Formulation and Optimization of Avanafil Biodegradable Polymeric Nanoparticles: A Single-Dose Clinical Pharmacokinetic Evaluation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation of AVA Biodegradable Polymeric NPs
2.3. Investigating the Effect of Formulation and Process Parameters Using the Box–Behnken Design
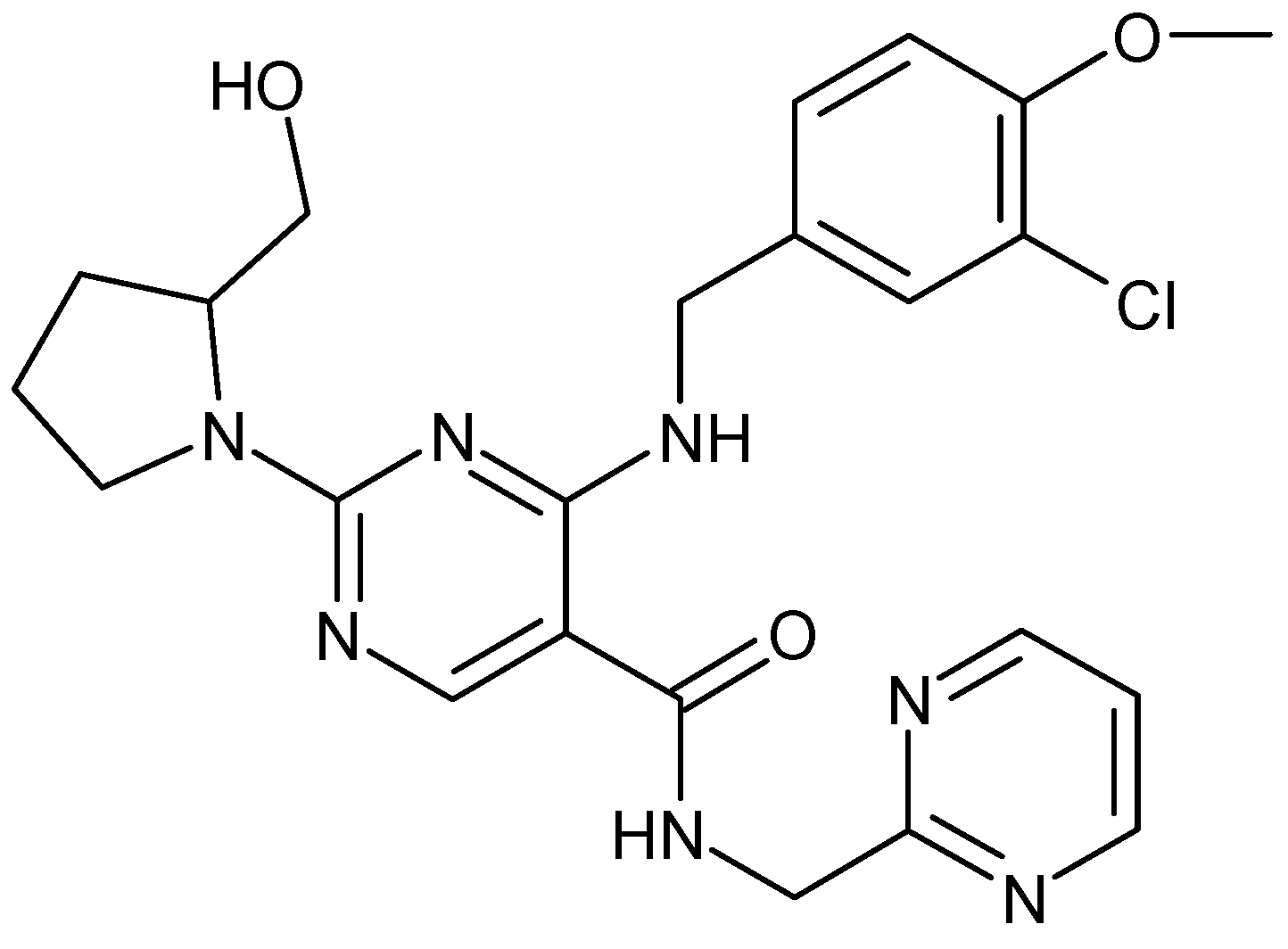
