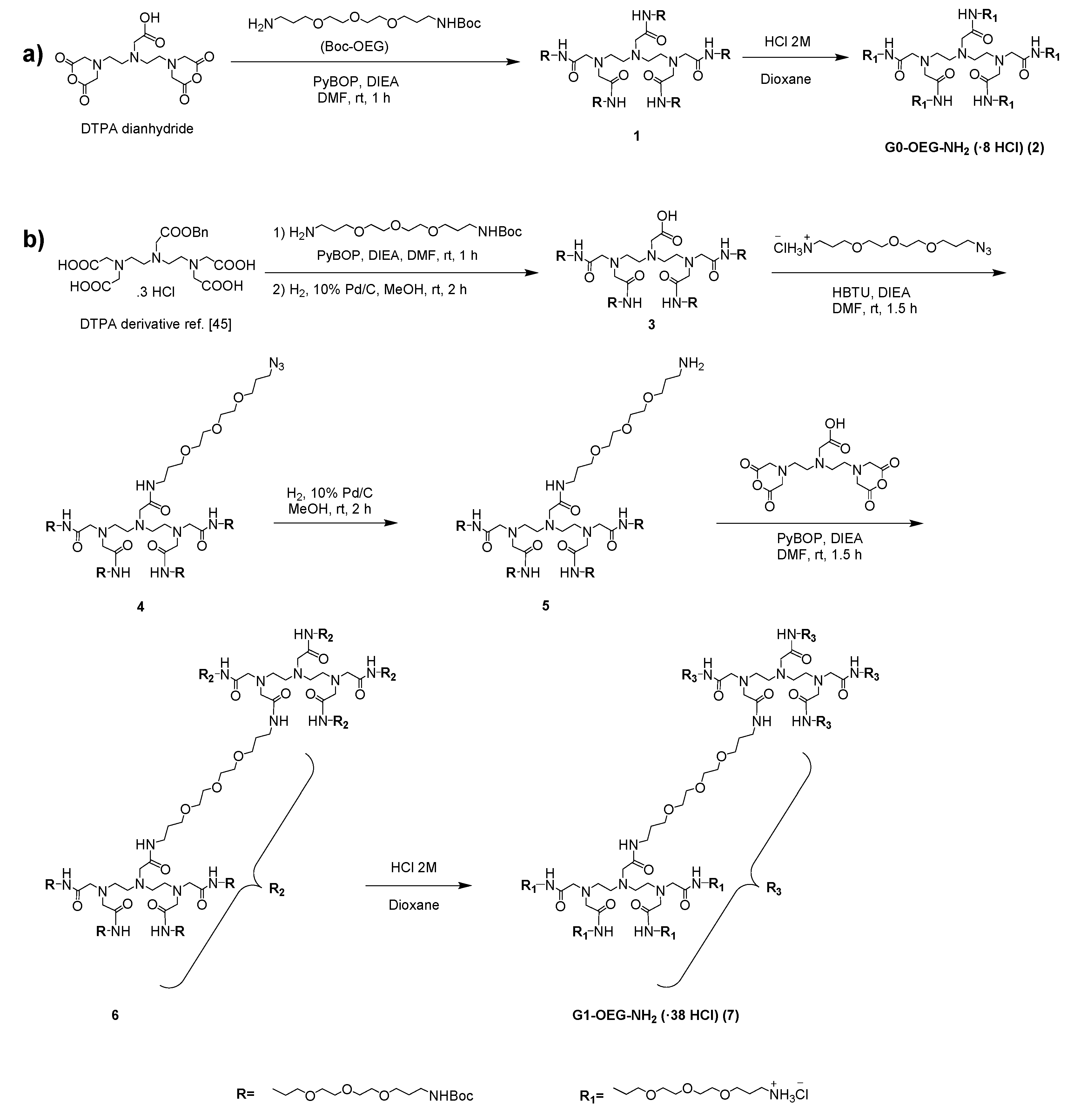
3.1. Synthesis of G0- and G1-OEG-NH2 Dendrimers
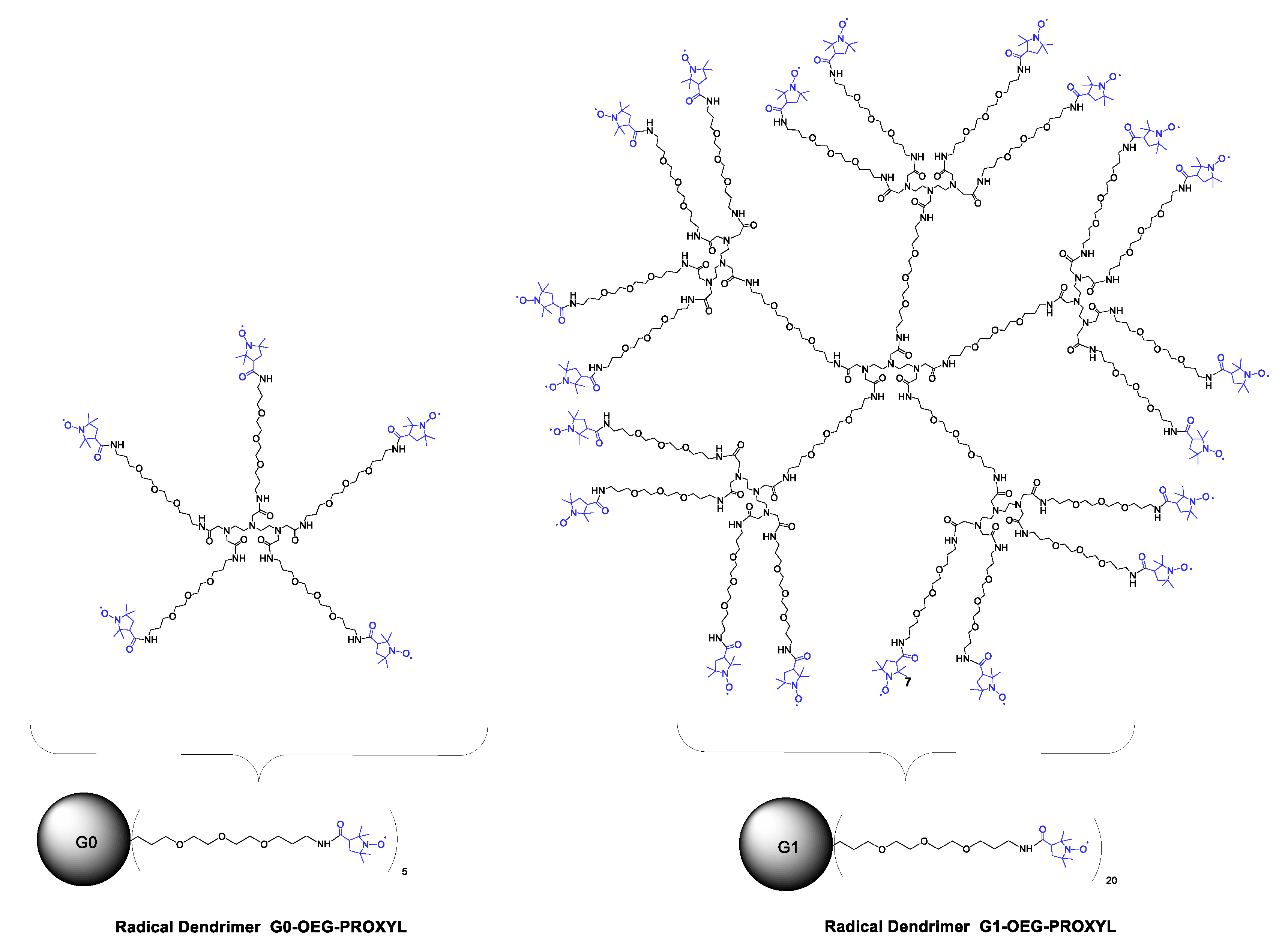
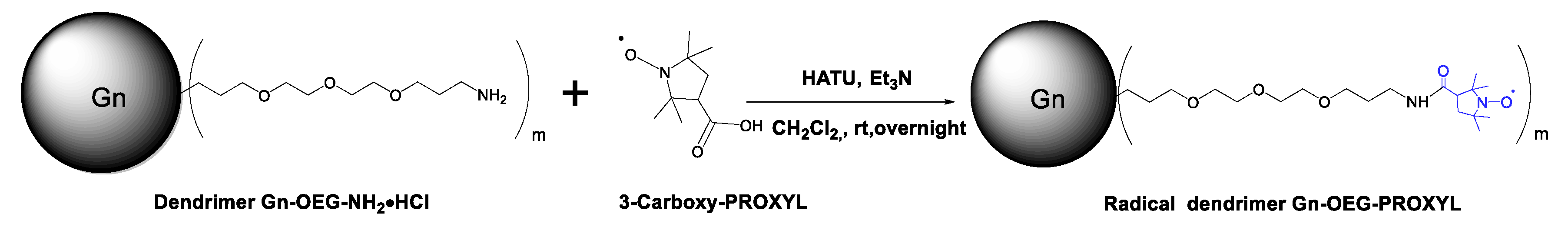
3.2. Synthesis and Characterization of G0- and G1-OEG-PROXYL Radical Dendrimers
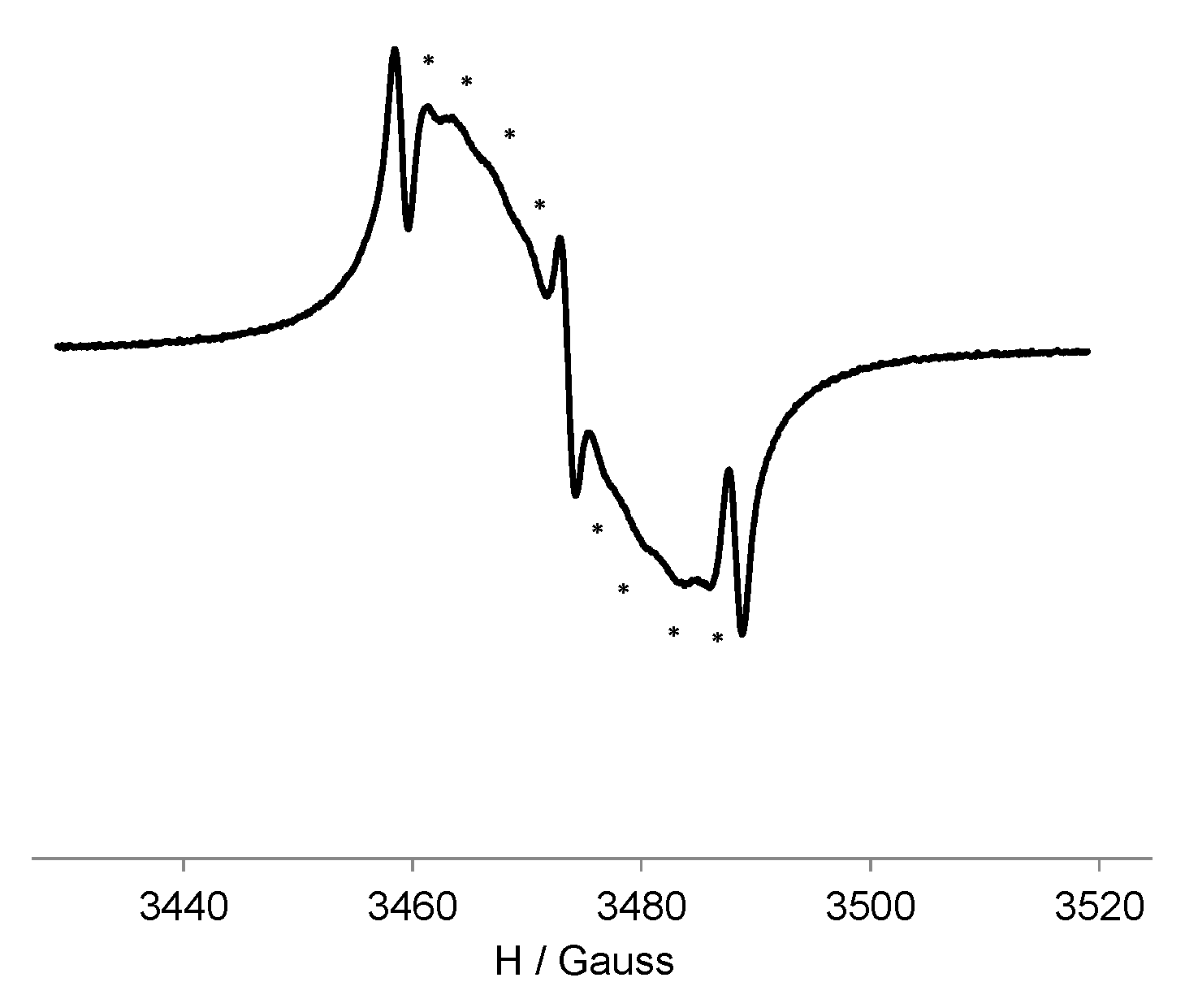
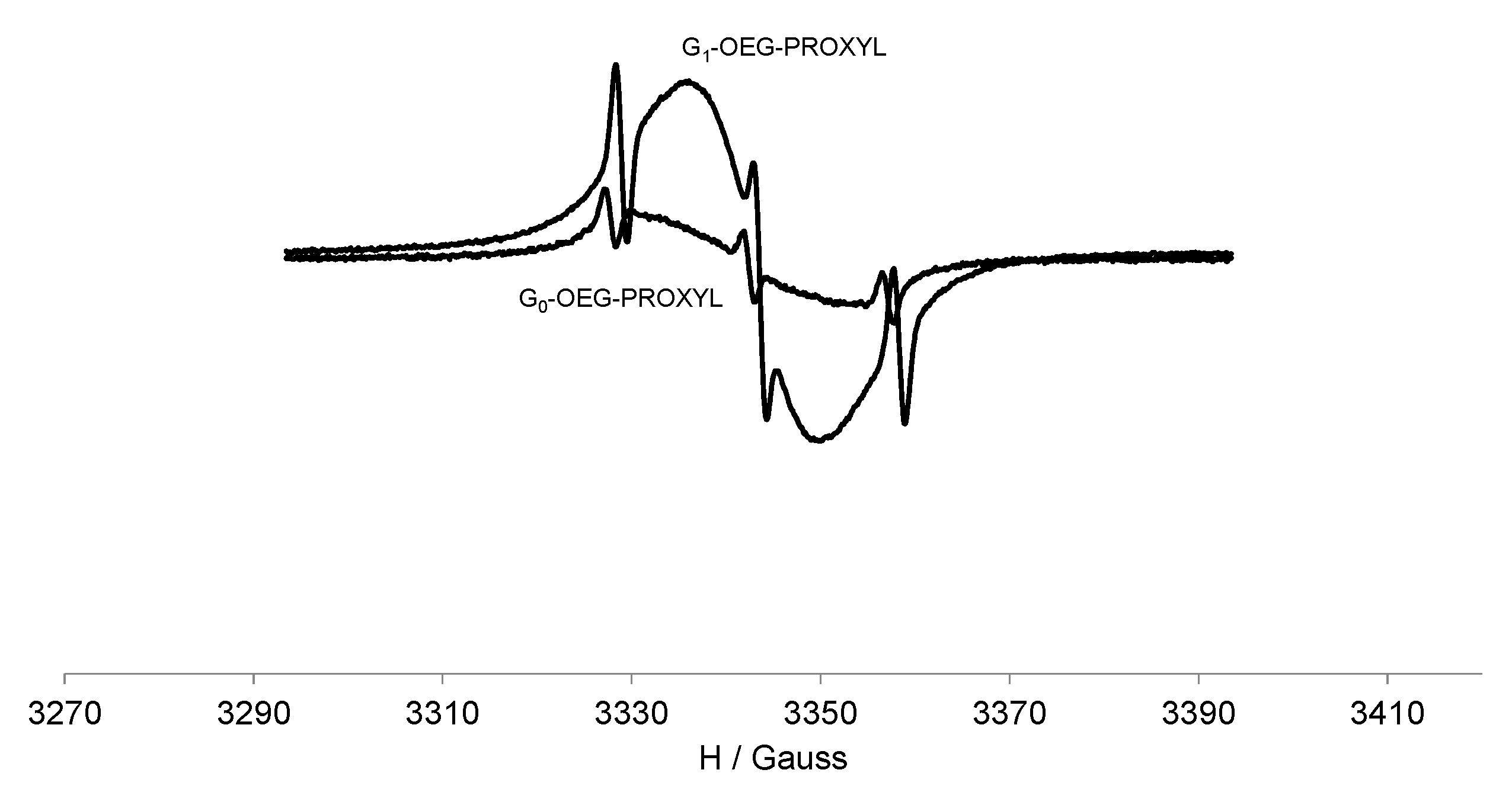
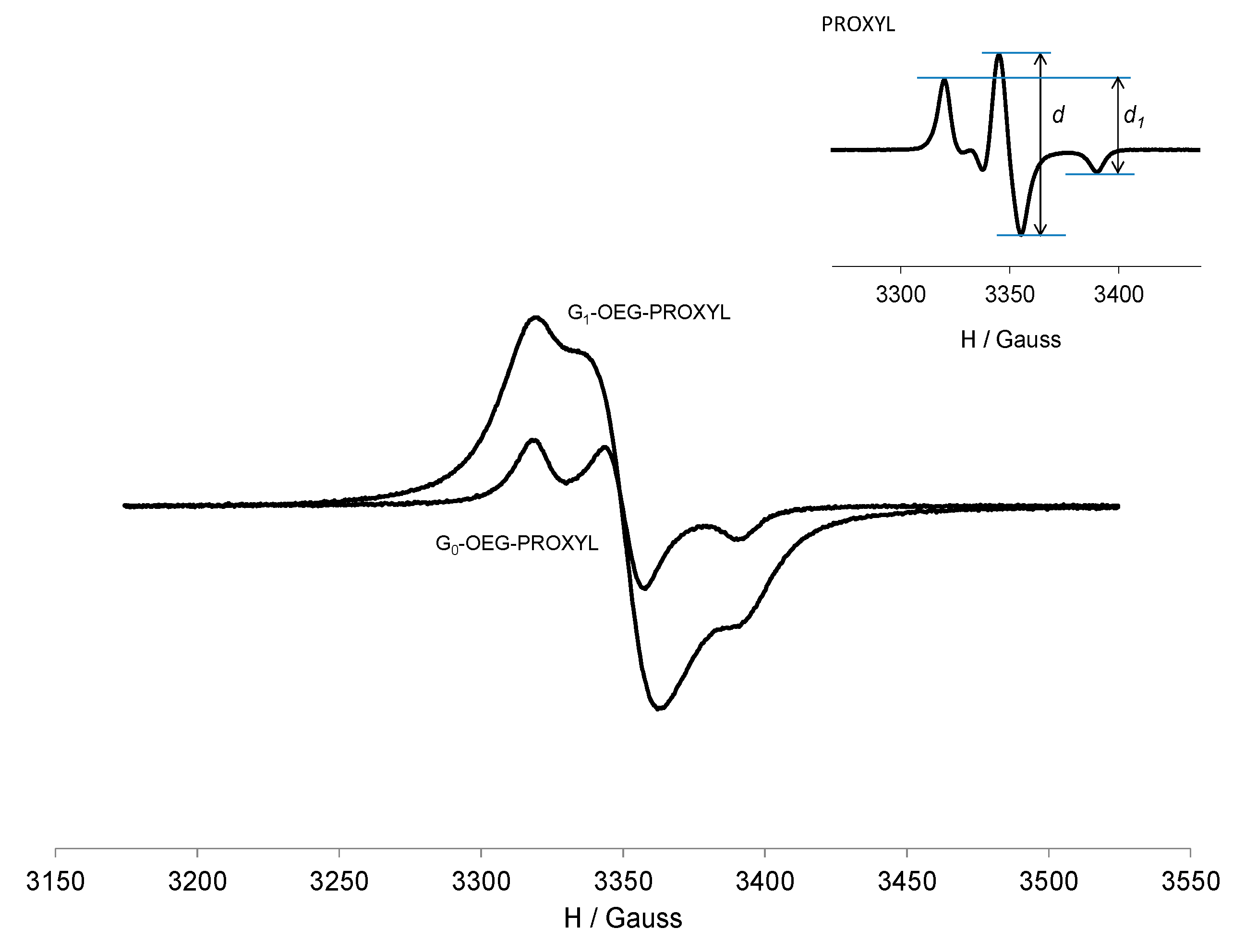
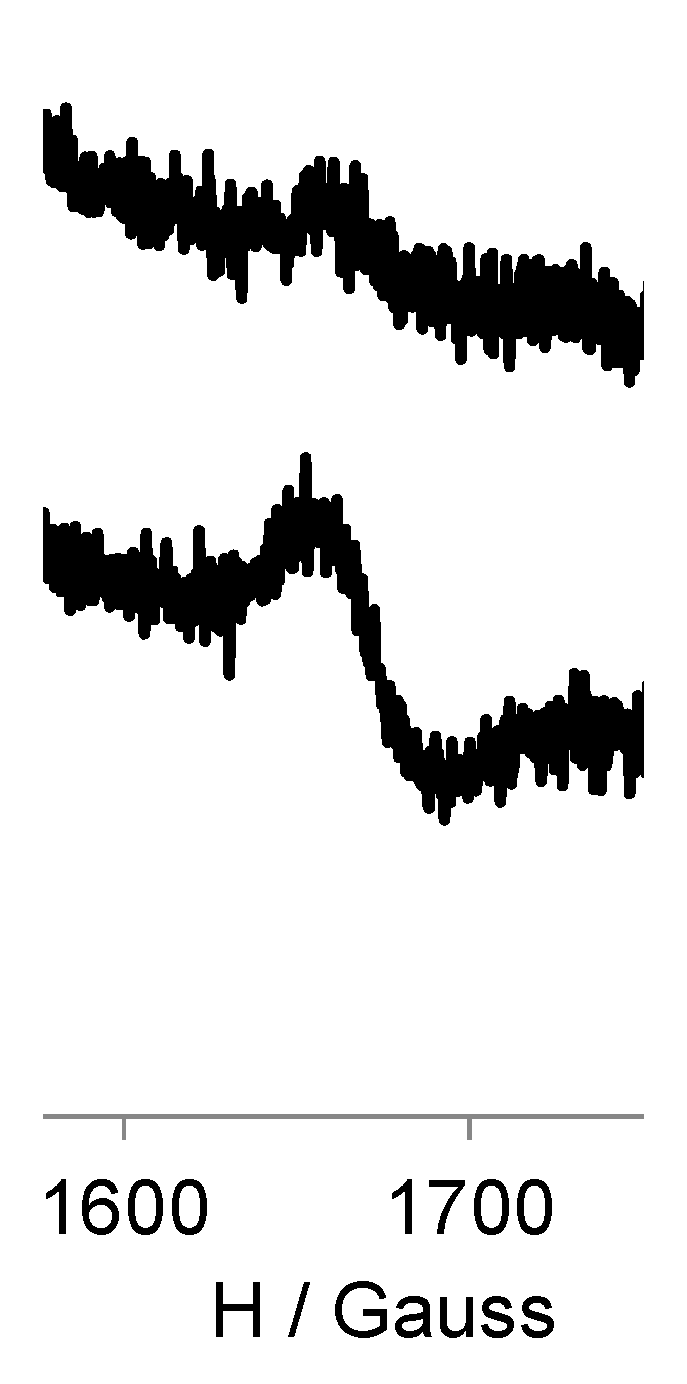
3.3. EPR Study of G0-OEG-PROXYL and G1-OEG-PROXYL
3.4. Relaxivity Measurements and Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoerr, V.; Faber, C. Magnetic resonance imaging characterization of microbial infections. J. Pharm. Biomed. Anal. 2014, 93, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palestro, C.J.; Love, C.; Miller, T.T. Diagnostic imaging tests and microbial infections. Cell. Microbiol. 2007, 9, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Jelicks, L.A.; Lisanti, M.P.; Machado, F.S.; Weiss, L.M.; Tanowitz, H.B.; Desruisseaux, M.S. Imaging of small-animal models of infectious diseases. Am. J. Pathol. 2013, 182, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Niska, J.A.; Meganck, J.A.; Pribaz, J.R.; Shahbazian, J.H.; Lim, E.; Zhang, N.; Rice, B.W.; Akin, A.; Ramos, R.I.; Berntal, N.M.; et al. Monitoring bacterial burden, inflammation and bone damage longitudinally using optical and μCT imaging in an orthopaedic implant infection in mice. PLoS ONE 2012, 7, e47397. [Google Scholar] [CrossRef]
- Gildehaus, F.J.; Haasters, F.; Drosse, I.; Wagner, E.; Zach, C.; Mutschler, W.; Cumming, P.; Bartenstein, P.; Schieker, M. Impact of indium-111 oxine labelling on viability of human mesenchymal stem cells in vitro, and 3D cell-tracking using SPECT/CT in vivo. Mol. Imaging Biol. 2011, 13, 1204–1214. [Google Scholar] [CrossRef]
- Gemmel, F.; Dumarey, N.; Welling, M. Future diagnostic agents. Semin. Nucl. Med. 2009, 39, 11–26. [Google Scholar] [CrossRef]
