Transporter-Mediated Delivery of Small Molecule Drugs to the Brain: A Critical Mechanism That Can Advance Therapeutic Development for Ischemic Stroke


Abstract
:1. Introduction
2. The Blood-Brain Barrier and the Neurovascular Unit
2.1. Endothelial Cell Junctions
2.2. Endogenous BBB Transporters
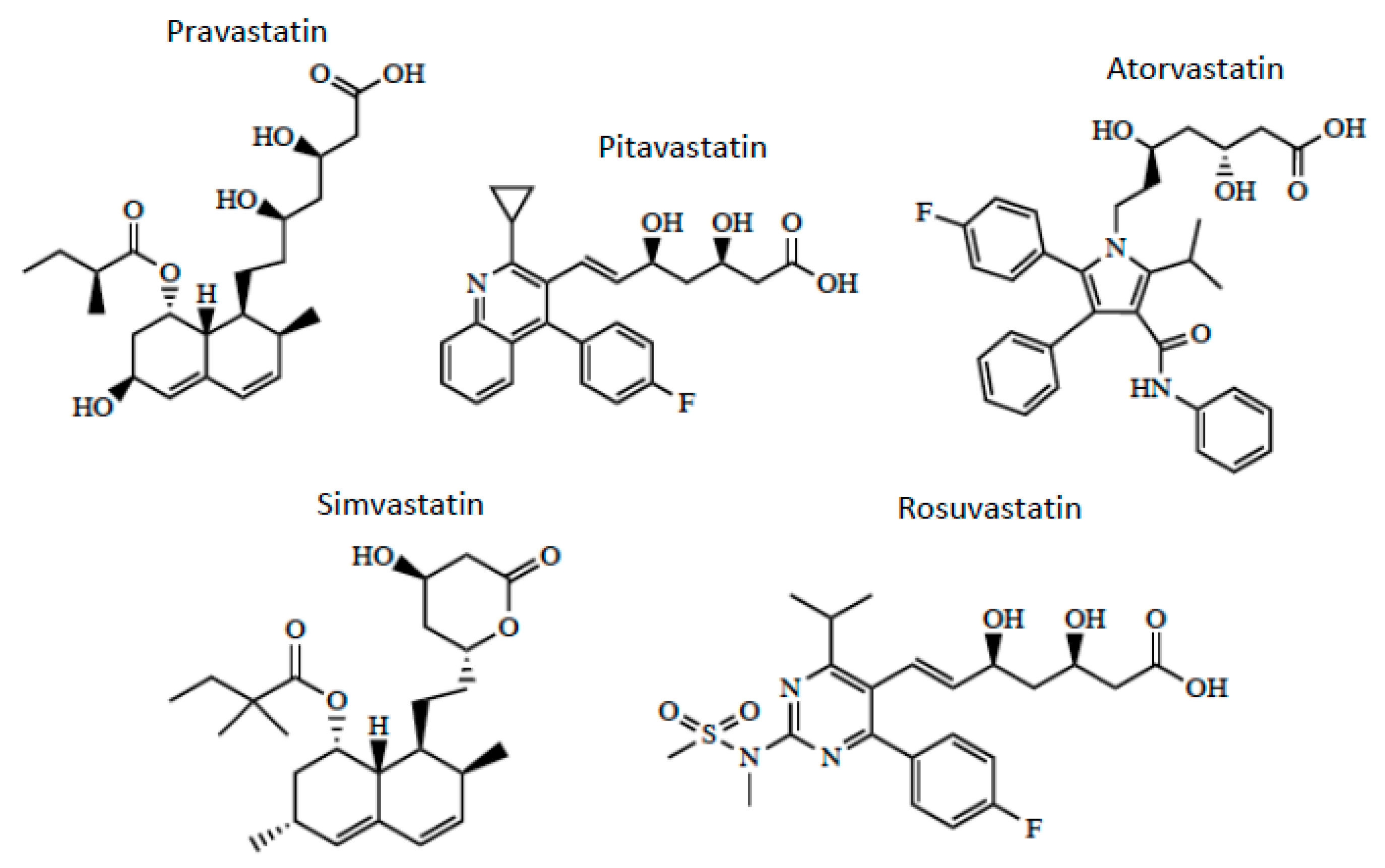
3. OATP/Oatp-Mediated Transport of Statins
4. Neuroprotective Properties of Statins in Stroke
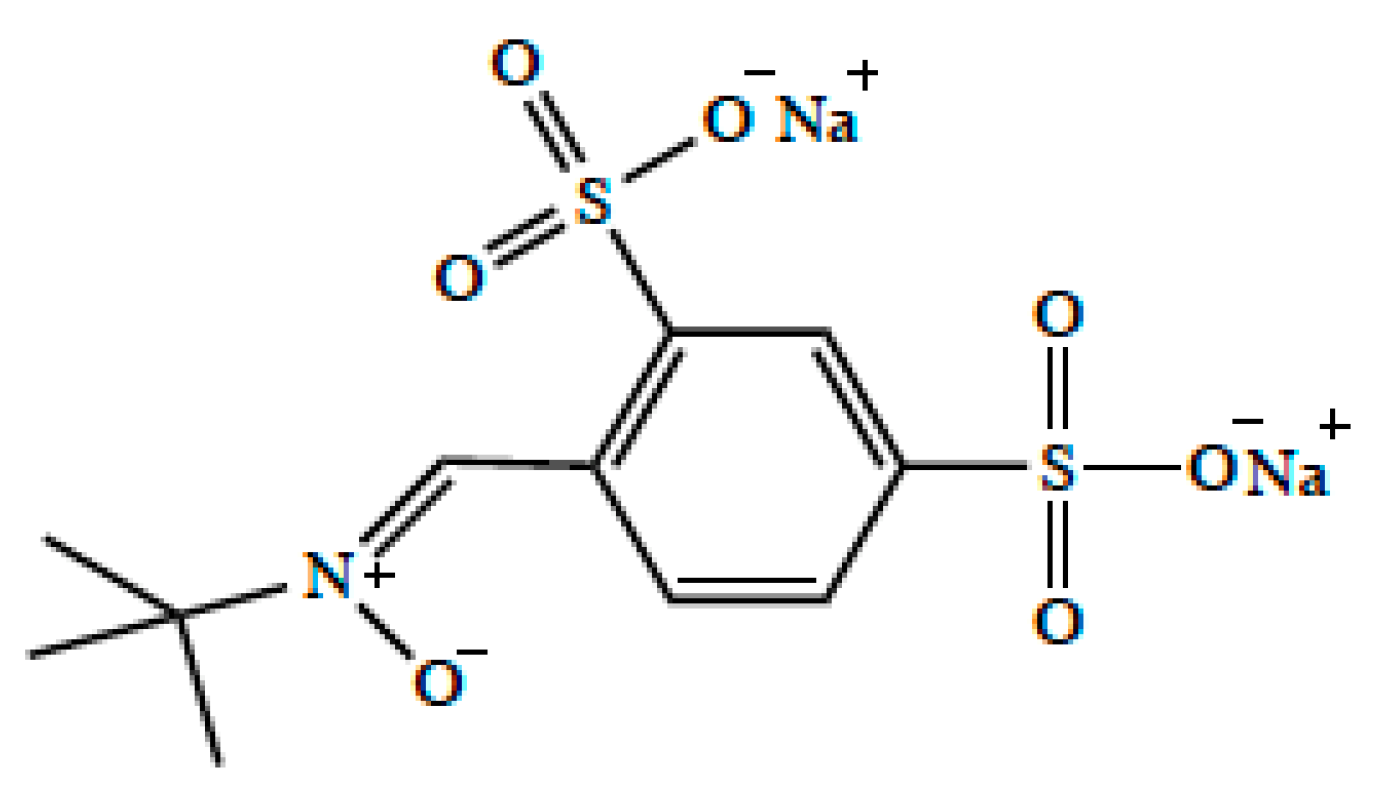
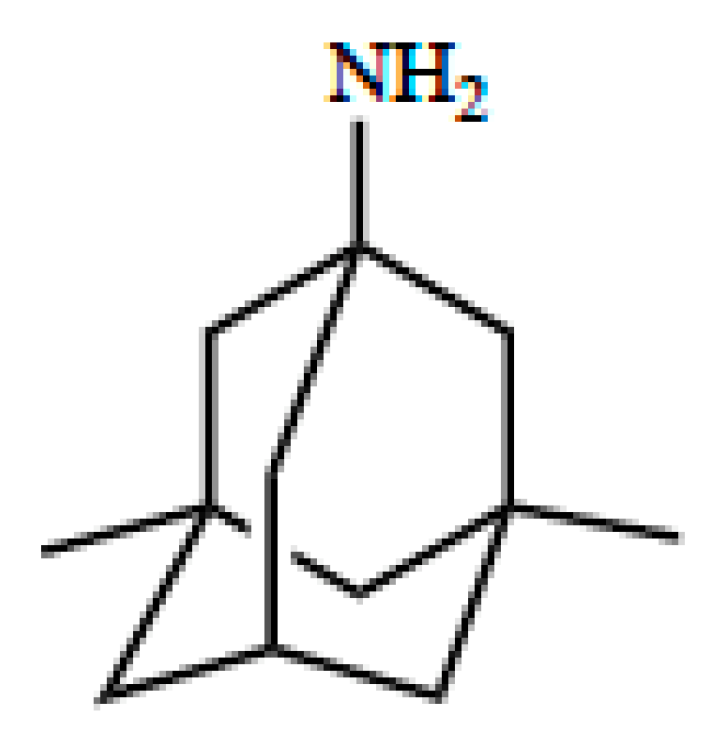
5. OCT/Oct-Mediated Transport of Memantine
6. Neuroprotective Properties of Memantine in Stroke
7. Conclusions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Levine, S.R.; Winn, H.R. Targeting ischemic penumbra: Part I—From pathophysiology to therapeutic strategy. J. Exp. Stroke Transl. Med. 2010, 3, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, N.W.; Campbell, B.C.V.; Oxley, T.J.; Chapot, R. Acute ischemic stroke: Time, penumbra, and reperfusion. Stroke 2014, 45, 640–644. [Google Scholar] [CrossRef] [Green Version]
- Brzica, H.; Abdullahi, W.; Ibbotson, K.; Ronaldson, P.T. Role of transporters in central nervous system drug delivery and blood-brain barrier protection: Relevance to treatment of stroke. J. Cent. Nerv. Syst. Dis. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Tymianski, M. Combining neuroprotection with endovascular treatment of acute stroke: Is there hope? Stroke 2017, 48, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.; Menon, B.K.; van Zwam, W.H.; Dippel, D.W.J.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.L.M.; van der Lugt, A.; de Miquel, M.A.; et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised Trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 h after stroke with a mismatch between deficit and infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.W.; Marks, M.P.; Lansberg, M.G. Thrombectomy for stroke with selection by perfusion imaging. N. Engl. J. Med. 2018, 378, 1849–1850. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Konstas, A.-A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nour, M.; Scalzo, F.; Liebeskind, D.S. Ischemia-reperfusion injury in stroke. Interv. Neurol. 2013, 1, 185–199. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Logsdon, A.F.; Turner, R.C.; Rosen, C.L.; Huber, J.D. Aging, the metabolic syndrome, and ischemic stroke: Redefining the approach for studying the blood-brain barrier in a complex neurological disease. Adv. Pharmacol. 2014, 71, 411–449. [Google Scholar] [CrossRef]
- Zhao, Z.; Cheng, M.; Maples, K.R.; Ma, J.Y.; Buchan, A.M. NXY-059, a novel free radical trapping compound, reduces cortical infarction after permanent focal cerebral ischemia in the rat. Brain Res. 2001, 909, 46–50. [Google Scholar] [CrossRef]
- Sydserff, S.G.; Borelli, A.R.; Green, A.R.; Cross, A.J. Effect of NXY-059 on infarct volume after transient or permanent middle cerebral artery occlusion in the rat; Studies on dose, plasma concentration and therapeutic time window. Br. J. Pharmacol. 2002, 135, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.W.; Duffin, K.J.; Green, A.R.; Ridley, R.M. NXY-059, a free radical—Trapping agent, substantially lessens the functional disability resulting from cerebral ischemia in a primate species. Stroke 2001, 32, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culot, M.; Mysiorek, C.; Renftel, M.; Roussel, B.D.; Hommet, Y.; Vivien, D.; Cecchelli, R.; Fenart, L.; Berezowski, V.; Dehouck, M.-P.; et al. Cerebrovascular protection as a possible mechanism for the protective effects of nxy-059 in preclinical models: An in vitro study. Brain Res. 2009, 1294, 144–152. [Google Scholar] [CrossRef]