2.4. AVA-NPs Characterization
2.4.1. Particle Size Analysis and Zeta Potential
2.4.2. AVA-NPs EE%
2.5. Optimization of AVA PLGA Nanoparticles
2.6. Examination of Optimized AVA-NPs Morphology
2.7. Optimized AVA-NPs In-Vitro Permeation Study
2.8. Single Dose Clinical Pharmacokinetic Investigation of AVA-PLGA NPs in Healthy Human Volunteers
2.9. AVA Human Plasma Analysis
2.10. Pharmacokinetic Data Analysis
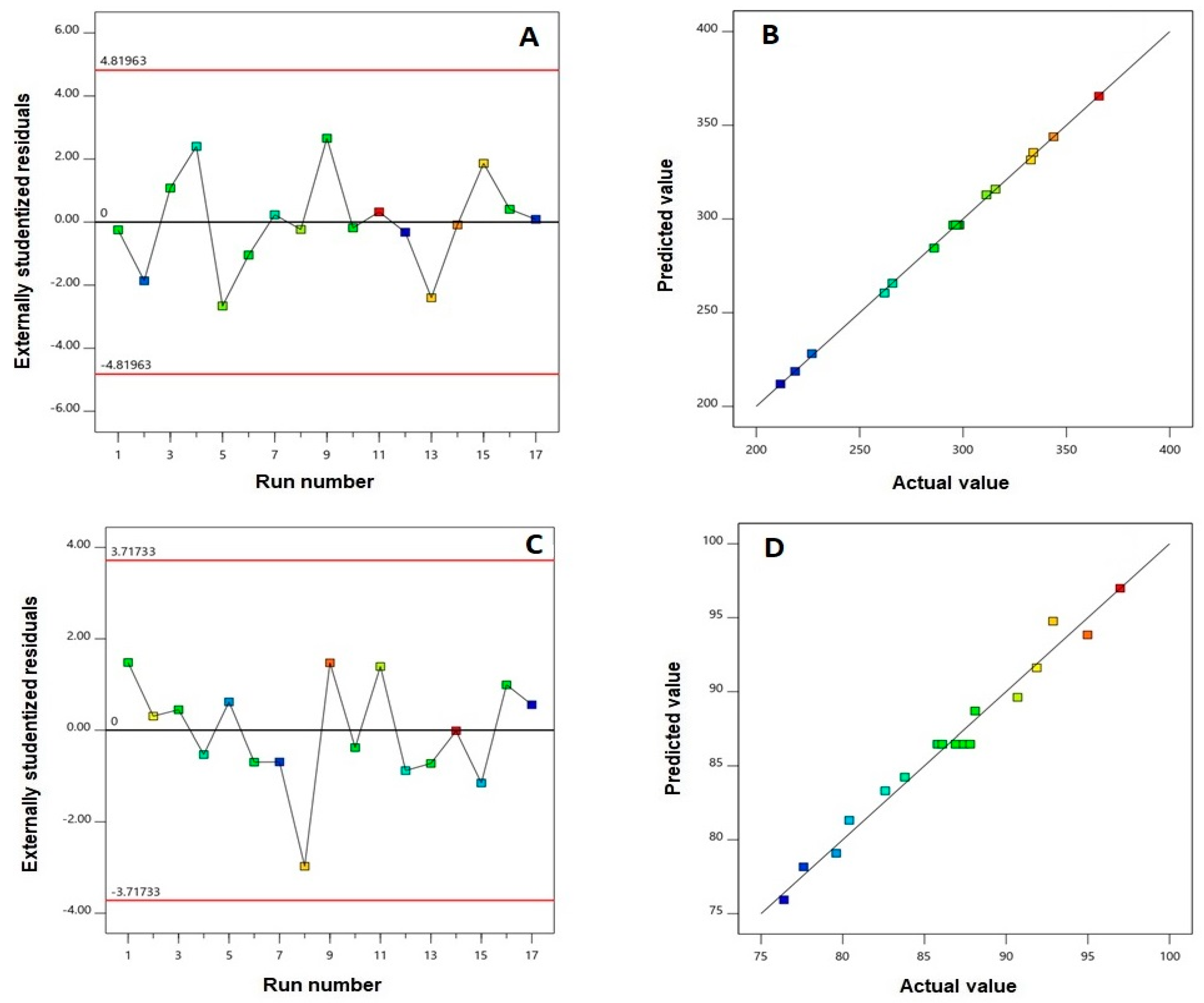
3. Results
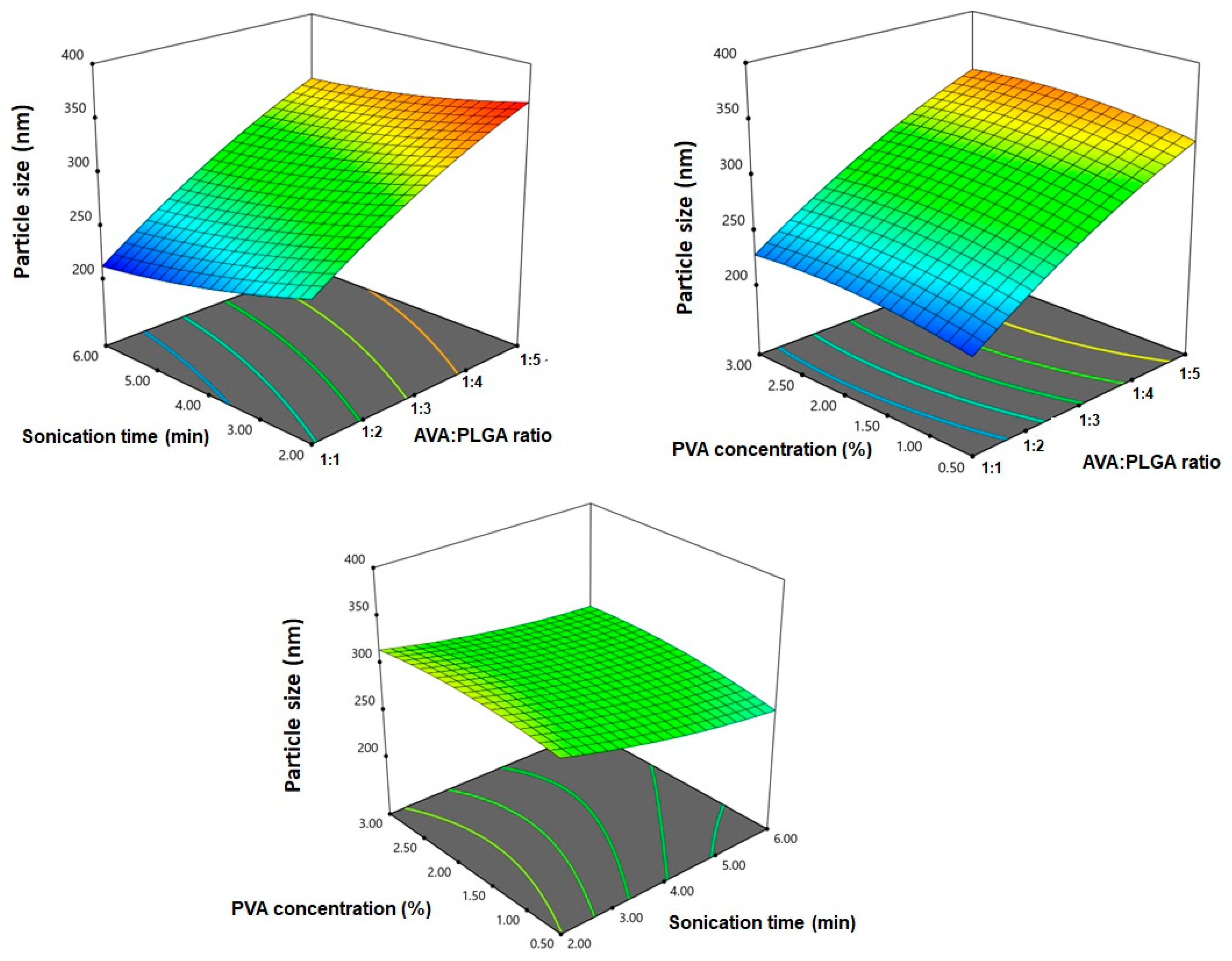
3.1. Effect of Variables on Particle Size
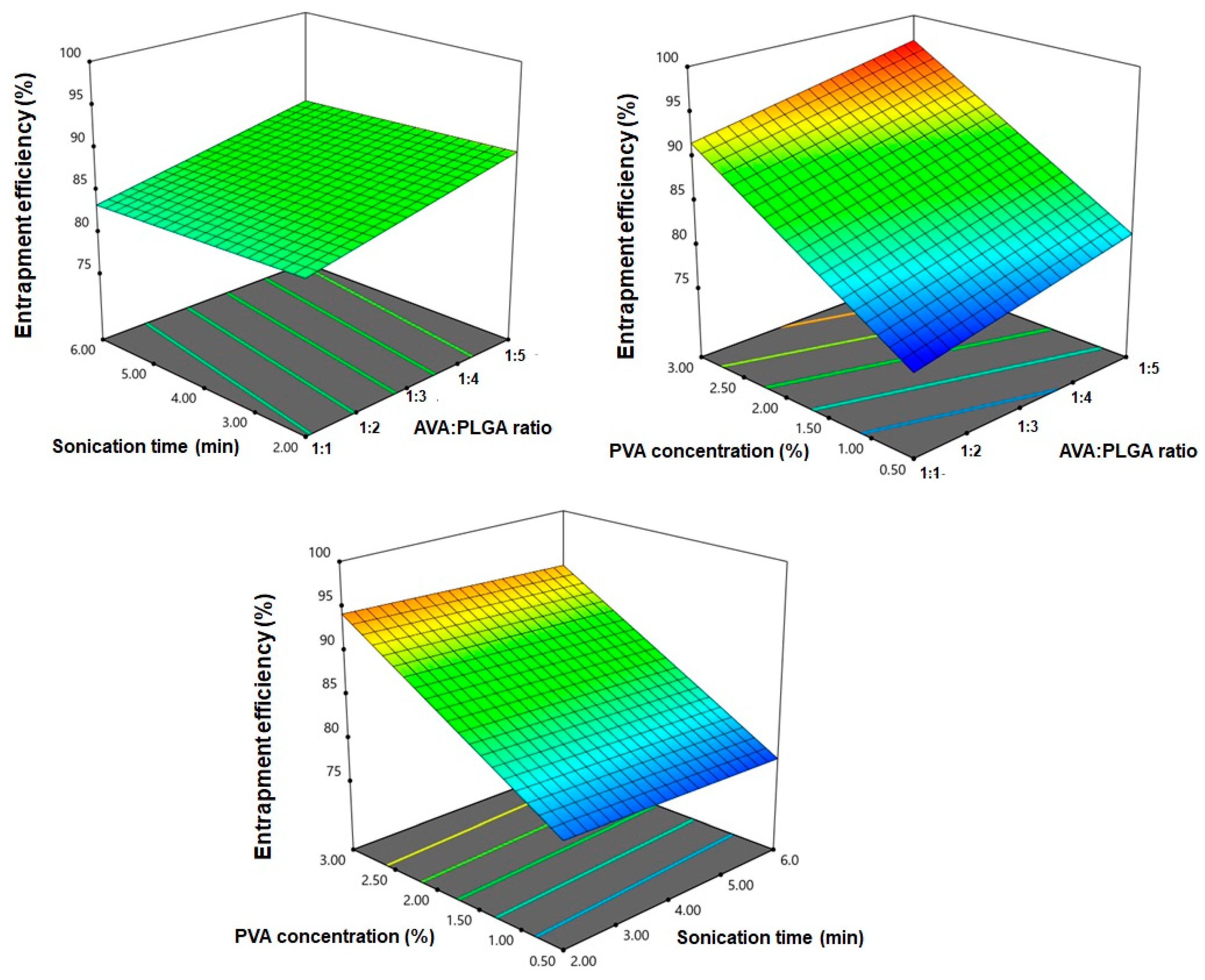
3.2. Effect of Design Variables on EE%
3.3. Optimization of AVA PLGA NPs
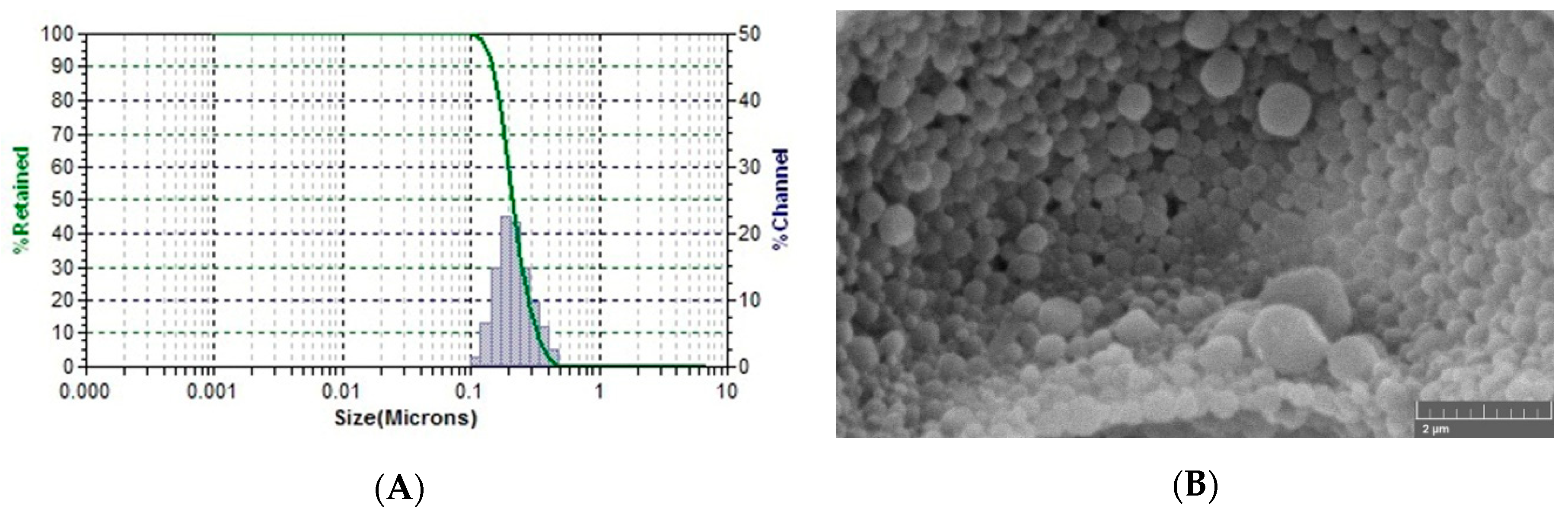
3.4. Optimized AVA-NPs SEM Morphology