- Soldatos, T.; Durand, D.J.; Subhawong, T.K.; Carrino, J.A.; Chhabra, A. Magnetic resonance imaging of musculoskeletal infections: Systematic diagnostic assessment and key points. Acad. Radiol. 2012, 19, 1434–1443. [Google Scholar] [CrossRef]
- Radermacher, K.A.; Beghein, N.; Boutry, S.; Laurent, S.; Vander Elst, L.; Muller, R.N.; Jordan, B.F.; Gallez, B. In vivo detection of inflammation using pegylated iron oxide particles targeted at E-selectin: A multimodal approach using MR imaging and EPR spectroscopy. Investig. Radiol. 2009, 44, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar] [CrossRef]
- Calcagno, C.; Ramachandran, S.; Millon, A.; Robson, P.M.; Mani, V.; Fayad, Z. Gadolinium-based contrast agents for vessel wall magnetic resonance imaging (MRI) of atherosclerosis. Curr. Cardiovasc. Imaging Rep. 2013, 6, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Wermuth, P.J.; Jimenez, S.A. Gadolinium compounds signaling through TLR4 and TLR7 in normal human macrophages: Establishment of a proinflammatory phenotype and implications for the pathogenesis of nephrogenic systemic fibrosis. J. Immunol. 2012, 189, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Cabello, J.; Barnett, B.P.; Bottomley, P.A.; Bulte, J.W. Fluorine (19F) MRS and MRI in biomedicine. NMR Biomed. 2011, 24, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Baraki, H.; Zinne, N.; Wedekind, D.; Meier, M.; Bleich, A.; Glage, S.; Hedrich, H.J.; Kutschka, I.; Haverich, A. Magnetic resonance imaging of soft tissue infection with iron oxide labeled granulocytes in a rat model. PLoS ONE 2012, 7, e51770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.M.; Lee, S.H.; Kang, H.Y.; Baek, S.Y.; Kim, S.M.; Shin, M.J. Assessment of musculoskeletal infection in rats to determine usefulness of SPIO-enhanced MRI. AJR Am. J. Roentgenol. 2007, 189, 542–548. [Google Scholar] [CrossRef]
