
In Combo Studies for the Optimization of 5-Aminoanthranilic Acid Derivatives as Potential Multitarget Drugs for the Management of Metabolic Syndrome
 , , , ,
, , , ,  , ,
, ,  and
and 
Abstract
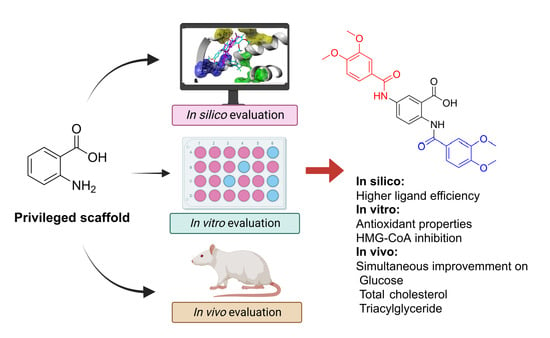
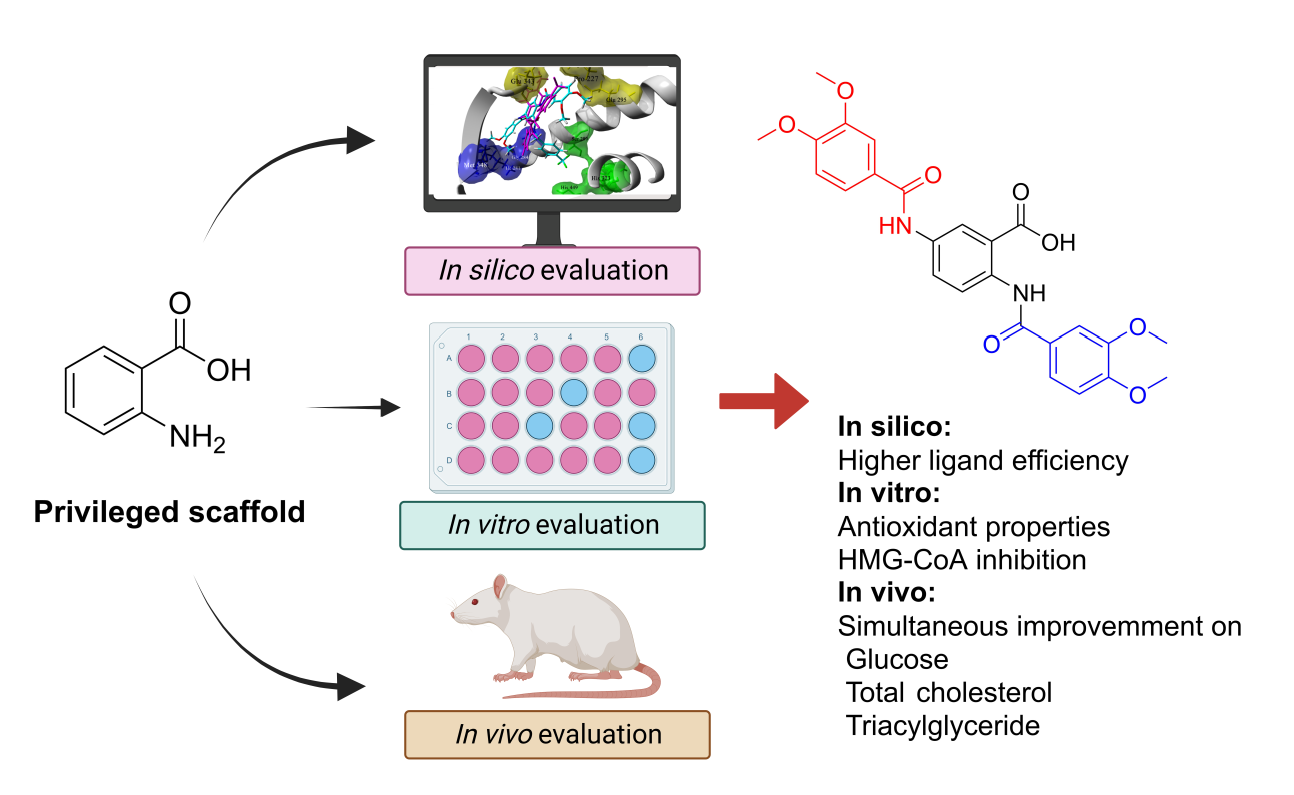
:
1. Introduction
2. Results
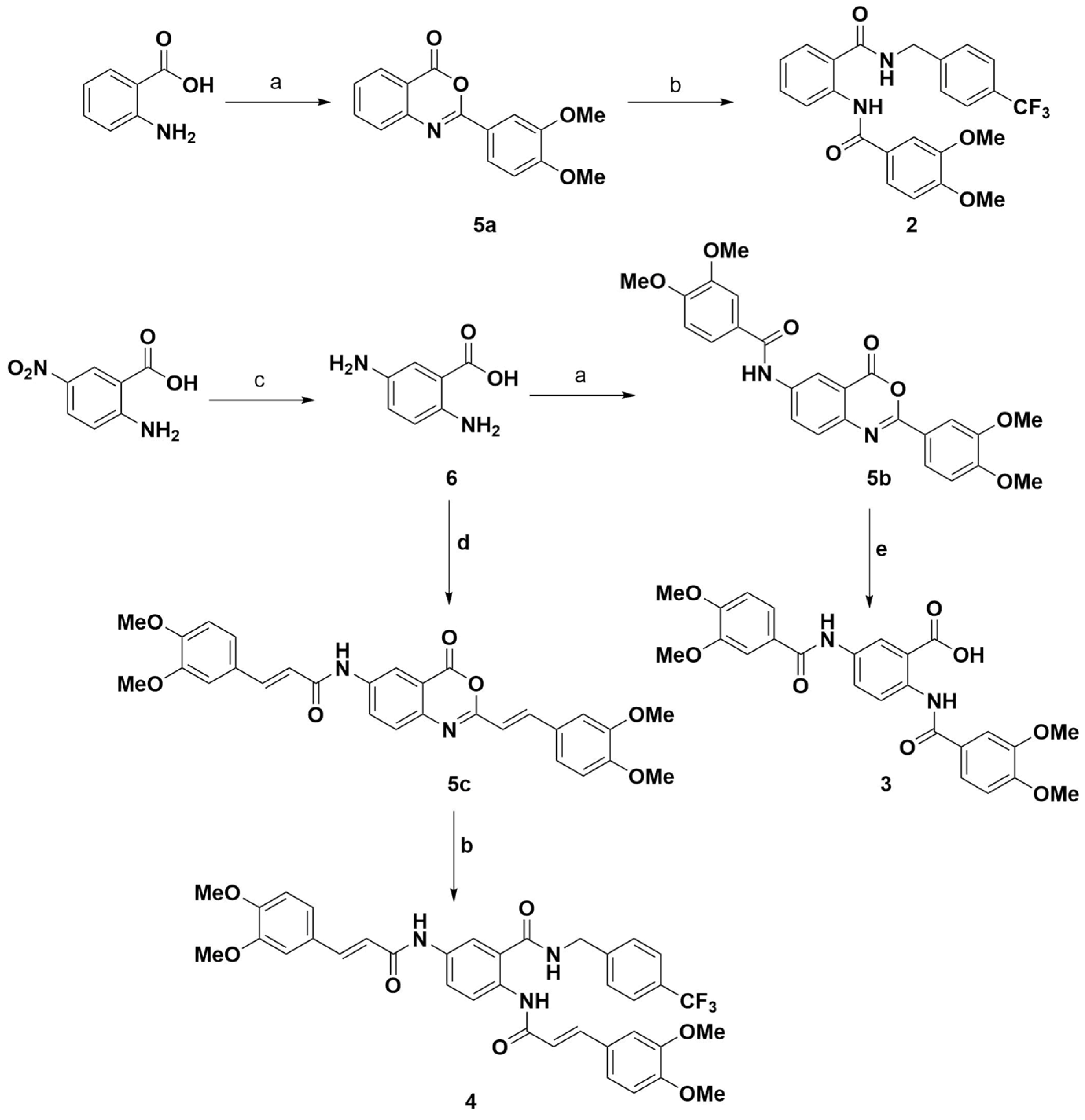
2.1. Preparation of Compounds 2–4
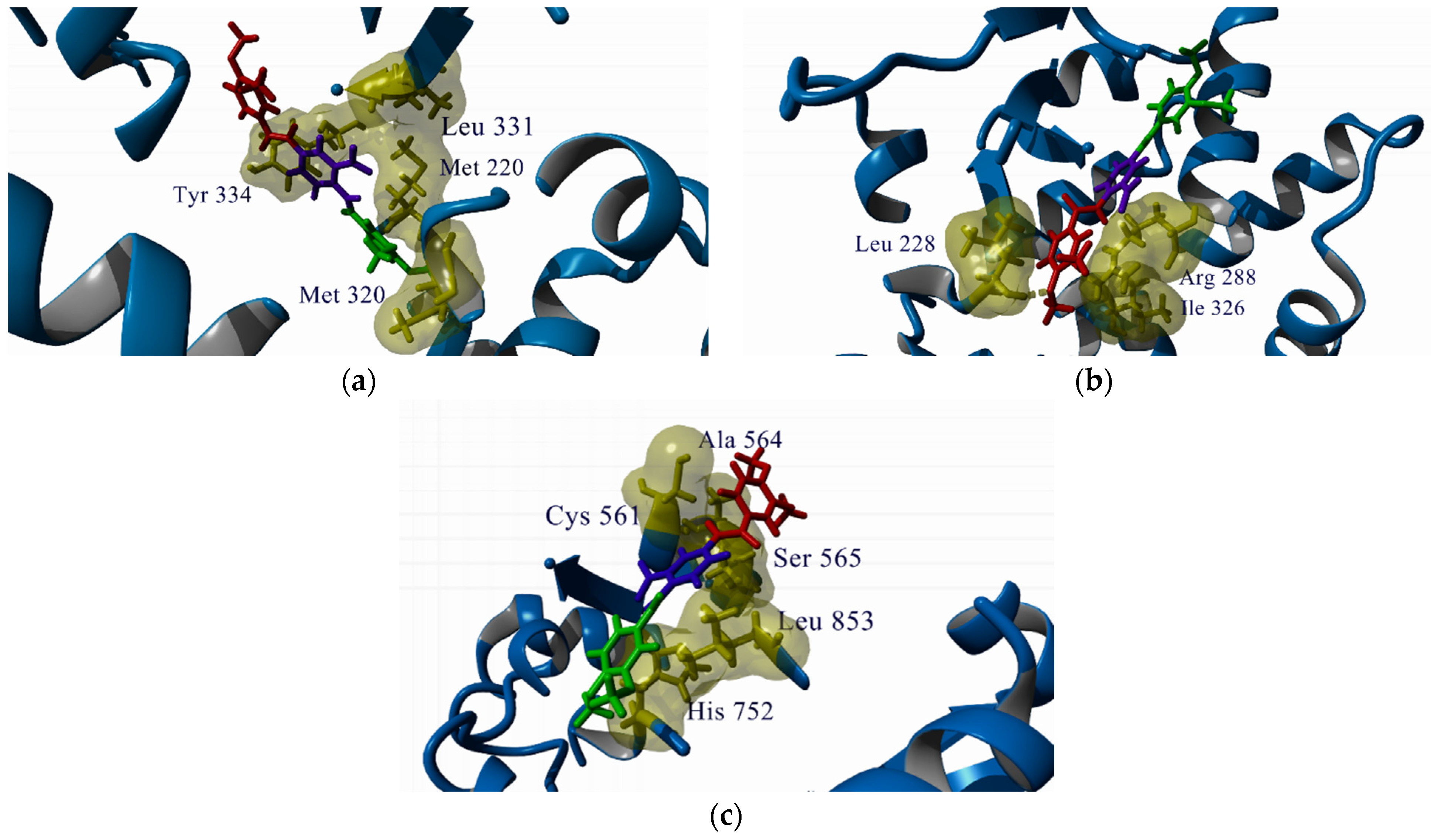
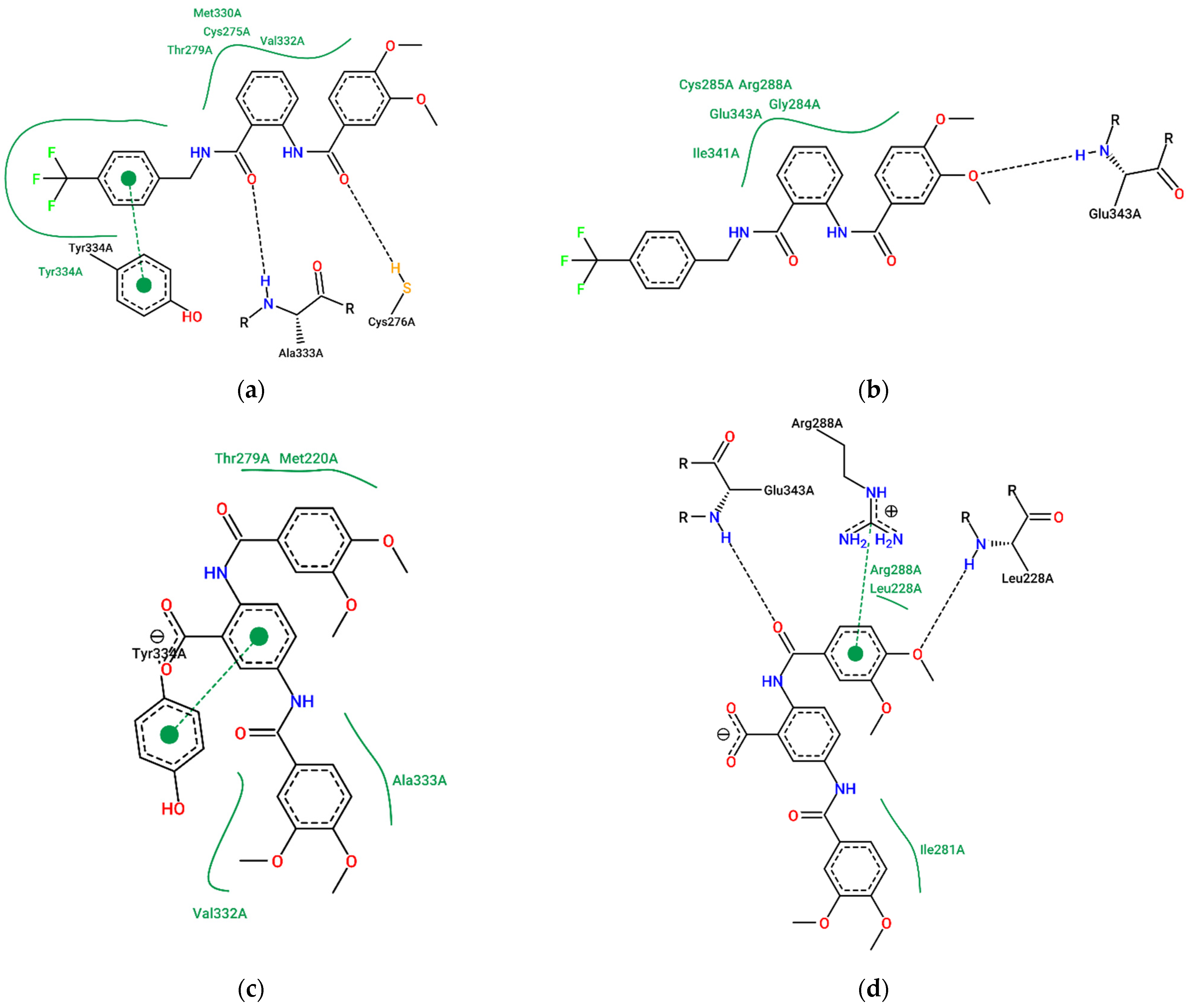
2.2. In Silico Studies
2.3. In Vivo and In Vitro Studies
3. Discussion
4. Materials and Methods
4.1. In Silico Studies
Docking Studies
4.2. Chemistry
4.2.1. General Procedure for the Preparation of Benzoxazinone Derivatives 5a–c
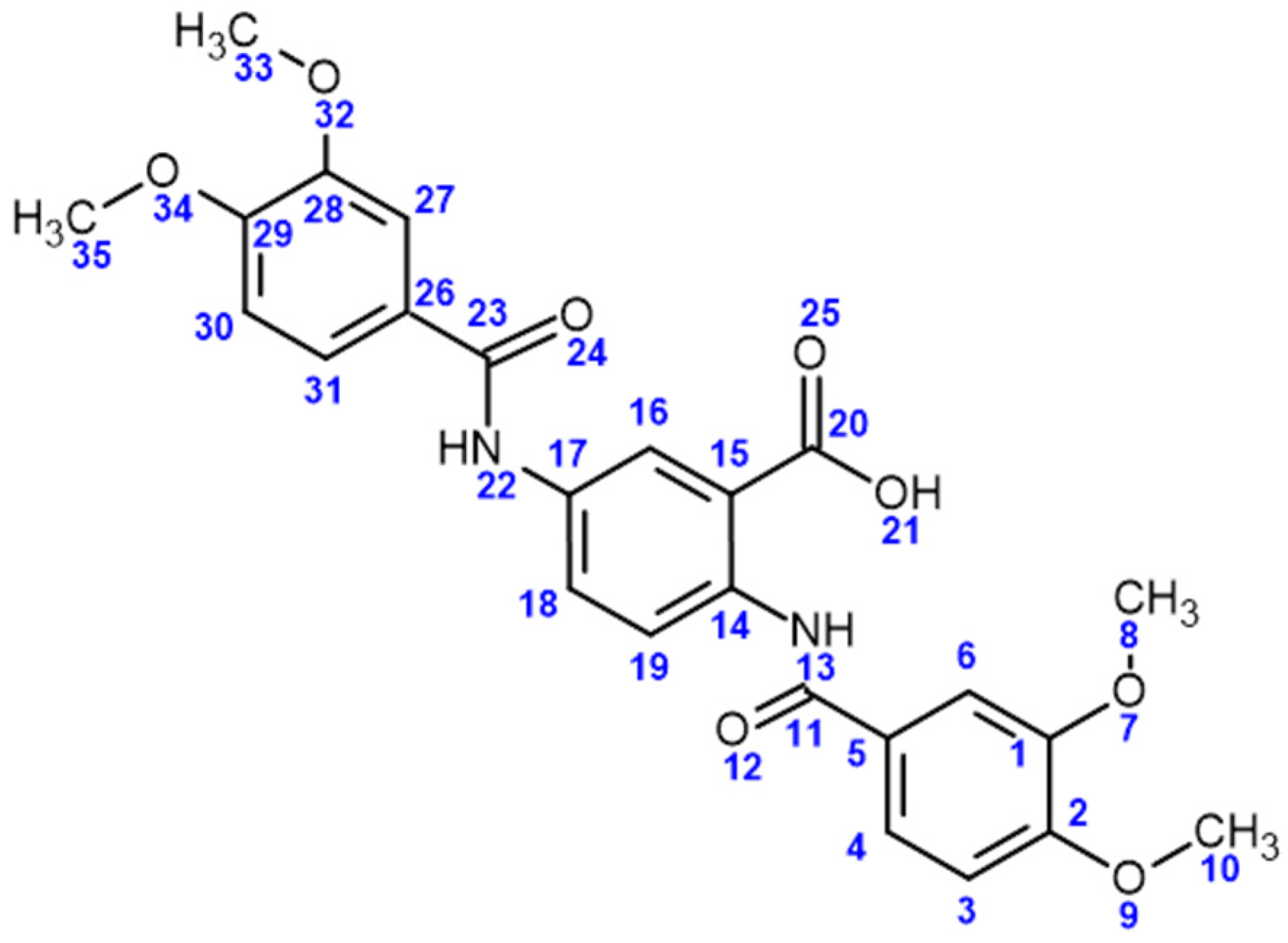
4.2.2. 2,5-Bis(3,4-dimethoxybenzamido)benzoic Acid (Compound 3)
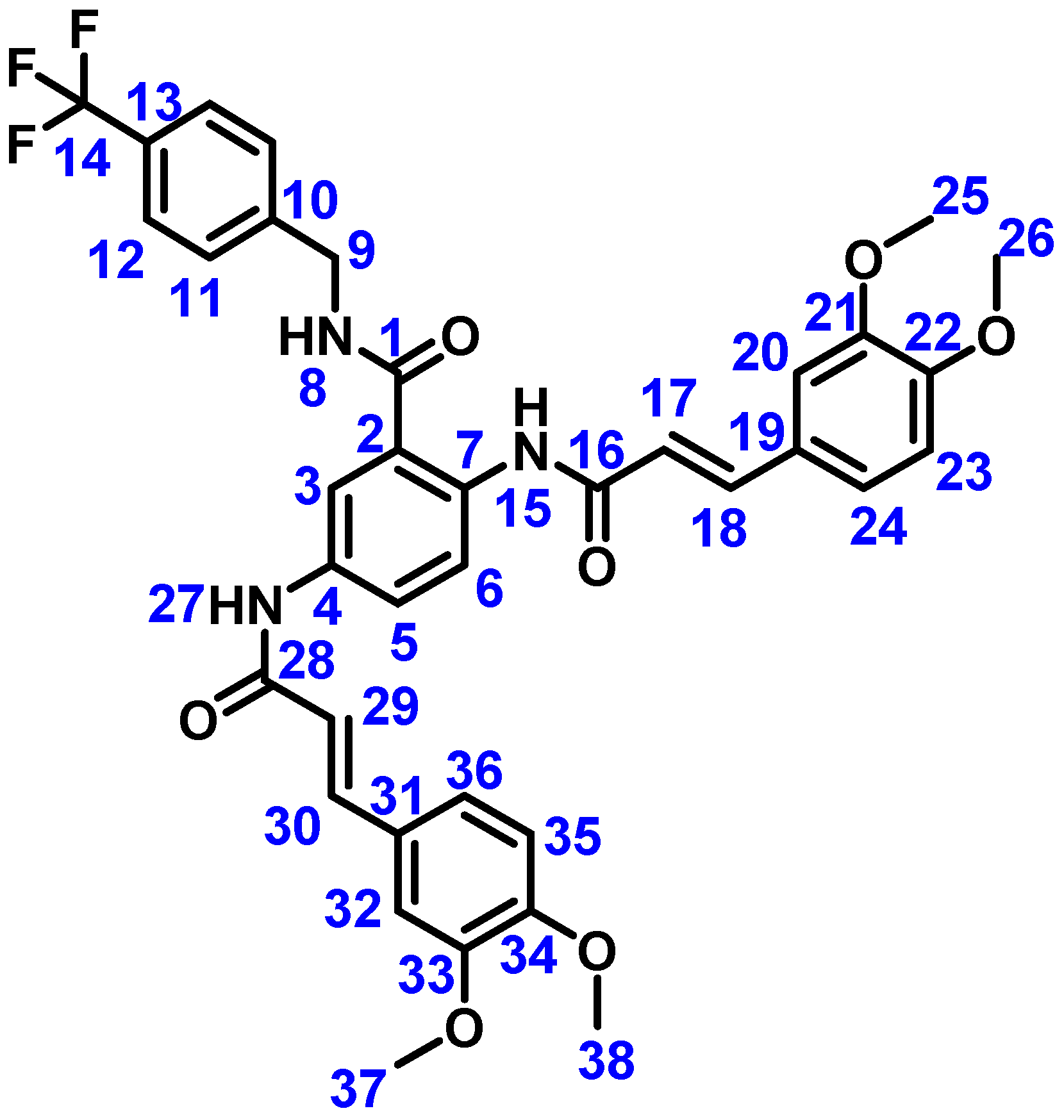
4.2.3. (2E,2′E)-N,N′-(2-((4-(Trifluoromethyl)benzyl)carbamoyl)-1,4-phenylene)bis(3-(3,4-dimethoxyphenyl)acrylamide) (Compound 4)
4.3. In Vivo Evaluation in Metabolic Syndrome
4.3.1. Animals
4.3.2. MetS Induction and Treatment with the Tested Compounds
4.3.3. Triacylglycerides, Cholesterol, and Glucose Analysis
4.4. In Vitro Evaluation of HMG-CoA Reductase Inhibition and Antioxidant Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, X.; Adams, H.; Kubena, K.; Guo, S. Etiology of metabolic syndrome and dietary intervention. Int. J. Mol. Sci. 2019, 20, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Zuin, M.; Rigatelli, G.; Bilato, C.; Cervellati, C.; Zuliani, G.; Roncon, L. Prognostic Role of Metabolic Syndrome in COVID-19 Patients: A Systematic Review Meta-Analysis. Viruses 2021, 13, 1938. [Google Scholar] [CrossRef]
- Bansal, R.; Gubbi, S.; Muniyappa, R. Metabolic Syndrome and COVID 19: Endocrine-Immune-Vascular Interactions Shapes Clinical Course. Endocrinology 2020, 161, bqaa112. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R.; Buse, J.; Ferrannini, E.; Stern, M. The Metabolic Syndrome: Time for a Critical Appraisal. Diabetes Care 2005, 28, 2289–2304. [Google Scholar] [CrossRef] [Green Version]
- Oladejo, A.O. Overview of the metabolic syndrome; an emerging pandemic of public health significance. Ann. Ibadan Postgrad. Med. 2011, 9, 78–82. [Google Scholar]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Fahed, G.; Aoun, L.; Zerdan, M.B.; Allam, S.; Zerdan, M.B.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome: Therapeutic considerations. Handb. Exp. Pharmacol. 2005, 170, 107–133. [Google Scholar]
- Grundy, S.M. Drug therapy of the metabolic syndrome: Minimizing the emerging crisis in polypharmacy. Nat. Rev. Drug Discov. 2006, 5, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Masnoon, N.; Shakib, S.; Kalisch-Ellett, L.; Caughey, G.E. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017, 17, 230. [Google Scholar] [CrossRef]
