
Selective COX-2 Inhibitors: Road from Success to Controversy and the Quest for Repurposing

 , ,
, ,  and
and
Abstract
:1. Introduction
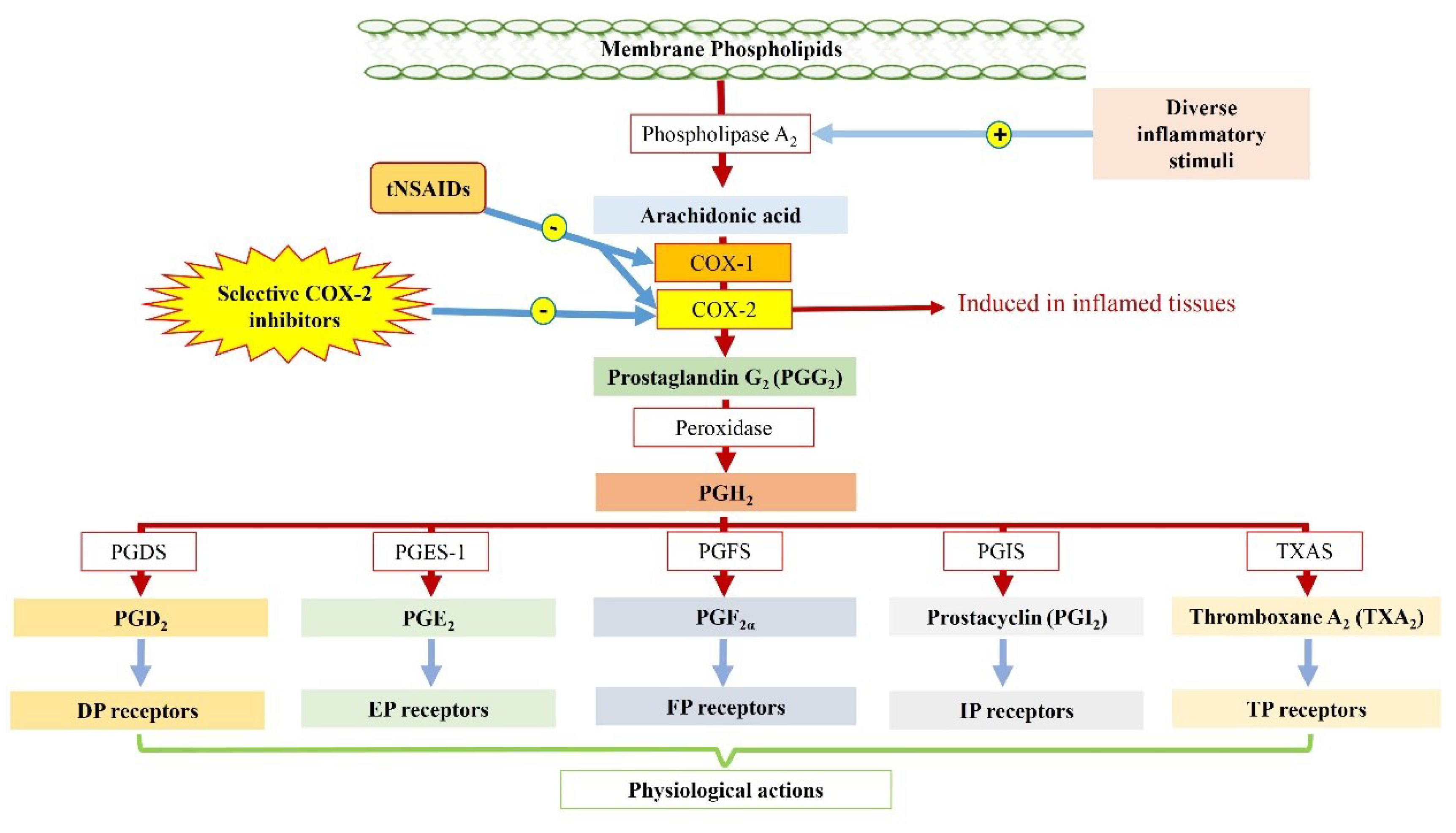
2. Purposeful Development of Coxibs and Their Success: The COX-2 Saga
3. Controversy in the COX-2 Saga: The Downfall
4. The Quest of Repurposing and Efforts from Medicinal Chemists
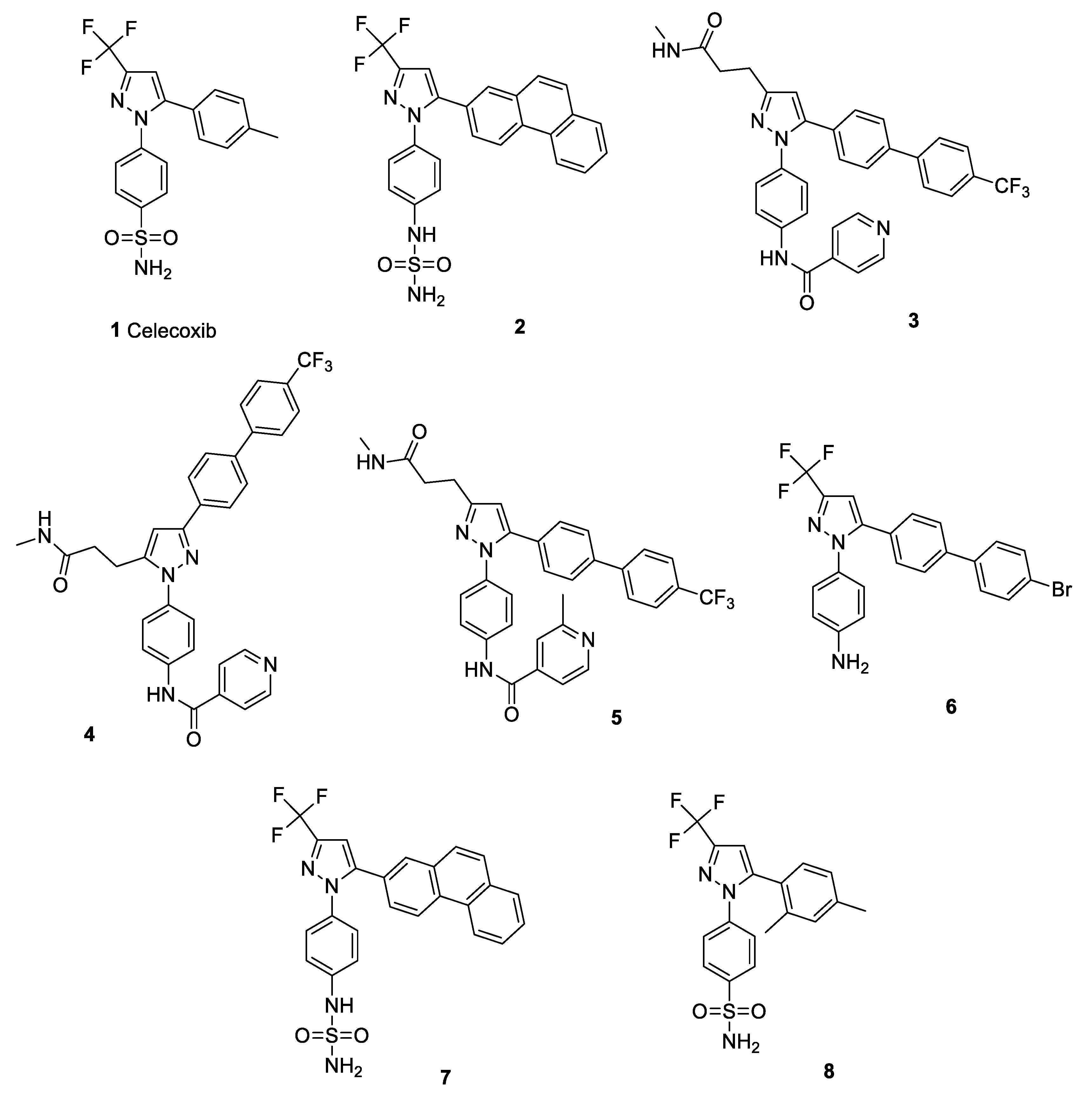
4.1. Celecoxib (Celebrex)
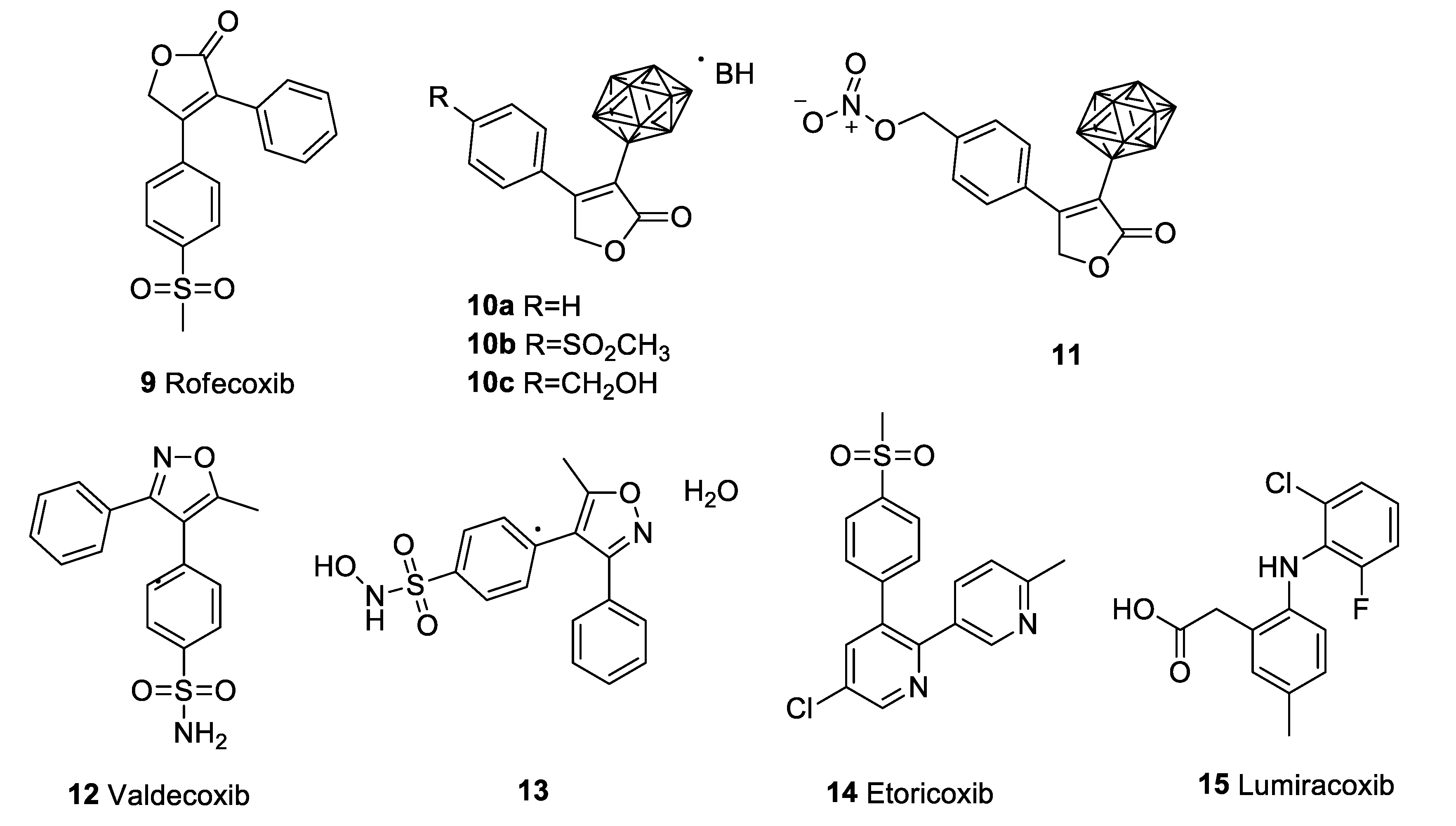
4.2. Rofecoxib (Vioxx)
4.3. Valdecoxib (Bextra)
4.4. Etoricoxib (Arcoxia)
4.5. Lumiracoxib (Prexige)
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-Hydroxydopamine |
| AA | Arachidonic acid |
| APC | Adenoma Prevention with Celecoxib |
| APPROVe | Adenomatous Polyp Prevention on Vioxx |
| CABG-II | Coronary Artery Bypass Surgery |
| COVID-19 | Coronavirus disease 2019 |
| COX | Cyclooxygenase |
| EMEA | European Medicines Agency |
| MCI | Mild cognitive impairment |
| MDR1 | Multi-drug resistance 1 |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NSCLC | Non-small-cell lung cancer |
| OA | Osteoarthritis |
| PG | Prostaglandin |
| PTGS | Prostaglandin-endoperoxide synthase |
| RA | Rheumatoid arthritis |
| T2DM | Type 2 diabetes mellitus |
| tNSAIDs | Traditional non-steroidal anti-inflammatory drugs |
| TxA2 | Thromboxane A2 |
| VIGOR | Vioxx Gastrointestinal Outcomes Research |
References