- Lin, C.; Cai, S.; Feng, J. Positive Contrast Imaging of SPIO Nanoparticles. J. Nanomater. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Margerum, L.D.; Campion, B.K.; Koo, M.; Shargill, N.; Lai, J.J.; Marumoto, A.; Sontum, P.C. Gadolinium(III) DO3A macrocycles and polyethylene glycol coupled to dendrimers. Effect of molecular weight on physical and biological properties of macromolecular magnetic resonance imaging contrast agents. J. Alloys Comp. 1997, 249, 185–190. [Google Scholar] [CrossRef]
- Kojima, C.; Turkbey, B.; Ogawa, M.; Bernardo, M.; Regino, C.A.S.; Bryant, L.H.; Choyke, P.L.; Kono, K.; Kobayashi, H. Dendrimer-based MRI contrast agents: The effects of PEGylation on relaxivity and pharmacokinetics. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C.T.; Simonsen, H.; Liptrot, M.; Søgaard, L.V.; Lundgren, J.D.; Ostergaard, C.; Frimodt-Møller, N.; Rowland, I.J. In vivo study of experimental pneumococcal meningitis using magnetic resonance imaging. BMC Med. Imaging 2008, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.; McCabe, C.; Gettinby, G.; Bradley, B.; Condon, B.; Kennedy, P.G. Magnetic resonance imaging to assess blood–brain barrier damage in murine trypanosomiasis. Am. J. Trop. Med. Hyg. 2011, 84, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Lelievre, B.; Legras, P.; Godon, C.; Franconi, F.; Saint-Andre, J.P.; Bouchara, J.P.; Diquet, B. Experimental models of disseminated scedosporiosis with cerebral involvement. J. Pharmacol. Exp. Ther. 2013, 345, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.S.; Kim, S.H.; Cai, Q.Y.; Kim, S.Y.; Kim, H.O.; Lee, H.J.; Kim, E.A.; Yoon, S.E.; Yun, K.J.; Yoon, K.H. Inflammation-specific T1 imaging using anti-intercellular adhesion molecule 1 antibody-conjugated gadolinium diethylenetriaminepentaacetic acid. Mol. Imaging 2007, 6, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dear, J.W.; Kobayashi, H.; Jo, S.K.; Holly, M.K.; Hu, X.; Yuen, P.S.; Brechbiel, M.W.; Star, R.A. Dendrimer-enhanced MRI as a diagnostic and prognostic biomarker of sepsis-induced