- Lees, K.R.; Sharma, A.K.; Barer, D.; Ford, G.A.; Kostulas, V.; Cheng, Y.F.; Odergren, T. Tolerability and pharmacokinetics of the nitrone NXY-059 in patients with acute stroke. Stroke 2001, 32, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Lees, K.R.; Barer, D.; Ford, G.A.; Hacke, W.; Kostulas, V.; Sharma, A.K.; Odergren, T.; SA-NXY-0004 investigators. Tolerability of NXY-059 at higher target concentrations in patients with acute stroke. Stroke 2003, 34, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Kågedal, M.; Nilsson, D.; Huledal, G.; Reinholdsson, I.; Cheng, Y.-F.; Asenblad, N.; Pekar, D.; Borgå, O. A study of organic acid transporter mediated pharmacokinetic interaction between NXY-059 and cefuroxime. J. Clin. Pharmacol. 2007, 47, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Davis, T.P. Targeting transporters: Promoting blood-brain barrier repair in response to oxidative stress injury. Brain Res. 2015, 1623, 39–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional expression of P-glycoprotein and organic anion transporting polypeptides at the blood-brain barrier: Understanding transport mechanisms for improved cns drug delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.E. Blood-brain barrier na transporters in ischemic stroke. Adv. Pharmacol. 2014, 71, 113–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Wallace, B.K.; Yuen, N.; Jenkins, D.P.; Wulff, H.; O’Donnell, M.E. Blood-brain barrier KCa3.1 channels: Evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke 2015, 46, 237–244. [Google Scholar] [CrossRef]
- Yuen, N.Y.; Chechneva, O.V.; Chen, Y.-J.; Tsai, Y.-C.; Little, L.K.; Dang, J.; Tancredi, D.J.; Conston, J.; Anderson, S.E.; O’Donnell, M.E. Exacerbated brain edema in a rat streptozotocin model of hyperglycemic ischemic stroke: Evidence for involvement of blood-brain barrier Na-K-Cl cotransport and Na/H exchange. J. Cereb. Blood Flow Metab. 2019, 39, 1678–1692. [Google Scholar] [CrossRef]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J. Pharmacol. Exp. Ther. 2009, 328, 487–495. [Google Scholar] [CrossRef]
- Shah, K.K.; Boreddy, P.R.; Abbruscato, T.J. Nicotine pre-exposure reduces stroke-induced glucose transporter-1 activity at the blood-brain barrier in mice. Fluids Barriers CNS 2015, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Spudich, A.; Kilic, E.; Xing, H.; Kilic, U.; Rentsch, K.M.; Wunderli-Allenspach, H.; Bassetti, C.L.; Hermann, D.M. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat. Neurosci. 2006, 9, 487–488. [Google Scholar] [CrossRef]
- Cen, J.; Liu, L.; Li, M.-S.; He, L.; Wang, L.-J.; Liu, Y.-Q.; Liu, M.; Ji, B.-S. Alteration in P-Glycoprotein at the blood-brain barrier in the early period of MCAO in rats. J. Pharm. Pharmacol. 2013, 65, 665–672. [Google Scholar] [CrossRef]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal changes in P-Glycoprotein levels in brain and peripheral tissues following ischemic stroke in rats. J. Exp. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Daneman, R. Formation and maintenance of the BBB. Mech. Dev. 2015, 138, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- DiNapoli, V.A.; Huber, J.D.; Houser, K.; Li, X.; Rosen, C.L. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 2008, 29, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Sakadžić, S.; Lee, J.; Boas, D.A.; Ayata, C. High-resolution in vivo optical imaging of stroke injury and repair. Brain Res. 2015, 1623, 174–192. [Google Scholar] [CrossRef] [Green Version]