- Davies, L.E.; Spiers, G.; Kingston, A.; Todd, A.; Adamson, J.; Hanratty, B. Adverse Outcomes of Polypharmacy in Older People: Systematic Review of Reviews. J. Am. Med. Dir. Assoc. 2020, 21, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z.; Huang, S.T.; Wen, Y.W.; Chen, L.K.; Hsiao, F.Y. Combined Effects of Frailty and Polypharmacy on Health Outcomes in Older Adults: Frailty Outweighs Polypharmacy. J. Am. Med. Dir. Assoc. 2021, 22, 606.e7–606.e18. [Google Scholar] [CrossRef] [PubMed]
- Alwhaibi, M.; Balkhi, B.; Alhawassi, T.M.; Alkofide, H.; Alduhaim, N.; Alabdulali, R.; Drweesh, H.; Sambamoorthi, U. Polypharmacy among patients with diabetes: A cross-sectional retrospective study in a tertiary hospital in Saudi Arabia. BMJ Open 2018, 8, e020852. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Kabir, A.; Muth, A. Polypharmacology: The science of multi-targeting molecules. Pharmacol. Res. 2022, 176, 106055. [Google Scholar] [CrossRef]
- Zheng, H.; Fridkin, M.; Youdim, M. From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy. Pharmaceuticals 2014, 7, 113–135. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tejeda, J.F.; Sánchez-Ruiz, J.F.; Salazar, J.R.; Loza-Mejía, M.A. A Definition of “Multitargeticity”: Identifying Potential Multitarget and Selective Ligands Through a Vector Analysis. Front. Chem. 2020, 8, 176. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csermely, P.; Ágoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef]
- Katselou, M.; Matralis, A.; Kourounakis, A. Multi-Target Drug Design Approaches for Multifactorial Diseases: From Neurodegenerative to Cardiovascular Applications. Curr. Med. Chem. 2014, 21, 2743–2787. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Loza-Mejía, M.; Salazar, J.; Sánchez-Tejeda, J. In Silico Studies on Compounds Derived from Calceolaria: Phenylethanoid Glycosides as Potential Multitarget Inhibitors for the Development of Pesticides. Biomolecules 2018, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Saldívar-González, F.I.; Gómez-García, A.; Chávez-Ponce de León, D.E.; Sánchez-Cruz, N.; Ruiz-Rios, J.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Inhibitors of DNA Methyltransferases From Natural Sources: A Computational Perspective. Front. Pharmacol. 2018, 9, 1144. [Google Scholar] [CrossRef] [Green Version]
- Speck-Planche, A.; Kleandrova, V.V.; Scotti, M.T. Fragment-based approach for the in silico discovery of multi-target insecticides. Chemom. Intell. Lab. Syst. 2012, 111, 39–45. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Natalia Dias Soeiro Cordeiro, M.; Guilarte-Montero, L.; Yera-Bueno, R. Current Computational Approaches Towards the Rational Design of New Insecticidal Agents. Curr. Comput. Aided-Drug Des. 2011, 7, 304–314. [Google Scholar] [CrossRef]
- Navarrete-Vázquez, G.; Alaniz-Palacios, A.; Hidalgo-Figueroa, S.; González-Acevedo, C.; Ávila-Villarreal, G.; Estrada-Soto, S.; Webster, S.P.; Medina-Franco, J.L.; López-Vallejo, F.; Guerrero-Álvarez, J.; et al. Discovery, synthesis and in combo studies of a tetrazole analogue of clofibric acid as a potent hypoglycemic agent. Bioorg. Med. Chem. Lett. 2013, 23, 3244–3247. [Google Scholar] [CrossRef] [PubMed]