acute renal failure in aged mice. Kidney Int. 2005, 67, 2159–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Tang, Z.; Wang, S.; Hui, H.; Gong, L.; Lu, Y.; Xue, Z.; Liao, H.; Chen, F.; Yang, F.; et al. The role of imaging in the detection and management of COVID-19: A review. IEEE Rev. Biomed. Eng. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.F.; Lloveras, V.; Zhang, S.; Liko, F.; Veciana, J.; Muñoz-Gómez, J.L.; Vidal-Gancedo, J. Fully Water-Soluble Polyphosphorhydrazone-Based Radical Dendrimers Functionalized with Tyr-PROXYL Radicals as Metal-Free MRI T1 Contrast Agents. ACS Appl. Bio Mater. 2020, 3, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Brasch, R.C.; London, D.A.; Wesbey, G.E.; Tozer, T.N.; Nitecki, D.E.; Williams, R.D.; Doemeny, J.; Tuck, L.D.; Lallemand, D.P. Work in progress: Nuclear magnetic resonance study of a paramagnetic nitroxide contrast agent for enhancement of renal structures in experimental animals. Radiology 1983, 147, 773–779. [Google Scholar] [CrossRef]
- Brasch, R.C.; Nitecki, D.E.; Brant-Zawadzki, M.; Enzmann, D.R.; Wesbey, G.E.; Tozer, T.N.; Tuck, L.D.; Cann, C.E.; Fike, J.R.; Sheldon, P. Brain nuclear magnetic resonance imaging enhanced by a paramagnetic nitroxide contrast agent: Preliminary report. Am. J. Roentgenol. 1983, 141, 1019–1023. [Google Scholar] [CrossRef]
- Rosen, G.M.; Griffeth, L.K.; Brown, M.A.; Drayer, B.P. Intrathecal administration of nitroxides as potential contrast agents for MR imaging. Radiology 1987, 163, 239–243. [Google Scholar] [CrossRef]
- Bosman, A.W.; Janssen, R.A.J.; Meijer, E.W. Five Generations of Nitroxyl-Functionalized Dendrimers. Macromolecules 1997, 30, 3606–3611. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, Y.; Kurashima, F.; Kikuchi, C.; Anzai, J.-i.; Osa, T. Voltammetric behavior of poly (amidoamine) dendrimers containing nitroxyl radical end groups. Electrochem. Commun. 1999, 1, 305–308. [Google Scholar] [CrossRef]