- Herculano-Houzel, S. Scaling of brain metabolism with a fixed energy budget per neuron: Implications for neuronal activity, plasticity and evolution. PLoS ONE 2011, 6, 17514. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Ronaldson, P.T.; Davis, T.P. Hypoxic stress and inflammatory pain disrupt blood-brain barrier tight junctions: Implications for drug delivery to the central nervous system. AAPS J. 2017, 19, 910–920. [Google Scholar] [CrossRef]
- Dithmer, S.; Staat, C.; Müller, C.; Ku, M.-C.; Pohlmann, A.; Niendorf, T.; Gehne, N.; Fallier-Becker, P.; Kittel, Á.; Walter, F.R.; et al. Claudin peptidomimetics modulate tissue barriers for enhanced drug delivery. Ann. N. Y. Acad. Sci. 2017, 1397, 169–184. [Google Scholar] [CrossRef]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef]
- McCaffrey, G.; Seelbach, M.J.; Staatz, W.D.; Nametz, N.; Quigley, C.; Campos, C.R.; Brooks, T.A.; Davis, T.P. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J. Neurochem. 2008, 106, 2395–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, G.; Staatz, W.D.; Quigley, C.A.; Nametz, N.; Seelbach, M.J.; Campos, C.R.; Brooks, T.A.; Egleton, R.D.; Davis, T.P. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. J. Neurochem. 2007, 103, 2540–2555. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; McCaffrey, G.; Sanchez-Covarrubias, L.; Finch, J.D.; Demarco, K.M.; Quigley, C.E.; Davis, T.P.; Ronaldson, P.T. Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood-brain barrier during inflammatory pain. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H582–H593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarmann, A.; Deiss, A.; Prochaska, J.; Foerch, C.; Weksler, B.; Romero, I.; Couraud, P.-O.; Stoll, G.; Rieckmann, P.; Buttmann, M. Evaluation of soluble junctional adhesion Molecule-A as a biomarker of human brain endothelial barrier breakdown. PLoS ONE 2010, 5, e13568. [Google Scholar] [CrossRef]
- Wang, X.-S.; Fang, H.-L.; Chen, Y.; Liang, S.-S.; Zhu, Z.-G.; Zeng, Q.-Y.; Li, J.; Xu, H.-Q.; Shao, B.; He, J.-C.; et al. Idazoxan reduces blood-brain barrier damage during experimental autoimmune encephalomyelitis in mouse. Eur. J. Pharmacol. 2014, 736, 70–76. [Google Scholar] [CrossRef]
- Tornabene, E.; Helms, H.C.C.; Pedersen, S.F.; Brodin, B. Effects of oxygen-glucose deprivation (OGD) on barrier properties and mrna transcript levels of selected marker proteins in brain endothelial cells/astrocyte Co-Cultures. PLoS ONE 2019, 14, e0221103. [Google Scholar] [CrossRef]
- Alluri, H.; Grimsley, M.; Anasooya Shaji, C.; Varghese, K.P.; Zhang, S.L.; Peddaboina, C.; Robinson, B.; Beeram, M.R.; Huang, J.H.; Tharakan, B. Attenuation of blood-brain barrier breakdown and hyperpermeability by calpain inhibition. J. Biol. Chem. 2016, 291, 26958–26969. [Google Scholar] [CrossRef] [Green Version]
- Atallah, A.; Mhaouty-Kodja, S.; Grange-Messent, V. Chronic depletion of gonadal testosterone leads to blood-brain barrier dysfunction and inflammation in male mice. J. Cereb. Blood Flow Metab. 2017, 37, 3161–3175. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Lampugnani, M.G.; Dejana, E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 2007, 120, S1–S6. [Google Scholar] [CrossRef]
- Williams, M.J.; Lowrie, M.B.; Bennett, J.P.; Firth, J.A.; Clark, P. Cadherin-10 is a novel blood-brain barrier adhesion molecule in human and mouse. Brain Res. 2005, 1058, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Ziegler, N.; Godet, M.; Guilbert, T.; Liebner, S.; Couraud, P.-O. The Wnt/Planar cell polarity signaling pathway contributes to the integrity of tight junctions in brain endothelial cells. J. Cereb. Blood Flow Metab. 2014, 34, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Hombach-Klonisch, S.; Miller, D.W. Modulation of Wnt/β-catenin signaling promotes blood-brain barrier phenotype in cultured brain endothelial cells. Sci. Rep. 2019, 9, 19718. [Google Scholar] [CrossRef] [Green Version]