- Colin-Lozano, B.; Torres-Gomez, H.; Hidalgo-Figueroa, S.; Chávez-Silva, F.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Navarrete-Vazquez, G. Synthesis, In Vitro, In Vivo and In Silico Antidiabetic Bioassays of 4-Nitro(thio)phenoxyisobutyric Acids Acting as Unexpected PPARγ Modulators: An In Combo Study. Pharmaceuticals 2022, 15, 102. [Google Scholar] [PubMed]
- Guzmán-Ávila, R.; Flores-Morales, V.; Paoli, P.; Camici, G.; Ramírez-Espinosa, J.J.; Cerón-Romero, L.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Yolanda Rios, M.; Villalobos-Molina, R.; et al. Ursolic acid derivatives as potential antidiabetic agents: In vitro, in vivo, and in silico studies. Drug Dev. Res. 2018, 79, 70–80. [Google Scholar] [CrossRef]
- Navarrete-Vázquez, G.; Morales-Vilchis, M.G.; Estrada-Soto, S.; Ramírez-Espinosa, J.J.; Hidalgo-Figueroa, S.; Nava-Zuazo, C.; Tlahuext, H.; Leon-Rivera, I.; Medina-Franco, J.L.; López-Vallejo, F.; et al. Synthesis of 2-{2-[(α/β-naphthalen-1-ylsulfonyl)amino]-1,3-thiazol-4-yl} acetamides with 11β-hydroxysteroid dehydrogenase inhibition and in combo antidiabetic activities. Eur. J. Med. Chem. 2014, 74, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bacci, A.; Corsi, F.; Runfola, M.; Sestito, S.; Piano, I.; Manera, C.; Saccomanni, G.; Gargini, C.; Rapposelli, S. Design, Synthesis, and In Vitro Evaluation of Novel 8-Amino-Quinoline Combined with Natural Antioxidant Acids. Pharmaceuticals 2022, 15, 688. [Google Scholar] [CrossRef] [PubMed]
- Suliman, R.S.; Alghamdi, S.S.; Ali, R.; Aljatli, D.; Aljammaz, N.A.; Huwaizi, S.; Suliman, R.; Kahtani, K.M.; Albadrani, G.M.; Barhoumi, T.; et al. The Role of Myrrh Metabolites in Cancer, Inflammation, and Wound Healing: Prospects for a Multi-Targeted Drug Therapy. Pharmaceuticals 2022, 15, 944. [Google Scholar] [CrossRef]
- Bortolami, M.; Pandolfi, F.; Tudino, V.; Messore, A.; Madia, V.N.; De Vita, D.; Di Santo, R.; Costi, R.; Romeo, I.; Alcaro, S.; et al. Design, Synthesis, and In Vitro, In Silico and In Cellulo Evaluation of New Pyrimidine and Pyridine Amide and Carbamate Derivatives as Multi-Functional Cholinesterase Inhibitors. Pharmaceuticals 2022, 15, 673. [Google Scholar] [CrossRef]
- González-Álvarez, H.; Bravo-Jiménez, A.; Martínez-Arellanes, M.; Gamboa-Osorio, G.O.; Chávez-Gutiérrez, E.; González-Hernández, L.A.; Gallardo-Ignacio, K.; Quintana-Romero, O.J.; Ariza-Castolo, A.; Guerra-Araiza, C.; et al. In Silico-Based Design and In Vivo Evaluation of an Anthranilic Acid Derivative as a Multitarget Drug in a Diet-Induced Metabolic Syndrome Model. Pharmaceuticals 2021, 14, 914. [Google Scholar] [CrossRef]
- Schmidt, J.; Rotter, M.; Weiser, T.; Wittmann, S.; Weizel, L.; Kaiser, A.; Heering, J.; Goebel, T.; Angioni, C.; Wurglics, M.; et al. A Dual Modulator of Farnesoid X Receptor and Soluble Epoxide Hydrolase To Counter Nonalcoholic Steatohepatitis. J. Med. Chem. 2017, 60, 7703–7724. [Google Scholar] [CrossRef]
- Heitel, P.; Faudone, G.; Helmstädter, M.; Schmidt, J.; Kaiser, A.; Tjaden, A.; Schröder, M.; Müller, S.; Schierle, S.; Pollinger, J.; et al. A triple farnesoid X receptor and peroxisome proliferator-activated receptor α/δ activator reverses hepatic fibrosis in diet-induced NASH in mice. Commun. Chem. 2020, 3, 174. [Google Scholar] [CrossRef]