- Sharma, V.; Bhatia, P.; Alam, O.; Javed Naim, M.; Nawaz, F.; Ahmad Sheikh, A.; Jha, M. Recent Advancement in the Discovery and Development of COX-2 Inhibitors: Insight into Biological Activities and SAR Studies (2008–2019). Bioorganic Chem. 2019, 89, 103007. [Google Scholar] [CrossRef] [PubMed]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological Basis for the Cardiovascular Consequences of COX-2 Inhibition: Therapeutic Challenges and Opportunities. J. Clin. Investig. 2006, 116, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A. Proposed Mechanisms. Am. J. Physiol. 1983, 245, G601–G623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkey, C.J. COX-2 Inhibitors. Lancet Lond. Engl. 1999, 353, 307–314. [Google Scholar] [CrossRef]
- Garavito, R.M.; Malkowski, M.G.; DeWitt, D.L. The Structures of Prostaglandin Endoperoxide H Synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 129–152. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Garg, S.K.; Kumar, R.; Motiwala, H.F.; Jadhavar, P.S. Progress in COX-2 Inhibitors: A Journey so Far. Curr. Med. Chem. 2010, 17, 1563–1593. [Google Scholar] [CrossRef]
- Deb, P.K.; Mailabaram, R.P.; Saadh, M.J.; Al-Jaidi, B. Molecular Basis of Binding Interactions of NSAIDs and Computer-Aided Drug Design Approaches in the Pursuit of the Development of Cyclooxygenase-2 (COX-2) Selective Inhibitors; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Seibert, K.; Masferrer, J.; Zhang, Y.; Gregory, S.; Olson, G.; Hauser, S.; Leahy, K.; Perkins, W.; Isakson, P. Mediation of Inflammation by Cyclooxygenase-2. Agents Actions. Suppl. 