- Steiner, E.; Enzmann, G.U.; Lyck, R.; Lin, S.; Rüegg, M.A.; Kröger, S.; Engelhardt, B. The heparan sulfate proteoglycan agrin contributes to barrier properties of mouse brain endothelial cells by stabilizing adherens junctions. Cell Tissue Res. 2014, 358, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Willis, C.L.; Camire, R.B.; Brule, S.A.; Ray, D.E. Partial recovery of the damaged rat blood-brain barrier is mediated by adherens junction complexes, extracellular matrix remodeling and macrophage infiltration following focal astrocyte loss. Neuroscience 2013, 250, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the atp binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Polli, J.W.; Olson, K.L.; Chism, J.P.; John-Williams, L.S.; Yeager, R.L.; Woodard, S.M.; Otto, V.; Castellino, S.; Demby, V.E. An unexpected synergist role of p-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-Chloro-4-[(3-Fluorobenzyl)Oxy]Phenyl}-6-[5-({[2-(Methylsulfonyl)Ethyl]Amino}methyl)-2-Furyl]-4-quinazolinamine; GW572016). Drug Metab. Dispos. 2009, 37, 439–442. [Google Scholar] [CrossRef]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug resistance-associated proteins: Expression and function in the central nervous system. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronaldson, P.T.; Persidsky, Y.; Bendayan, R. Regulation of ABC membrane transporters in glial cells: Relevance to the pharmacotherapy of brain HIV-1 infection. Glia 2008, 56, 1711–1735. [Google Scholar] [CrossRef] [PubMed]
- Lötsch, J.; Schmidt, R.; Vetter, G.; Schmidt, H.; Niederberger, E.; Geisslinger, G.; Tegeder, I. Increased CNS uptake and enhanced antinociception of Morphine-6-Glucuronide in rats after inhibition of P-Glycoprotein. J. Neurochem. 2002, 83, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Jekerle, V.; Klinkhammer, W.; Scollard, D.A.; Breitbach, K.; Reilly, R.M.; Piquette-Miller, M.; Wiese, M. In vitro and in vivo evaluation of WK-X-34, a novel inhibitor of P-Glycoprotein and BCRP, using radio imaging techniques. Int. J. Cancer 2006, 119, 414–422. [Google Scholar] [CrossRef]
- Foran, E.; Kwon, D.Y.; Nofziger, J.H.; Arnold, E.S.; Hall, M.D.; Fischbeck, K.H.; Burnett, B.G. CNS uptake of bortezomib is enhanced by P-Glycoprotein inhibition: Implications for spinal muscular atrophy. Neurobiol. Dis. 2016, 88, 118–124. [Google Scholar] [CrossRef] [Green Version]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.M.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved brain penetration and antitumor efficacy of temozolomide by inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef]
- Laramy, J.K.; Kim, M.; Parrish, K.E.; Sarkaria, J.N.; Elmquist, W.F. Pharmacokinetic assessment of cooperative efflux of the multitargeted kinase inhibitor ponatinib across the blood-brain barrier. J. Pharmacol. Exp. Ther. 2018, 365, 249–261. [Google Scholar] [CrossRef]
- Kalvass, J.C.; Polli, J.W.; Bourdet, D.L.; Feng, B.; Huang, S.-M.; Liu, X.; Smith, Q.R.; Zhang, L.K.; Zamek-Gliszczynski, M.J. International transporter consortium. Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin. Pharmacol. Ther. 2013, 94, 80–94. [Google Scholar] [CrossRef]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting P-Glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [CrossRef]