3.5. In-Vitro Diffusion Study of Optimized AVA-NPs
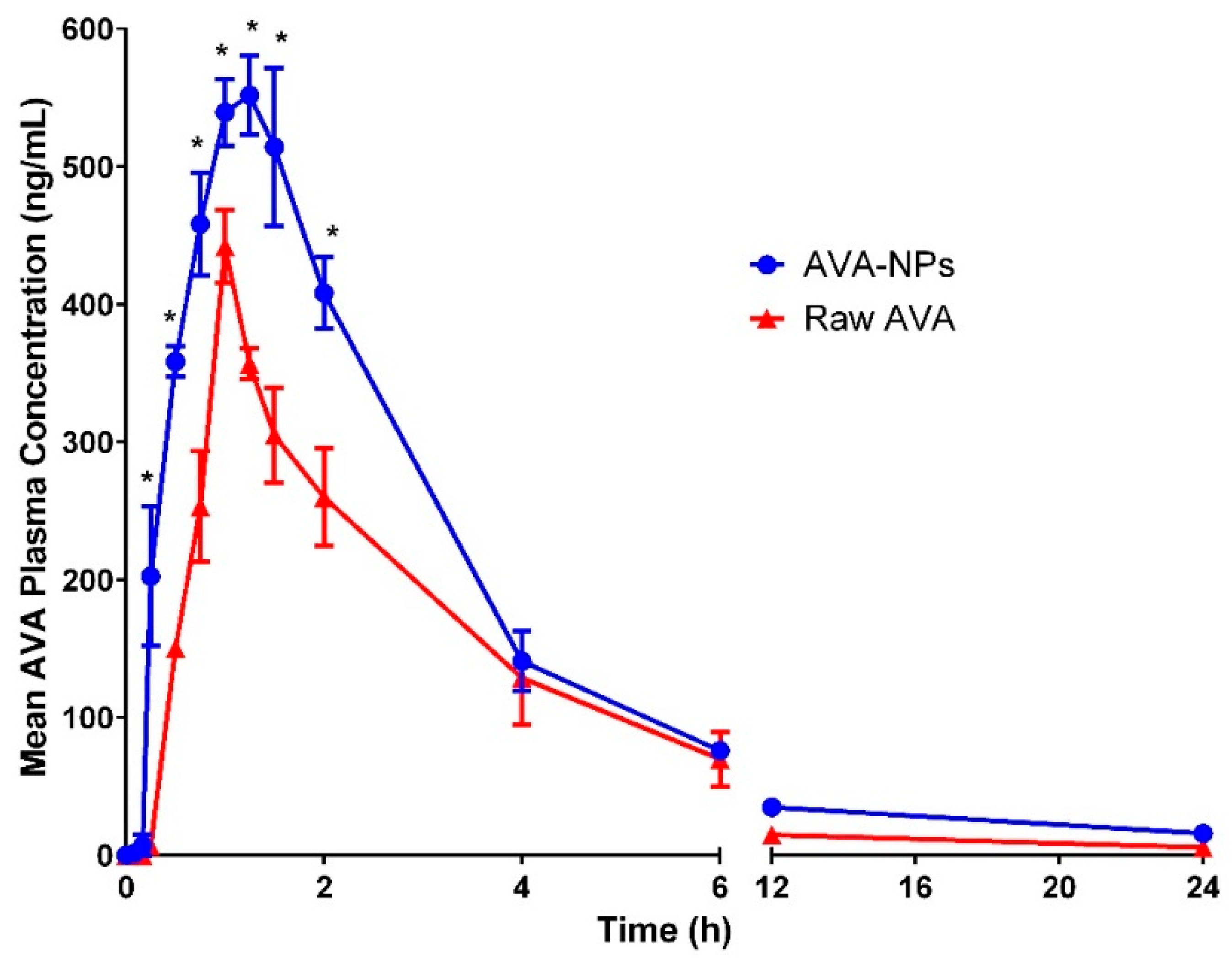
3.6. Clinical Investigation of AVA Formulation in Healthy Human Volunteers
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Food and Drug Administration. Drug Approval Package STENDRA (Avanafil) Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202276Orig1s000TOC.cfm (accessed on 30 December 2018).