- Winalski, C.S.; Shortkroff, S.; Mulkern, R.V.; Schneider, E.; Rosen, G.M. Magnetic resonance relaxivity of dendrimer-linked nitroxides. Magn. Reson. Med. 2002, 48, 965–972. [Google Scholar] [CrossRef]
- Maliakal, A.J.; Turro, N.J.; Bosman, A.W.; Cornel, J.; Meijer, E.W. Relaxivity studies on dinitroxide and polynitroxyl functionalized dendrimers: Effect of electron exchange and structure on paramagnetic relaxation enhancement. J. Phys. Chem. A 2003, 107, 8467–8475. [Google Scholar] [CrossRef]
- Francese, G.; Dunand, F.A.; Loosli, C.; Merbach, A.E.; Decurtins, S. Functionalization of PAMAM dendrimers with nitronyl nitroxide radicals as models for the outer-sphere relaxation in dentritic potential MRI contrast agents. Magn. Reson. Chem. 2003, 41, 81–83. [Google Scholar] [CrossRef]
- Sebby, K.B.; Walter, E.D.; Usselman, R.J.; Cloninger, M.J.; Singel, D.J. End-Group Distributions of Multiple Generations of Spin-Labeled PAMAM Dendrimers. J. Phys. Chem. B 2011, 115, 4613–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimono, S.; Tamura, R.; Ikuma, N.; Takahashi, H.; Sakai, N.; Yamauchi, J. Characterization of the Chiral Paramagnetic Multispin System Built on a Cyclotriphosphazene Scaffold. Chem. Lett. 2004, 33, 932–933. [Google Scholar] [CrossRef]
- Shimono, S.; Takahashi, H.; Sakai, N.; Tamura, R.; Ikuma, N.; Yamauchi, J. Use of Cyclotriphosphazene as a Molecular Scaffold for Building Chiral Multispin Systems. Mol. Cryst. Liq. Cryst. 2005, 440, 37–52. [Google Scholar] [CrossRef]
- Fidan, I.; Önal, E.; Yerli, Y.; Luneau, D.; Ahsen, V.; Hirel, C. Synthethic Acces to a Pure Polyradical Architecture: Nucleophilic Insertion of Nitronyl Nitroxide on a Cyclotriphosphazene Scaffold. ChemPlusChem 2017, 82, 1384–1389. [Google Scholar] [CrossRef]
- Fidan, I.; Önal, E.; Yerli, Y.; Luneau, D.; Ahsen, V.; Hirel, C. Synthesis and Straightforward Quantification Methods of Imino Nitroxide-Based Hexaradical Architecture on a Cyclotriphosphazene Scaffold. Inorg. Chem. 2016, 55, 11447–11453. [Google Scholar] [CrossRef]