- Cannon, R.E.; Peart, J.C.; Hawkins, B.T.; Campos, C.R.; Miller, D.S. Targeting Blood-brain barrier sphingolipid signaling reduces basal p-glycoprotein activity and improves drug delivery to the brain. Proc. Natl. Acad. Sci. USA 2012, 109, 15930–15935. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, T.A.; Campos, C.R.; Cannon, R.E.; Miller, D.S. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. J. Cereb. Blood Flow Metab. 2013, 33, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Davis, T.P. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, P.T.; Finch, J.D.; Demarco, K.M.; Quigley, C.E.; Davis, T.P. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J. Pharmacol. Exp. Ther. 2011, 336, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J.; Sanchez-Covarrubias, L.; Slosky, L.M.; Zhang, Y.; Laracuente, M.; Ronaldson, P.T. Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: Relevance to CNS drug delivery. J. Cereb. Blood Flow Metab. 2014, 34, 699–707. [Google Scholar] [CrossRef]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Statins and neuroprotection: A prescription to move the field forward. Ann. N. Y. Acad. Sci. 2010, 1199, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar]
- Bronger, H.; König, J.; Kopplow, K.; Steiner, H.-H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Brzica, H.; Ibbotson, K.; Davis, T.P.; Ronaldson, P.T. Bone morphogenetic Protein-9 increases the functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like Kinase-1 receptor. J. Cereb. Blood Flow Metab. 2017, 37, 2340–2345. [Google Scholar] [CrossRef]
- Lin, C.-J.; Tai, Y.; Huang, M.-T.; Tsai, Y.-F.; Hsu, H.-J.; Tzen, K.-Y.; Liou, H.-H. Cellular localization of the organic cation transporters, OCT1 and OCT2, in brain microvessel endothelial cells and its implication for MPTP transport across the blood-brain barrier and MPTP-induced dopaminergic toxicity in rodents. J. Neurochem. 2010, 114, 717–727. [Google Scholar] [CrossRef]
- Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.J.; Hu, T.; Wang, J. Polyspecific organic cation transporters and their impact on drug intracellular levels and pharmacodynamics. Pharmacol. Res. 2016, 111, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.C.; Short, J.L.; Nicolazzo, J.A. Memantine transport across the mouse blood-brain barrier is mediated by a cationic influx H+ antiporter. Mol. Pharm. 2013, 10, 4491–4498. [Google Scholar] [CrossRef] [PubMed]
- Huck, J.H.J.; Freyer, D.; Böttcher, C.; Mladinov, M.; Muselmann-Genschow, C.; Thielke, M.; Gladow, N.; Bloomquist, D.; Mergenthaler, P.; Priller, J. De novo expression of dopamine D2 receptors on microglia after stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1804–1811. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.S.; Ali, M.; Tabassum, H.; Parveen, S.; Parvez, S. Pramipexole prevents ischemic cell death via mitochondrial pathways in ischemic stroke. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardai, S.; Dobolyi, A.; Pál, G.; Skopál, J.; Pintér, N.; Lakatos, K.; Merkely, B.; Nagy, Z. Selegiline promotes NOTCH-JAGGED signaling in astrocytes of the peri-infarct region and improves the functional integrity of the neurovascular unit in a rat model of focal ischemia. Restor. Neurol. Neurosci. 2015, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bennet, L.; McGregor, A.L. Delayed varenicline administration reduces inflammation and improves forelimb use following experimental stroke. J. Stroke Cerebrovasc. Dis. 2017, 26, 2778–2787. [Google Scholar] [CrossRef]
- Brzica, H.; Abdullahi, W.; Reilly, B.G.; Ronaldson, P.T. Sex-specific differences in organic anion transporting polypeptide 1a4 (Oatp1a4) functional expression at the blood–brain barrier in sprague–dawley rats. Fluids Barriers CNS 2018, 15, 25. [Google Scholar] [CrossRef]
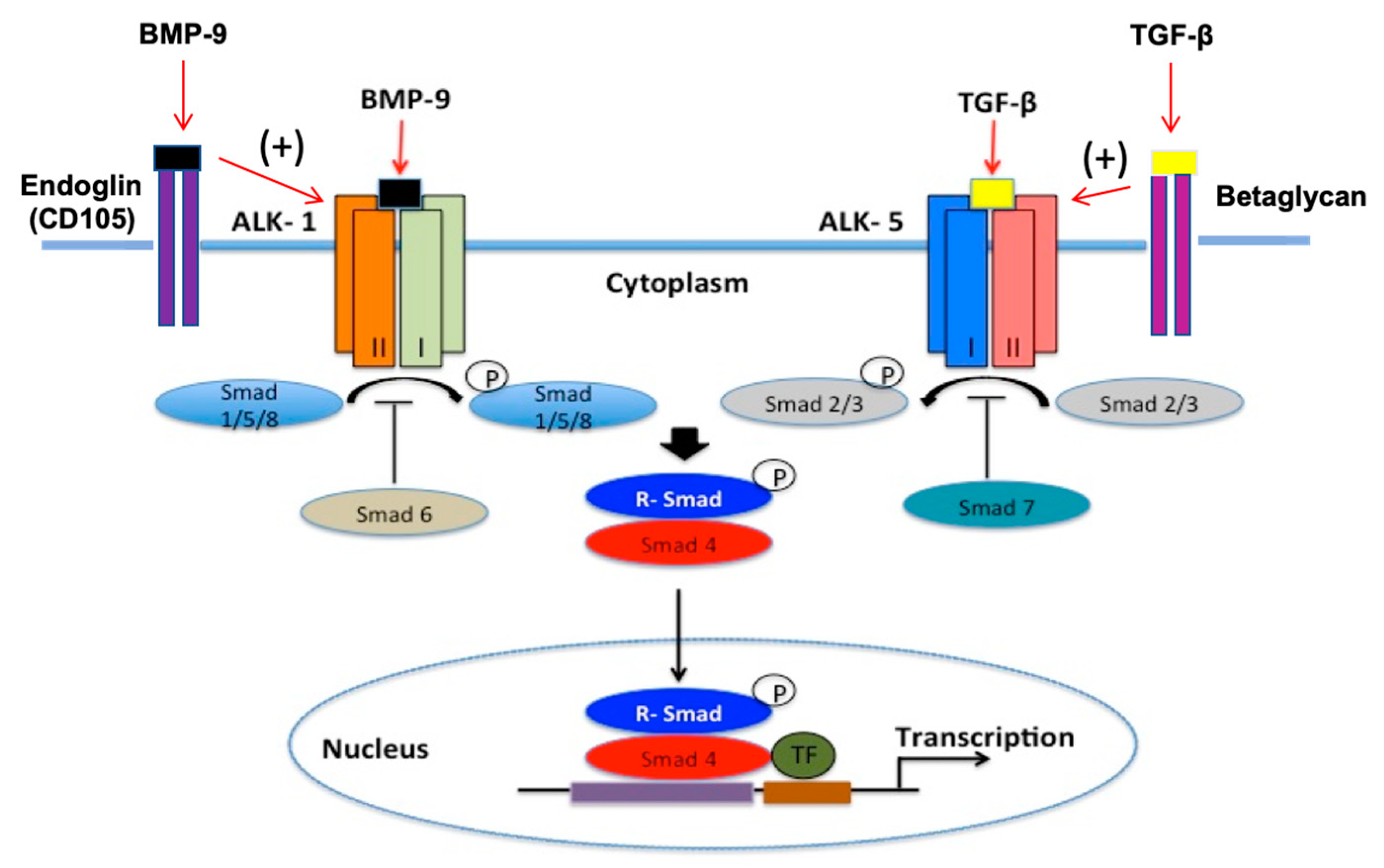