- European Medicines Agency. CHMP Assessment Report International Non-Proprietary Name: Avanafil. Available online: www.ema.europa.eu (accessed on 6 October 2018).
- Gur, S.; Sikka, S.C.; Hellstrom, W.J. Novel phosphodiesterase-5 (PDE5) inhibitors in the alleviation of erectile dysfunction due to diabetes and ageing-induced oxidative stress. Expert Opin. Investig. Drugs 2008, 17, 855–864. [Google Scholar] [CrossRef]
- Sanford, M. Avanafil: A review of its use in patients with erectile dysfunction. Drugs Aging 2013, 30, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Moschos, M.M.; Nitoda, E. Pathophysiology of visual disorders induced by phosphodiesterase inhibitors in the treatment of erectile dysfunction. Drug Des. Dev. Ther. 2016, 10, 3407–3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, R.M.; Evans, J.D. Avanafil for treatment of erectile dysfunction: Review of its potential. Vasc. Health Risk Manag. 2012, 8, 517–523. [Google Scholar] [PubMed] [Green Version]
- Jung, J.; Choi, S.; Cho, S.H.; Ghim, J.L.; Hwang, A.; Kim, U.; Kim, B.S.; Koguchi, A.; Miyoshi, S.; Okabe, H.; et al. Tolerability and pharmacokinetics of avanafil, a phosphodiesterase type 5 inhibitor: A single- and multiple-dose, double-blind, randomized, placebo-controlled, dose-escalation study in healthy Korean male volunteers. Clin. Ther. 2010, 32, 1178–1187. [Google Scholar] [CrossRef]
- Katz, E.G.; Tan, R.B.; Rittenberg, D.; Hellstrom, W.J. Avanafil for erectile dysfunction in elderly and younger adults: Differential pharmacology and clinical utility. Ther. Clin. Risk Manag. 2014, 10, 701–711. [Google Scholar]
- Wan, F.; Maltesen, M.J.; Andersen, S.K.; Bjerregaard, S.; Foged, C.; Rantanen, J.; Yang, M. One-Step Production of Protein-Loaded PLGA Microparticles via Spray Drying Using 3-Fluid Nozzle. Pharm. Res. 2014, 31, 1967–1977. [Google Scholar] [CrossRef]
- Ando, S.; Putnam, D.; Pack, D.W.; Langer, R. PLGA microspheres containing plasmid DNA: Preservation of supercoiled DNA via cryopreparation and carbohydrate stabilization. J. Pharm. Sci. 1999, 88, 126–130. [Google Scholar] [CrossRef]
- Amir Kalvanagh, P.; Ebtekara, M.; Kokhaei, P.; Soleimanjahi, H. Preparation and Characterization of PLGA Nanoparticles Containing Plasmid DNA Encoding Human IFN-lambda-1/IL-29. Iran. J. Pharm. Res. 2019, 18, 156–167. [Google Scholar]
- Ahmed, O.A.; Hussein, A.K.; Mady, F.M. Optimisation of microstructured biodegradable finasteride formulation for depot parenteral application. J. Microencapsul. 2016, 33, 229–238. [Google Scholar] [CrossRef]
- Yoo, H.S.; Lee, K.H.; Oh, J.E.; Park, T.G. In vitro and in vivo anti-tumor activities of nanoparticles based on doxorubicin-PLGA conjugates. J. Control Release 2000, 68, 419–431. [Google Scholar] [CrossRef]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Kim, C.S.; Saylor, D.M.; Koo, D. Polymer degradation and drug delivery in PLGA-based drug–polymer applications: A review of experiments and theories. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 1692–1716. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.V.; Kala, M.; Nivsarkar, M. Formulation and optimization of doxorubicin loaded polymeric nanoparticles using Box-Behnken design: Ex-vivo stability and in-vitro activity. Eur. J. Pharm. Sci. 2017, 100, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Bodmeier, R.; McGinity, J.W. The preparation and evaluation of drug-containing poly(dl-lactide) microspheres formed by the solvent evaporation method. Pharm. Res. 1987, 4, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, H.; Davis, S.S.; O’Hagan, D.T. The preparation and characterization of poly(lactide-co-glycolide) microparticles. II. The entrapment of a model protein using a (water-in-oil)-in-water emulsion solvent evaporation technique. Pharm. Res. 1993, 10, 362–368. [Google Scholar] [CrossRef]
- Ogawa, Y.; Yamamoto, M.; Okada, H.; Yashiki, T.; Shimamoto, T. A New Technique to Efficiently Entrap Leuprolide Acetate into Microcapsules of Polylactic Acid or Copoly(Lactic/Glycolic) Acid. Chem. Pharm. Bull. 1988, 36, 1095–1103. [Google Scholar] [CrossRef]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control Release 2004, 99, 271–280. [Google Scholar] [CrossRef]