- Merk, D.; Gabler, M.; Gomez, R.C.; Flesch, D.; Hanke, T.; Kaiser, A.; Lamers, C.; Werz, O.; Schneider, G.; Schubert-Zsilavecz, M. Anthranilic acid derivatives as novel ligands for farnesoid X receptor (FXR). Bioorg. Med. Chem. 2014, 22, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.I.; Chen, J.; Du, M.; Hogan, M.; Kincaid, S.; Nelson, F.C.; Venkatesan, A.M.; Wehr, T.; Zask, A.; DiJoseph, J.; et al. The discovery of anthranilic acid-based MMP inhibitors. Part 2: SAR of the 5-position and P1(1) groups. Bioorg. Med. Chem. Lett. 2001, 11, 2189–2192. [Google Scholar] [CrossRef]
- Han, S.H.; Suh, H.S.; Jo, H.; Oh, Y.; Mishra, N.K.; Han, S.; Kim, H.S.; Jung, Y.H.; Lee, B.M.; Kim, I.S. Synthesis and anti-inflammatory evaluation of N-sulfonyl anthranilic acids via Ir(III)-catalyzed C–H amidation of benzoic acids. Bioorg. Med. Chem. Lett. 2017, 27, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Lamers, C.; Weber, J.; Flesch, D.; Gabler, M.; Proschak, E.; Schubert-Zsilavecz, M. Anthranilic acid derivatives as nuclear receptor modulators—Development of novel PPAR selective and dual PPAR/FXR ligands. Bioorg. Med. Chem. 2015, 23, 499–514. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; Abdel-Aziz, A.A.M.; Bua, S.; Nocentini, A.; AlSaif, N.A.; Almehizia, A.A.; Alanazi, M.M.; Hefnawy, M.M.; Supuran, C.T. New anthranilic acid-incorporating N-benzenesulfonamidophthalimides as potent inhibitors of carbonic anhydrases I, II, IX, and XII: Synthesis, in vitro testing, and in silico assessment. Eur. J. Med. Chem. 2019, 181, 111573. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-García, C.; Fuentes-Venado, C.E.; Guerra-Araiza, C.; Segura-Uribe, J.; Chávez-Gutiérrez, E.; Farfán-García, E.D.; Estrada Cruz, N.A.; Pinto-Almazán, R. Sex differences in the performance of cognitive tasks in a murine model of metabolic syndrome. Eur. J. Neurosci. 2020, 52, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Panchal, S.K.; Brown, L. Rodent Models for Metabolic Syndrome Research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, M.K.J.; Hausner, S.H.; Marik, J.; Abbey, C.K.; Marshall, J.F.; Sutcliffe, J.L. High-throughput in vivo screening of targeted molecular imaging agents. Proc. Natl. Acad. Sci. USA 2009, 106, 17904–17909. [Google Scholar] [CrossRef] [Green Version]
- Speak, A.O.; Swiatkowska, A.; Karp, N.A.; Arends, M.J.; Adams, D.J.; van der Weyden, L. A high-throughput in vivo screening method in the mouse for identifying regulators of metastatic colonization. Nat. Protoc. 2017, 12, 2465–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bumrungpert, A.; Lilitchan, S.; Tuntipopipat, S.; Tirawanchai, N.; Komindr, S. Ferulic Acid Supplementation Improves Lipid Profiles, Oxidative Stress, and Inflammatory Status in Hyperlipidemic Subjects: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Nutrients 2018, 10, 713. [Google Scholar] [CrossRef] [Green Version]