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Sankar, S.; Mahooti-Brooks, N.; Centrella, M.; McCarthy, T.L.; Madri, J.A. Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor beta 2. J. Biol. Chem. 1995, 270, 13567–13572. [Google Scholar] [CrossRef] [Green Version]
- Lebrin, F.; Goumans, M.-J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-Beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.H.; Bernabeu, C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J. Cell. Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef]
- Lebrin, F.; Deckers, M.; Bertolino, P.; Ten Dijke, P. TGF-beta receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Curado, F.; Spuul, P.; Egaña, I.; Rottiers, P.; Daubon, T.; Veillat, V.; Duhamel, P.; Leclercq, A.; Gontier, E.; Génot, E. ALK5 and ALK1 play antagonistic roles in transforming growth factor β-Induced podosome formation in aortic endothelial cells. Mol. Cell. Biol. 2014, 34, 4389–4403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Salmon, R.M.; Upton, P.D.; Morrell, N.W.; Li, W. Regulation of bone morphogenetic protein 9 (BMP9) by redox-dependent proteolysis. J. Biol. Chem. 2014, 289, 31150–31159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Salmon, R.M.; Jiang, H.; Morrell, N.W. Regulation of the ALK1 ligands, BMP9 and BMP10. Biochem. Soc. Trans. 2016, 44, 1135–1141. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Brzica, H.; Hirsch, N.A.; Reilly, B.G.; Ronaldson, P.T. Functional expression of organic anion transporting polypeptide 1a4 is regulated by transforming growth factor-β/Activin receptor-like kinase 1 signaling at the blood-brain barrier. Mol. Pharmacol. 2018, 94, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.L.; An, H.; D’Angelo, G.; Ponisio, R.; Bushard, P.; Vo, K.D.; Powers, W.J.; Lin, W.; Lee, J.-M. Preexisting statin use is associated with greater reperfusion in hyperacute ischemic stroke. Stroke 2011, 42, 1307–1313. [Google Scholar] [CrossRef]
- Ishikawa, H.; Wakisaka, Y.; Matsuo, R.; Makihara, N.; Hata, J.; Kuroda, J.; Ago, T.; Kitayama, J.; Nakane, H.; Kamouchi, M.; et al. Influence of statin pretreatment on initial neurological severity and short-term functional outcome in acute ischemic stroke patients: The fukuoka stroke registry. Cerebrovasc. Dis. 2016, 42, 395–403. [Google Scholar] [CrossRef]
- Malhotra, K.; Safouris, A.; Goyal, N.; Arthur, A.; Liebeskind, D.S.; Katsanos, A.H.; Sargento-Freitas, J.; Ribo, M.; Molina, C.; Chung, J.-W.; et al. Association of statin pretreatment with collateral circulation and final infarct volume in acute ischemic stroke patients: A meta-analysis. Atherosclerosis 2019, 282, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Amarenco, P.; Bogousslavsky, J.; Callahan, A.; Goldstein, L.B.; Hennerici, M.; Rudolph, A.E.; Sillesen, H.; Simunovic, L.; Szarek, M.; Welch, K.M.A.; et al. High-dose atorvastatin after stroke or transient ischemic attack. N. Engl. J. Med. 2006, 355, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Saver, J.L.; Wu, Y.-L.; Tang, S.-C.; Lee, J.-D.; Rao, N.M.; Wang, H.-H.; Jeng, J.-S.; Lee, T.-H.; Chen, P.-C.; et al. Utilization of statins beyond the initial period after stroke and 1-year risk of recurrent stroke. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Bustamante, A.; García-Matas, S.; Martínez-Zabaleta, M.; Jiménez, C.; de la Torre, J.; Rubio, F.R.; Segura, T.; Masjuán, J.; Cánovas, D.; et al. Combination of thrombolysis and statins in acute stroke is safe: Results of the STARS randomized trial (stroke treatment with acute reperfusion and simvastatin). Stroke 2016, 47, 2870–2873. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Nombela, F.; Castellanos, M.; Rodriguez-Yáñez, M.; García-Gil, M.; Leira, R.; Lizasoain, I.; Serena, J.; Vivancos, J.; Moro, M.A.; et al. Statin treatment withdrawal in ischemic stroke: A controlled randomized study. Neurology 2007, 69, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Gertz, K.; Dirnagl, U.; Böhm, M.; Nickenig, G.; Endres, M. Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res. 2002, 942, 23–30. [Google Scholar] [CrossRef]