- Lloveras, V.; Badetti, E.; Wurst, K.; Vidal-Gancedo, J. Synthesis, X-Ray Structure, Magnetic Properties, and a Study of Intra/Intermolecular Radical-Radical Interactions of a Triradical TEMPO Compound. ChemPhysChem 2015, 16, 3302–3307. [Google Scholar] [CrossRef]
- Badetti, E.; Lloveras, V.; Wurst, K.; Sebastián, R.M.; Caminade, A.-M.; Majoral, J.-P.; Veciana, J.; Vidal-Gancedo, J. Synthesis and Structural Characterization of a Dendrimer Model Compound Based on a Cyclotriphosphazene Core with TEMPO Radicals as Substituents. Org. Lett. 2013, 15, 3490–3493. [Google Scholar] [CrossRef]
- Badetti, E.; Lloveras, V.; Muñoz-Gómez, J.L.; Sebastián, R.M.; Caminade, A.M.; Majoral, J.P.; Veciana, J.; Vidal-Gancedo, J. Radical Dendrimers: A Family of Five Generations of Phosphorus Dendrimers Functionalized with TEMPO Radicals. Macromolecules 2014, 47, 7717–7724. [Google Scholar] [CrossRef] [Green Version]
- Rajca, A.; Wang, Y.; Boska, M.; Paletta, J.T.; Olankitwanit, A.; Swanson, M.A.; Mitchell, D.G.; Eaton, S.S.; Eaton, G.R.; Rajca, S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2012, 134, 15724–15727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niidome, T.; Gokuden, R.; Watanabe, K.; Mori, T.; Naganuma, T.; Utsumi, H.; Ichikawa, K.; Katayama, Y. Nitroxyl radicals-modified dendritic poly(l-lysine) as a contrast agent for Overhauser-enhanced MRI. J. Biomater. Sci. Polym. Ed. 2014, 25, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.A.; Al-Abd, A.M. Thermoresponsive dendrimers based on oligoethylene glycols: Design, synthesis and cytotoxic activity against MCF-7 breast cancer cells. Eur. J. Med. Chem. 2013, 69, 848–854. [Google Scholar] [CrossRef]
- Wang, L.; Kiemle, D.J.; Boyle, C.J.; Connors, E.L.; Gitsov, I. “Click” Synthesis of Intrinsically Hydrophilic Dendrons and Dendrimers Containing Metal Binding Moieties at Each Branching Unit. Macromolecules 2014, 47, 2199–2213. [Google Scholar] [CrossRef]