- Senaphan, K.; Kukongviriyapan, U.; Sangartit, W.; Pakdeechote, P.; Pannangpetch, P.; Prachaney, P.; Greenwald, S.; Kukongviriyapan, V. Ferulic Acid Alleviates Changes in a Rat Model of Metabolic Syndrome Induced by High-Carbohydrate, High-Fat Diet. Nutrients 2015, 7, 6446–6464. [Google Scholar] [CrossRef] [PubMed]
- Ardiansyah; Ohsaki, Y.; Shirakawa, H.; Koseki, T.; Komai, M. Novel Effects of a Single Administration of Ferulic Acid on the Regulation of Blood Pressure and the Hepatic Lipid Metabolic Profile in Stroke-Prone Spontaneously Hypertensive Rats. J. Agric. Food Chem. 2008, 56, 2825–2830. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Peperidou, A.; Fotopoulos, I.; Hadjipavlou-Litina, D. Cinnamate Hybrids: A Unique Family of Compounds with Multiple Biological Activities. Curr. Pharm. Biotechnol. 2018, 19, 1019–1048. [Google Scholar] [CrossRef]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Venado, C.E.; Terán-Pérez, G.; Espinosa-Hernández, V.M.; Martínez-Herrera, E.; Segura-Uribe, J.J.; Mercadillo, R.E.; Pinto-Almazán, R.; Guerra-Araiza, C. Nutritional Status Influences Oxidative Stress and Insulin Resistance in Preschool Children. Metab. Syndr. Relat. Disord. 2021, 19, 513–523. [Google Scholar] [CrossRef]
- Beato, A.; Gori, A.; Boucherle, B.; Peuchmaur, M.; Haudecoeur, R. β-Carboline as a Privileged Scaffold for Multitarget Strategies in Alzheimer’s Disease Therapy. J. Med. Chem. 2021, 64, 1392–1422. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Waseem, M.; Subbarao, N. Discovery of multi-target mur enzymes inhibitors with anti-mycobacterial activity through a Scaffold approach. J. Biomol. Struct. Dyn. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Maccallini, C.; Amoia, P.; Amoroso, R. Multitarget PPARγ agonists as innovative modulators of the metabolic syndrome. Eur. J. Med. Chem. 2019, 173, 261–273. [Google Scholar] [CrossRef]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Colín-Lozano, B.; Estrada-Soto, S.; Chávez-Silva, F.; Gutiérrez-Hernández, A.; Cerón-Romero, L.; Giacoman-Martínez, A.; Almanza-Pérez, J.C.; Hernández-Núñez, E.; Wang, Z.; Xie, X.; et al. Design, Synthesis and in Combo Antidiabetic Bioevaluation of Multitarget Phenylpropanoic Acids. Molecules 2018, 23, 340. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Guo, Y.; Yan, J.; Luo, Z.; Luo, H.-B.; Yan, M.; Huang, L.; Li, X. Design, Synthesis, and Evaluation of Multitarget-Directed Resveratrol Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2013, 56, 5843–5859. [Google Scholar] [CrossRef] [PubMed]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARα Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Takei, K.; Arulmozhiraja, S.; Sladek, V.; Matsuo, N.; Han, S.; Matsuzaka, T.; Sekiya, M.; Tokiwa, T.; Shoji, M.; et al. Molecular association model of PPARα and its new specific and efficient ligand, pemafibrate: Structural basis for SPPARMα. Biochem. Biophys. Res. Commun. 2018, 499, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.A.; Tan, L.; Yang, H.; Im, Y.-G.; Im, Y.J. Structures of PPARγ complexed with lobeglitazone and pioglitazone reveal key determinants for the recognition of antidiabetic drugs. Sci. Rep. 2017, 7, 16837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtera, A.; Miyamae, Y.; Yoshida, K.; Maejima, K.; Akita, T.; Kakizuka, A.; Irie, K.; Masuda, S.; Kambe, T.; Nagao, M. Identification of a New Type of Covalent PPARγ Agonist using a Ligand-Linking Strategy. ACS Chem. Biol. 2015, 10, 2794–2804. [Google Scholar] [CrossRef]