- Tewes, F.; Munnier, E.; Antoon, B.; Ngaboni Okassa, L.; Cohen-Jonathan, S.; Marchais, H.; Douziech-Eyrolles, L.; Soucé, M.; Dubois, P.; Chourpa, I. Comparative study of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur. J. Pharm. Biopharm. 2007, 66, 488–492. [Google Scholar] [CrossRef]
- El-Say, K.M.; Ahmed, O.A.A.; Mohamed, A.I.; Safo, M.K.; Omar, A.M. Zein-alpha lipoic acid-loaded nanoparticles to enhance the oral bioavailability of dapoxetine: Optimization and clinical pharmacokinetic evaluation. Int. J. Nanomed. 2019, 14, 7461–7473. [Google Scholar] [CrossRef] [Green Version]
- Fahmy UA, A.B. Stability indicating HPLC method for analysis of avanafil using diode array detector. Int. J. Adv. Pharm. Biol. Chem. 2016, 5, 59–64. [Google Scholar]
- Ahmed, O.A.A. Development and single dose clinical pharmacokinetics investigation of novel zein assisted-alpha lipoic acid nanoencapsulation of vardenafil. Sci. Rep. 2018, 8, 15802. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahallawi, A.M.; Abdelbary, A.A.; Aburahma, M.H. Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam. Int. J. Pharm. 2015, 485, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Némati, F.; Dubernet, C.; Fessi, H.; Colin de Verdière, A.; Poupon, M.F.F.; Puisieux, F.; Couvreur, P.; De Verdière, A.C.; Poupon, M.F.F.; Puisieux, F.; et al. Reversion of multidrug resistance using nanoparticles in vitro: Influence of the nature of the polymer. Int. J. Pharm. 1996, 138, 237–246. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [Green Version]
- Coelho, J.F.; Ferreira, P.C.; Alves, P.; Cordeiro, R.; Fonseca, A.C.; Góis, J.R.; Gil, M.H. Drug delivery systems: Advanced technologies potentially applicable in personalized treatments. EPMA J. 2010, 1, 164–209. [Google Scholar] [CrossRef] [Green Version]
- Hosny, K.M.K.M.; Ahmed, O.A.A.O.A.A.; Fahmy, U.A.; Alkhalidi, H.M.H.M. Nanovesicular systems loaded with a recently approved second generation type-5 phospodiesterase inhibitor (avanafil): I. Plackett-Burman screening and characterization. J. Drug Deliv. Sci. Technol. 2018, 43, 154–159. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.S.; Ahmed, O.A.A. Optimized nano-transfersomal films for enhanced sildenafil citrate transdermal delivery: Ex vivo and in vivo evaluation. Drug Des. Dev. Ther. 2016, 10, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Koopaei, M.N.; Khoshayand, M.R.; Mostafavi, S.H.; Amini, M.; Khorramizadeh, M.R.; Tehrani, M.J.; Atyabi, F.; Dinarvand, R. Docetaxel loaded PEG-PLGA nanoparticles: Optimized drug loading, in-vitro cytotoxicity and in-vivo antitumor effect. Iran. J. Pharm. Res. 2014, 13, 819–834. [Google Scholar]
- Zhang, K.; Tang, X.; Zhang, J.; Lu, W.; Lin, X.; Zhang, Y.; Tian, B.; Yang, H.; He, H. PEG-PLGA copolymers: Their structure and structure-influenced drug delivery applications. J. Control Release 2014, 183, 77–86. [Google Scholar] [CrossRef]
- Haggag, Y.; Abdel-Wahab, Y.; Ojo, O.; Osman, M.; El-Gizawy, S.; El-Tanani, M.; Faheem, A.; McCarron, P. Preparation and in vivo evaluation of insulin-loaded biodegradable nanoparticles prepared from diblock copolymers of PLGA and PEG. Int. J. Pharm. 2016, 499, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Jain, S.K. Formulation and optimization of temozolomide nanoparticles by 3 factor 2 level factorial design. Biomatter 2013, 3, e25102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Madan, P.; Lin, S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2015, 11, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Madani, F.; Esnaashari, S.S.; Mujokoro, B.; Dorkoosh, F.; Khosravani, M.; Adabi, M. Investigation of effective parameters on size of paclitaxel loaded PLGA nanoparticles. Adv. Pharm. Bull. 2018, 8, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweers, M.L.T.; Grijpma, D.W.; Engbers, G.H.M.; Feijen, J. The Preparation of Monodisperse Biodegradable Polyester Nanoparticles with a Controlled Size. J. Biomed. Mater. Res. Part B Appl. Biomater. 2003, 66, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Duxfield, L.; Sultana, R.; Wang, R.; Englebretsen, V.; Deo, S.; Swift, S.; Rupenthal, I.; Al-Kassas, R. Development of gatifloxacin-loaded cationic polymeric nanoparticles for ocular drug delivery. Pharm. Dev. Technol. 2016, 21, 172–179. [Google Scholar] [CrossRef]