- Pulido, D.; Albericio, F.; Royo, M. Controlling Multivalency and Multimodality: Up to Pentamodal Dendritic Platforms Based on Diethylenetriaminepentaacetic Acid Cores. Org. Lett. 2014, 16, 1318–1321. [Google Scholar] [CrossRef]
- Simón-Gracia, L.; Pulido, D.; Sevrin, C.; Grandfils, C.; Albericio, F.; Royo, M. Biocompatible, multifunctional, and well-defined OEG-based dendritic platforms for biomedical applications. Org. Biomol. Chem. 2013, 11, 4109–4121. [Google Scholar] [CrossRef] [PubMed]
- Kunz, T.K.; Wolf, M.O. Electrodeposition and properties of TEMPO functionalized polythiophene thin films. Polym. Chem. 2011, 2, 640–644. [Google Scholar] [CrossRef]
- Badetti, E.; Caminade, A.-M.; Majoral, J.-P.; Moreno-Mañas, M.; Sebastián, R.M. Palladium(0) Nanoparticles Stabilized by Phosphorus Dendrimers Containing Coordinating 15-Membered Triolefinic Macrocycles in Periphery. Langmuir 2008, 24, 2090–2101. [Google Scholar] [CrossRef]
- Blais, J.-C.; Turrin, C.-O.; Caminade, A.-M.; Majoral, J.-P. MALDI TOF Mass Spectrometry for the Characterization of Phosphorus-Containing Dendrimers. Scope and Limitations. Anal. Chem. 2000, 72, 5097–5105. [Google Scholar] [CrossRef]
- Hudson, A.; Luckhurst, G.R. Electron resonance spectrum of a triradical. Mol. Phys. 1967, 13, 409–416. [Google Scholar] [CrossRef]
- Likhtenstein, G.I. Spin Labeling Methods in Molecular Biology; Wiley: New York, NY, USA, 1976. [Google Scholar]
- Likhtenstein, G.I. Biophysical Labeling Methods in Molecular Biology; Cambridge University Press: New York, NY, USA, 1993. [Google Scholar]
- Lloveras, V.; Badetti, E.; Wurst, K.; Chechik, V.; Veciana, J.; Vidal-Gancedo, J. Magnetic and Electrochemical Properties of a Diradical TEMPO-substituted Disulfide in Solution, in a Crystal and anchored on Au(111) forming a SAM. Chem. Eur. J. 2016, 22, 1805–1815. [Google Scholar] [CrossRef] [Green Version]