- Fagan, S.C.; Hess, D.C.; Hohnadel, E.J.; Pollock, D.M.; Ergul, A. Targets for vascular protection after acute ischemic stroke. Stroke 2004, 35, 2220–2225. [Google Scholar] [CrossRef]
- Dziedzic, T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev. Neurother. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Murphy, M.P.; Mancuso, C.; Head, E. Atorvastatin treatment in a dog preclinical model of alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int. J. Neuropsychopharmacol. 2012, 15, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, X.; Dong, L.; Wen, Y.; Cui, L. The many roles of statins in ischemic stroke. Curr. Neuropharmacol. 2014, 12, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chopp, M.; Jia, L.; Cui, Y.; Lu, M.; Zhang, Z.G. Atorvastatin extends the therapeutic window for tpa to 6 h after the onset of embolic stroke in rats. J. Cereb. Blood Flow Metab. 2009, 29, 1816–1824. [Google Scholar] [CrossRef]
- Chaturvedi, M.; Kaczmarek, L. Mmp-9 inhibition: A therapeutic strategy in ischemic stroke. Mol. Neurobiol. 2014, 49, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, L.R.; Mitton, M.W.; Arvik, B.M.; Doraiswamy, P.M. Statin-associated memory loss: Analysis of 60 case reports and review of the literature. Pharmacotherapy 2003, 23, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Van der Most, P.J.; Dolga, A.M.; Nijholt, I.M.; Luiten, P.G.M.; Eisel, U.L.M. Statins: Mechanisms of neuroprotection. Prog. Neurobiol. 2009, 88, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-associated side effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Kitamura, A.; Okura, T.; Deguchi, Y. Memantine transport by a proton-coupled organic cation antiporter in HCMEC/D3 Cells, an in vitro human blood-brain barrier model. Drug Metab. Pharmacokinet. 2015, 30, 182–187. [Google Scholar] [CrossRef]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2010, 38, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J.; Ronaldson, P.T. Drug delivery to the ischemic brain. Adv. Pharmacol. 2014, 71, 165–202. [Google Scholar] [CrossRef] [Green Version]
- López-Valdés, H.E.; Clarkson, A.N.; Ao, Y.; Charles, A.C.; Carmichael, S.T.; Sofroniew, M.V.; Brennan, K.C. Memantine enhances recovery from stroke. Stroke 2014, 45, 2093–2100. [Google Scholar] [CrossRef]
- Kornhuber, J.; Weller, M.; Schoppmeyer, K.; Riederer, P. Amantadine and memantine are nmda receptor antagonists with neuroprotective properties. J. Neural Transm. Suppl. 1994, 43, 91–104. [Google Scholar]
- Seif el Nasr, M.; Peruche, B.; Rossberg, C.; Mennel, H.D.; Krieglstein, J. Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur. J. Pharmacol. 1990, 185, 19–24. [Google Scholar] [CrossRef]
- Agarwal, R.; Shukla, G.S. Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem. Res. 1999, 24, 1507–1514. [Google Scholar] [CrossRef]
- Albekairi, T.H.; Vaidya, B.; Patel, R.; Nozohouri, S.; Villalba, H.; Zhang, Y.; Lee, Y.S.; Al-Ahmad, A.; Abbruscato, T.J. Brain delivery of a potent opioid receptor agonist, biphalin during ischemic stroke: Role of organic anion transporting polypeptide (OATP). Pharmaceutics 2019, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shah, K.; Wang, H.; Karamyan, V.T.; Abbruscato, T.J. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J. Pharmacol. Exp. Ther. 2011, 339, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Goal | |