- Pickavance, L.C.; Tadayyon, M.; Widdowson, P.S.; Buckingham, R.E.; Wilding, J.P.H. Therapeutic index for rosiglitazone in dietary obese rats: Separation of efficacy and haemodilution. Br. J. Pharmacol. 1999, 128, 1570–1576. [Google Scholar] [CrossRef]
- Collino, M.; Aragno, M.; Castiglia, S.; Miglio, G.; Tomasinelli, C.; Boccuzzi, G.; Thiemermann, C.; Fantozzi, R. Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. Br. J. Pharmacol. 2010, 160, 1892–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.-F.; Jhao, Y.-T.; Chiu, C.-H.; Sun, L.-H.; Chou, T.-K.; Shiue, C.-Y.; Cheng, C.-Y.; Ma, K.-H. Bezafibrate Exerts Neuroprotective Effects in a Rat Model of Sporadic Alzheimer’s Disease. Pharmaceuticals 2022, 15, 109. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M.; Vodeničarovová, M. Effects of low and high doses of fenofibrate on protein, amino acid, and energy metabolism in rat. Int. J. Exp. Pathol. 2020, 101, 171–182. [Google Scholar] [CrossRef]
- Cao, N.T.; Nguyen, N.A.; Park, C.M.; Cha, G.S.; Park, K.D.; Yun, C.-H. A Novel Statin Compound from Monacolin J Produced Using CYP102A1-Catalyzed Regioselective C-Hydroxylation. Pharmaceuticals 2021, 14, 981. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Ryu, S.H.; Park, S.H.; Cha, G.S.; Kim, D.H.; Kim, K.H.; Hong, A.W.; Ahn, T.; Pan, J.G.; Joung, Y.H.; et al. Chimeric cytochromes P450 engineered by domain swapping and random mutagenesis for producing human metabolites of drugs. Biotechnol. Bioeng. 2014, 111, 1313–1322. [Google Scholar] [CrossRef]
- Mendieta, A.; Jiménez, F.; Garduño-Siciliano, L.; Mojica-Villegas, A.; Rosales-Acosta, B.; Villa-Tanaca, L.; Chamorro-Cevallos, G.; Medina-Franco, J.L.; Meurice, N.; Gutiérrez, R.U.; et al. Synthesis and highly potent hypolipidemic activity of alpha-asarone- and fibrate-based 2-acyl and 2-alkyl phenols as HMG-CoA reductase inhibitors. Bioorg. Med. Chem. 2014, 22, 5871–5882. [Google Scholar] [CrossRef]
- Rodríguez-Páez, L.; Juárez-Sanchez, M.; Antúnez-Solís, J.; Baeza, I.; Wong, C. α-Asarone inhibits HMG-CoA reductase, lowers serum LDL-cholesterol levels and reduces biliary CSI in hypercholesterolemic rats. Phytomedicine 2003, 10, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.F.; Yuan, J.; Roy, S.; Imig, J.D. Simvastatin and tempol protect against endothelial dysfunction and renal injury in a model of obesity and hypertension. Am. J. Physiol. Physiol. 2009, 298, F86–F94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Liu, Y.; Zhang, Q.; Ma, B.; Yang, Z.; Liu, L.; Yao, D.; Cui, G.; Sun, J.; Wu, Z. Metabolomic analysis of simvastatin and fenofibrate intervention in high-lipid diet-induced hyperlipidemia rats. Acta Pharmacol. Sin. 2014, 35, 1265–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Han, K.; Park, C.-Y. The triglyceride glucose index is a simple and low-cost marker associated with atherosclerotic cardiovascular disease: A population-based study. BMC Med. 2020, 18, 361. [Google Scholar] [CrossRef]