1995, 46, 41–50. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-2 Chronology. Gut 2005, 54, 1509–1514. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J. The COXIB Experience: A Look in the Rearview Mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Moncada, S.; Vane, J.R. Indomethacin and Aspirin Abolish Prostaglandin Release from the Spleen. Nat. New Biol. 1971, 231, 237–239. [Google Scholar] [CrossRef]
- Smith, J.B.; Willis, A.L. Aspirin Selectively Inhibits Prostaglandin Production in Human Platelets. Nat. New Biol. 1971, 231, 235–237. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Fu, J.Y.; Masferrer, J.L.; Seibert, K.; Raz, A.; Needleman, P. The Induction and Suppression of Prostaglandin H2 Synthase (Cyclooxygenase) in Human Monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [Google Scholar] [CrossRef]
- Kujubu, D.A.; Fletcher, B.S.; Varnum, B.C.; Lim, R.W.; Herschman, H.R. TIS10, a Phorbol Ester Tumor Promoter-Inducible MRNA from Swiss 3T3 Cells, Encodes a Novel Prostaglandin Synthase/Cyclooxygenase Homologue. J. Biol. Chem. 1991, 266, 12866–12872. [Google Scholar] [CrossRef]
- Xie, W.L.; Chipman, J.G.; Robertson, D.L.; Erikson, R.L.; Simmons, D.L. Expression of a Mitogen-Responsive Gene Encoding Prostaglandin Synthase Is Regulated by MRNA Splicing. Proc. Natl. Acad. Sci. USA 1991, 88, 2692–2696. [Google Scholar] [CrossRef] [Green Version]