- Afzal, V.; Brasch, R.C.; Nitecki, D.E.; Wolff, S. Nitroxyl spin label contrast enhancers for magnetic resonance imaging. Studies of acute toxicity and mutagenesis. Investig. Radiol. 1984, 19, 549–552. [Google Scholar] [CrossRef]
- Samuni, Y.; Gamson, J.; Samuni, A.; Yamada, K.; Russo, A.; Krishna, M.C.; Mitchell, J.B. Factors influencing nitroxide reduction and cytotoxicity in vitro. Antioxid. Redox Signal. 2004, 6, 587–595. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dendrimer | Area (Double Integral) Spectra at 300 K a | Ratio G1/G0 | Area (Double Integral) Spectra at 120 K a | Ratio G1/G0 |
|---|---|---|---|---|
| G0-OEG-PROXYL | 1.829 × 105 | 3.96 | 1.231 × 106 | 4.08 |
| G1-OEG-PROXYL | 7.246 × 105 | 5.028 × 106 |
| Compound | r17T (mM−1s−1) per Molecule | r17T (mM−1s−1) per Unit of Radical | r27T (mM−1s−1) per Molecule | r27T (mM−1s−1) per Unit of Radical |
|---|---|---|---|---|
| 3-carboxy PROXYL | 0.18 | 0.18 | 0.20 | 0.20 |
| G0-OEG-PROXYL | 0.91 | 0.18 | 0.95 | 0.19 |
| G1-OEG-PROXYL | 3.39 | 0.17 | 4.02 | 0.19 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Lloveras, V.; Pulido, D.; Liko, F.; Pinto, L.F.; Albericio, F.; Royo, M.; Vidal-Gancedo, J. Radical Dendrimers Based on Biocompatible Oligoethylene Glycol Dendrimers as Contrast Agents for MRI. Pharmaceutics 2020, 12, 772. https://doi.org/10.3390/pharmaceutics12080772
Zhang S, Lloveras V, Pulido D, Liko F, Pinto LF, Albericio F, Royo M, Vidal-Gancedo J. Radical Dendrimers Based on Biocompatible Oligoethylene Glycol Dendrimers as Contrast Agents for MRI. Pharmaceutics. 2020; 12(8):772. https://doi.org/10.3390/pharmaceutics12080772
Chicago/Turabian StyleZhang, Songbai, Vega Lloveras, Daniel Pulido, Flonja Liko, Luiz F. Pinto, Fernando Albericio, Miriam Royo, and José Vidal-Gancedo. 2020. "Radical Dendrimers Based on Biocompatible Oligoethylene Glycol Dendrimers as Contrast Agents for MRI" Pharmaceutics 12, no. 8: 772. https://doi.org/10.3390/pharmaceutics12080772