- Simental-Mendía, L.E.; Rodríguez-Morán, M.; Guerrero-Romero, F. The Product of Fasting Glucose and Triglycerides As Surrogate for Identifying Insulin Resistance in Apparently Healthy Subjects. Metab. Syndr. Relat. Disord. 2008, 6, 299–304. [Google Scholar] [CrossRef]
- Du, T.; Yuan, G.; Zhang, M.; Zhou, X.; Sun, X.; Yu, X. Clinical usefulness of lipid ratios, visceral adiposity indicators, and the triglycerides and glucose index as risk markers of insulin resistance. Cardiovasc. Diabetol. 2014, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, D.; Sánchez-Íñigo, L.; Pastrana-Delgado, J.; Fernández-Montero, A.; Martinez, J.A. Triglyceride–glucose index (TyG index) in comparison with fasting plasma glucose improved diabetes prediction in patients with normal fasting glucose: The Vascular-Metabolic CUN cohort. Prev. Med. 2016, 86, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Ostos, F.; Ramos-Lopez, O.; Blaak, E.E.; Astrup, A.; Martinez, J.A. The triglyceride-glucose index as an adiposity marker and a predictor of fat loss induced by a low-calorie diet. Eur. J. Clin. Investig. 2022, 52, e13674. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Souza, V.; César-Gomes, C.J.; Da Fonseca, L.J.S.; Guedes, G.D.S.; Smaniotto, S.; Rabelo, L.A. Aging Increases Susceptibility to High Fat Diet-Induced Metabolic Syndrome in C57BL/6 Mice: Improvement in Glycemic and Lipid Profile after Antioxidant Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 1987960. [Google Scholar] [CrossRef] [PubMed]
- Monserrat-Mesquida, M.; Quetglas-Llabrés, M.; Capó, X.; Bouzas, C.; Mateos, D.; Pons, A.; Tur, J.A.; Sureda, A. Metabolic Syndrome Is Associated with Oxidative Stress and Proinflammatory State. Antioxidants 2020, 9, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero-Serrano, E.M.; Delgado-Lista, J.; Peña-Orihuela, P.; Perez-Martinez, P.; Fuentes, F.; Marin, C.; Tunez, I.; Jose Tinahones, F.; Perez-Jimenez, F.; Roche, H.M.; et al. Oxidative stress is associated with the number of components of metabolic syndrome: LIPGENE study. Exp. Mol. Med. 2013, 45, e28. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Rottmann, S.; Aspillaga, A.A.; Pérez, D.D.; Vasquez, L.; Martinez, A.L.F.; Leighton, F. Juice and Phenolic Fractions of the Berry Aristotelia chilensis Inhibit LDL Oxidation in Vitro and Protect Human Endothelial Cells against Oxidative Stress. J. Agric. Food Chem. 2002, 50, 7542–7547. [Google Scholar] [CrossRef]