|---|---|---|
| Immediate | r-tPA (Alteplase) | |
| i.v. infusion Must be administered no more than 4.5 h after onset of stroke symptoms. | Fibrinolysis Restoration of perfusion of ischemic brain tissue | |
| EVT | ||
| Surgical approach where stent retriever is used to remove slot from occluded blood vessel. Must be conducted no more than 6 h after onset of stroke symptoms | Restore perfusion to ischemic grain tissue. EVT is commonly recommended for large vessel occlusion | |
| Preventative | Lifestyle changes | |
| Physical activity Smoking Cessation | 20-30 min of aerobic exercise 5 days a week improves cardiovascular health. Reduces negative effects of tobacco on cardiovascular health | |
| Anticoagulant/Antiplatelet Medications | ||
| Warfarin (anticoagulant) Aspirin (antiplatelet) | Reduction in time to thrombolysis leads to reduced stroke incidence | |
| Carotid Endarterectomy | ||
| Surgical procedure involving removal of atherosclerotic plaque from carotid arteries. | Reduction in risk of new or recurrent ischemic stroke. | |
| Cerebral Angioplasty or Stenting | ||
| Surgical procedure involving either a balloon catheter (angioplasty) or insertion of a mesh steel brace (stent) into an occluded artery. | Increase cerebral reperfusion and reduce risk of recurrent ischemic stroke. | |
| Rehabilitative | Pharmacotherapy | |
| Statins Antihypertensive Drugs ACE inhibitors | Improve neurocognitive recovery Reduce risk of recurrent stroke | |
| Supportive Care | ||
| Speech therapy Occupational therapy Physical therapy | Improve motor and cognitive functionality |
| Endogenous Substrates | Drug Substrates | |
|---|---|---|
| Steroid Hormones: | Antibiotics: | Beta-blockers: |
| DHEA-S Estradiol-17β-glucuronide Estrone-3-sulfate | Erythromycin Ciprofloxacin Gatifloxacin Rifamycin Levofloxacin/fluoroquinolones Tebipenem Pivoxil | Acebutolol Atenolol Celiprolol Labetalol Nadolol Sotalol Talinolol |
| Thyroid Hormones: | Prostaglandins: | Chemotherapeutics: |
| Reverse triiodothyronine (rT3) Thyroxine (T4) Triiodothyronine (T3) | Unaprostone | Methotrexate Imatinib Atrasentan |
| Prostaglandins: | HIV antivirals: | Dyes: |
| Prostaglandin E2 | Darunavir Lopinavir Saquinavir | Sulfobromophthalein (SBP) |
| Bile Acids: | Statins: | Paralytics: |
| Bilirubin Cholic acid Glycocholate Taurocholate Taurochenodeoxycholate (TCDC) Tauroursodeoxycholate (TUDC) | Rosuvastatin Atorvostatin Pitavastatin | Rocuronium |
| Other: | Cardiac Glycosides: | Opiates: |
| linear and cyclic peptides | Ouabain | Deltophorin II DPDPE (synthetic) |
| Antihistamines: | ||
| Fexofenadine | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.I.; Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Transporter-Mediated Delivery of Small Molecule Drugs to the Brain: A Critical Mechanism That Can Advance Therapeutic Development for Ischemic Stroke. Pharmaceutics 2020, 12, 154. https://doi.org/10.3390/pharmaceutics12020154
Williams EI, Betterton RD, Davis TP, Ronaldson PT. Transporter-Mediated Delivery of Small Molecule Drugs to the Brain: A Critical Mechanism That Can Advance Therapeutic Development for Ischemic Stroke. Pharmaceutics. 2020; 12(2):154. https://doi.org/10.3390/pharmaceutics12020154
Chicago/Turabian StyleWilliams, Erica I., Robert D. Betterton, Thomas P. Davis, and Patrick T. Ronaldson. 2020. "Transporter-Mediated Delivery of Small Molecule Drugs to the Brain: A Critical Mechanism That Can Advance Therapeutic Development for Ischemic Stroke" Pharmaceutics 12, no. 2: 154. https://doi.org/10.3390/pharmaceutics12020154