- Feng, S.; Huang, G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol®) from nanospheres of biodegradable polymers. J. Control Release 2001, 71, 53–69. [Google Scholar] [CrossRef]
- Chansiri, G.; Lyons, R.T.; Patel, M.V.; Hem, S.L. Effect of surface charge on the stability of oil/water emulsions during steam sterilization. J. Pharm. Sci. 1999, 88, 454–458. [Google Scholar] [CrossRef]
- Quellec, P.; Gref, R.; Perrin, L.; Dellacherie, E.; Sommer, F.; Verbavatz, J.M.; Alonso, M.J. Protein encapsulation within polyethylene glycol-coated nanospheres. I. Physicochemical characterization. J. Biomed. Mater. Res. 1998, 42, 45–54. [Google Scholar] [CrossRef]
- Labhasetwar, V.; Song, C.; Humphrey, W.; Shebuski, R.; Levy, R.J. Arterial uptake of biodegradable nanoparticles: Effect of surface modifications. J. Pharm. Sci. 1998, 87, 1229–1234. [Google Scholar] [CrossRef]
- Quintanar-Guerrero, D.; Fessi, H.; Allémann, E.; Doelker, E. Influence of stabilizing agents and preparative variables on the formation of poly(D,L-lactic acid) nanoparticles by an emulsification-diffusion technique. Int. J. Pharm. 1996, 143, 133–141. [Google Scholar] [CrossRef]
- Helgason, T.; Awad, T.S.; Kristbergsson, K.; McClements, D.J.; Weiss, J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J. Colloid Interface Sci. 2009, 334, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Salatin, S.; Barar, J.; Barzegar-Jalali, M.; Adibkia, K.; Kiafar, F.; Jelvehgari, M. Development of a nanoprecipitation method for the entrapment of a very water soluble drug into Eudragit RL nanoparticles. Res. Pharm. Sci. 2017, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Halayqa, M.; Domańska, U. PLGA biodegradable nanoparticles containing perphenazine or chlorpromazine hydrochloride: Effect of formulation and release. Int. J. Mol. Sci. 2014, 15, 23909–23923. [Google Scholar] [CrossRef]
- Derman, S. Caffeic Acid Phenethyl Ester Loaded PLGA Nanoparticles: Effect of Various Process Parameters on Reaction Yield, Encapsulation Efficiency, and Particle Size. J. Nanomater. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alhakamy, N.; Ahmed, O.; Aldawsari, H.; Alfaifi, M.; Eid, B.; Abdel-Naim, A.; Fahmy, U. Encapsulation of Lovastatin in Zein Nanoparticles Exhibits Enhanced Apoptotic Activity in HepG2 Cells. Int. J. Mol. Sci. 2019, 20, 5788. [Google Scholar] [CrossRef] [Green Version]
- Alzubaidi, A.F.A.; El-Helw, A.-R.M.; Ahmed, T.A.; Ahmed, O.A.A. The use of experimental design in the optimization of risperidone biodegradable nanoparticles: In vitro and in vivo study. Artif. Cells Nanomed. Biotechnol. 2017, 45, 313–320. [Google Scholar] [CrossRef]
- Fahmy, U.A.; Badr-Eldin, S.M.; Ahmed, O.A.A.; Aldawsari, H.M.; Tima, S.; Asfour, H.Z.; Al-Rabia, M.W.; Negm, A.A.; Sultan, M.H.; Madkhali, O.A.A.; et al. Intranasal niosomal in situ gel as a promising approach for enhancing flibanserin bioavailability and brain delivery: In vitro optimization and ex vivo/in vivo evaluation. Pharmaceutics 2020, 12, 485. [Google Scholar] [CrossRef]
- Al-Gethmy, H.A.; Fahmy, U.A.; Alhakamy, N.A.; Ahmed, O.A.A.; El-Say, K.M. Optimization of the factors affecting the absorption of vardenafil from oral disintegrating tablets: A clinical pharmacokinetic investigation. Pharmaceutics 2019, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Alhakamy, N.A.; Fahmy, U.A.; Badr-Eldin, S.M.; Ahmed, O.A.A.; Asfour, H.Z.; Aldawsari, H.M.; Algandaby, M.M.; Eid, B.G.; Abdel-Naim, A.B.; Awan, Z.A.; et al. Optimized icariin phytosomes exhibit enhanced cytotoxicity and apoptosis-inducing activities in ovarian cancer cells. Pharmaceutics 2020, 12, 346. [Google Scholar] [CrossRef]