- Quispe-Fuentes, I.; Vega-Gálvez, A.; Aranda, M. Evaluation of phenolic profiles and antioxidant capacity of maqui (Aristotelia chilensis) berries and their relationships to drying methods. J. Sci. Food Agric. 2018, 98, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente Muñoz, M.; de la Fuente Fernández, M.; Román-Carmena, M.; Iglesias de la Cruz, M.D.C.; Amor, S.; Martorell, P.; Enrique-López, M.; García-Villalón, A.L.; Inarejos-García, A.M.; Granado, M. Supplementation with Two New Standardized Tea Extracts Prevents the Development of Hypertension in Mice with Metabolic Syndrome. Antioxidants 2022, 11, 1573. [Google Scholar] [CrossRef]
- Pérez-Areales, F.J.; Garrido, M.; Aso, E.; Bartolini, M.; De Simone, A.; Espargaró, A.; Ginex, T.; Sabate, R.; Pérez, B.; Andrisano, V.; et al. Centrally Active Multitarget Anti-Alzheimer Agents Derived from the Antioxidant Lead CR-6. J. Med. Chem. 2020, 63, 9360–9390. [Google Scholar] [CrossRef]
- Apak, R.; Güçlü, K.; Özyürek, M.; Bektaşoğlu, B.; Bener, M. Cupric Ion Reducing Antioxidant Capacity Assay for Antioxidants in Human Serum and for Hydroxyl Radical Scavengers. In Advanced Protocols in Oxidative Stress II. Methods in Molecular Biology; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 215–239. ISBN 978-1-60761-411-1. [Google Scholar]
- Loza-Mejía, M.A.; Salazar, J.R. Sterols and triterpenoids as potential anti-inflammatories: Molecular docking studies for binding to some enzymes involved in inflammatory pathways. J. Mol. Graph. Model. 2015, 62, 18–25. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PPAR-α | LE | PPAR-γ | LE | HMG-CoA Reductase | LE |
|---|---|---|---|---|---|---|
| 1 | −151.6 | −3.3 | −165.2 | −3.6 | −156.2 | −3.4 |
| 2 | −119.8 | −3.6 | −109.8 | −3.3 | −135.2 | −4.1 |
| 3 | −121.8 | −3.5 | −125.4 | −3.6 | −171.4 | −4.8 |
| 4 | −132.7 | −2.6 | −144.5 | −2.9 | −211.5 | −4.2 |
| Reference ligands 1 | −112.4 | −4.0 | −109.0 | −3.9 | −156.1 | −3.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chávez-Gutiérrez, E.; Martínez-Arellanes, M.; Murillo-López, M.; Medina-Guzmán, M.F.; Mobarak-Richaud, L.; Pelcastre-Guzmán, K.; Quintana-Romero, O.J.; Ariza-Castolo, A.; Ayala-Moreno, M.d.R.; Salazar, J.R.; et al. In Combo Studies for the Optimization of 5-Aminoanthranilic Acid Derivatives as Potential Multitarget Drugs for the Management of Metabolic Syndrome. Pharmaceuticals 2022, 15, 1461. https://doi.org/10.3390/ph15121461
Chávez-Gutiérrez E, Martínez-Arellanes M, Murillo-López M, Medina-Guzmán MF, Mobarak-Richaud L, Pelcastre-Guzmán K, Quintana-Romero OJ, Ariza-Castolo A, Ayala-Moreno MdR, Salazar JR, et al. In Combo Studies for the Optimization of 5-Aminoanthranilic Acid Derivatives as Potential Multitarget Drugs for the Management of Metabolic Syndrome. Pharmaceuticals. 2022; 15(12):1461. https://doi.org/10.3390/ph15121461
Chicago/Turabian StyleChávez-Gutiérrez, Edwin, Matilda Martínez-Arellanes, Montserrat Murillo-López, María Fernanda Medina-Guzmán, Laila Mobarak-Richaud, Karen Pelcastre-Guzmán, Osvaldo Javier Quintana-Romero, Armando Ariza-Castolo, María del Rosario Ayala-Moreno, Juan Rodrigo Salazar, and et al. 2022. "In Combo Studies for the Optimization of 5-Aminoanthranilic Acid Derivatives as Potential Multitarget Drugs for the Management of Metabolic Syndrome" Pharmaceuticals 15, no. 12: 1461. https://doi.org/10.3390/ph15121461