- Fonseca, C.; Simões, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control Release 2002, 83, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.L.; Zhu, K.J. Bioadhesive fluorescent microspheres as visible carriers for local delivery of drugs. I: Preparation and characterization of insulin-loaded PCEFB/PLGA microspheres. J. Microencapsul. 2002, 19, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Tao, Q.; Zou, Y.; Zhang, F.; Guo, M.; Wang, Y.; Wang, H.; Zhou, Q.; Yu, S. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: Characterizations and mechanisms. J. Agric. Food Chem. 2011, 59, 9280–9289. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Mishra, G.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur. J. Pharm. Sci. 2009, 36, 580–590. [Google Scholar] [CrossRef]
- Yin, Y.S.; Chen, D.W.; Qiao, M.X.; Lu, Z.; Hu, H.Y. Preparation and evaluation of lectin-conjugated PLGA nanoparticles for oral delivery of thymopentin. J. Control Release 2006, 116, 337–345. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Levels | ||
|---|---|---|---|
| (−1) | (0) | (+1) | |
| X1: AVA:PLGA (w/w) | 1:1 | 1:3 | 1:5 |
| X2: Sonication time (min) | 2 | 4 | 6 |
| X3: PVA concentration (%) | 0.50 | 1.75 | 3.00 |
| Responses | Desirability constraints | ||
| Y1: Particle size (nm) | Minimize | ||
| Y2: EE (%) | Maximize | ||
| Experimental Run # | Independent Variables | Dependent Variables | |||
|---|---|---|---|---|---|
| AVA:PLGA (w/w) | Sonication Time (min) | PVA Concentration (%) | Particle Size (nm) ± SD | EE (%) ± SD | |
| 1 | 1:3 | 4.00 | 1.75 | 296.4 ± 3.34 | 87.8 ± 1.76 |
| 2 | 1:1 | 4.00 | 3.00 | 226.9 ± 4.99 | 91.9 ± 0.98 |
| 3 | 1:3 | 4.00 | 1.75 | 298.3 ± 2.43 | 86.9 ± 1.11 |
| 4 | 1:1 | 2.00 | 1.75 | 262.1 ± 3.15 | 83.8 ± 1.21 |
| 5 | 1:3 | 2.00 | 0.50 | 311.3 ± 4.67 | 79.6 ± 0.86 |
| 6 | 1:3 | 4.00 | 1.75 | 295.3 ± 2.73 | 85.8 ± 0.91 |
| 7 | 1:3 | 6.00 | 0.50 | 265.9 ± 3.92 | 77.6 ± 1.08 |
| 8 | 1:3 | 2.00 | 3.00 | 315.7 ± 3.65 | 92.9 ± 1.89 |
| 9 | 1:3 | 6.00 | 3.00 | 286.2 ± 4.66 | 94.9 ± 1.88 |
| 10 | 1:3 | 4.00 | 1.75 | 296.5 ± 3.11 | 86.1 ± 1.45 |
| 11 | 1:5 | 2.00 | 1.75 | 365.8 ± 4.98 | 90.7 ± 1.99 |
| 12 | 1:1 | 6.00 | 1.75 | 211.7 ± 2.87 | 82.6 ± 2.14 |
| 13 | 1:5 | 6.00 | 1.75 | 334.1 ± 5.34 | 88.1 ± 1.77 |
| 14 | 1:5 | 4.00 | 3.00 | 343.8 ± 4.98 | 96.9 ± 1.49 |
| 15 | 1:5 | 4.00 | 0.50 | 332.8 ± 4.66 | 80.4 ± 2.11 |
| 16 | 1:3 | 4.00 | 1.75 | 297.4 ± 3.56 | 87.4 ± 2.31 |
| 17 | 1:1 | 4.00 | 0.50 | 218.8 ± 3.39 | 76.4 ± 1.29 |
| Responses | Model | Sequential p-Value | Lack of Fit p-Value | R2 | Adjusted R2 | Predicted R2 | Adequate Precision | Significant Terms |
|---|---|---|---|---|---|---|---|---|
| Y1: Particle size (nm) | Quadratic | <0.0001 | 0.1394 | 0.9994 | 0.9987 | 0.9931 | 125.69 | X1, X2, X3, X1X2, X2X3, X12, X22, X32 |
| Y2: EE (%) | Linear | <0.0001 | 0.3808 | 0.9783 | 0.9733 | 0.9604 | 44.73 | X1, X3 |
| Variables | X1: AVA:PLGA (w:w) Ratio | X2: Sonication Time (min) | X3: PVA Concentration (%) |
|---|---|---|---|
| Optimum values | 1:1 | 6.00 | 3.00 |
| Predicted value | Observed value | Error % | |
| Particle size (nm) | 213.19 | 217.42 | 1.98 |
| EE (%) | 91.15 | 92.67 | 1.66 |
| Parameter | Raw AVA | AVA-NPs |
|---|---|---|
| ke | 0.12 ± 0.03 | 0.06 ± 0.01 |
| t1/2 | 6.05 ± 1.8 | 12.14 ± 3.81 |
| Tmax | 1 | 1.25 ± 0.25 |
| Cmax | 441.98 ± 26.7 | 576.3 ± 8.2 * |
| AUC 0-inf_obs | 1448.86 ± 166.2 | 2434.25 ± 179.22 * |
| MRT 0-inf_obs | 5.37 ± 0.95 | 9.24 ± 2.35 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldawsari, H.M.; Fahmy, U.A.; Abd-Allah, F.; Ahmed, O.A.A. Formulation and Optimization of Avanafil Biodegradable Polymeric Nanoparticles: A Single-Dose Clinical Pharmacokinetic Evaluation. Pharmaceutics 2020, 12, 596. https://doi.org/10.3390/pharmaceutics12060596
Aldawsari HM, Fahmy UA, Abd-Allah F, Ahmed OAA. Formulation and Optimization of Avanafil Biodegradable Polymeric Nanoparticles: A Single-Dose Clinical Pharmacokinetic Evaluation. Pharmaceutics. 2020; 12(6):596. https://doi.org/10.3390/pharmaceutics12060596
Chicago/Turabian StyleAldawsari, Hibah M., Usama A. Fahmy, Fathy Abd-Allah, and Osama A. A. Ahmed. 2020. "Formulation and Optimization of Avanafil Biodegradable Polymeric Nanoparticles: A Single-Dose Clinical Pharmacokinetic Evaluation" Pharmaceutics 12, no. 6: 596. https://doi.org/10.3390/pharmaceutics12060596