- Masferrer, J.L.; Zweifel, B.S.; Manning, P.T.; Hauser, S.D.; Leahy, K.M.; Smith, W.G.; Isakson, P.C.; Seibert, K. Selective Inhibition of Inducible Cyclooxygenase 2 in Vivo Is Antiinflammatory and Nonulcerogenic. Proc. Natl. Acad. Sci. USA 1994, 91, 3228–3232. [Google Scholar] [CrossRef] [Green Version]
- Prasit, P.; Riendeau, D. Chapter 21. Selective Cyclooxygenase-2 Inhibitors. In Annual Reports in Medicinal Chemistry; Bristol, J.A., Ed.; Academic Press: Cambridge, MA, USA, 1997; Volume 32, pp. 211–220. [Google Scholar] [CrossRef]
- Talley, J.J. Selective Inhibitors of Cyclooxygenase-2 (COX-2). Prog. Med. Chem. 1999, 36, 201–234. [Google Scholar] [CrossRef]
- FitzGerald, G.A.; Patrono, C. The Coxibs, Selective Inhibitors of Cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal Toxicity With Celecoxib vs Nonsteroidal Anti-Inflammatory Drugs for Osteoarthritis and Rheumatoid ArthritisThe CLASS Study: A Randomized Controlled Trial. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Schnitzer, T.J.; Burmester, G.R.; Mysler, E.; Hochberg, M.C.; Doherty, M.; Ehrsam, E.; Gitton, X.; Krammer, G.; Mellein, B.; Matchaba, P.; et al. Comparison of Lumiracoxib with Naproxen and Ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), Reduction in Ulcer Complications: Randomised Controlled Trial. Lancet Lond. Engl. 2004, 364, 665–674. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Bradford, W.D.; Kleit, A.N.; Nietert, P.J.; Steyer, T.; McIlwain, T.; Ornstein, S. How Direct-to-Consumer Television Advertising for Osteoarthritis Drugs Affects Physicians’ Prescribing Behavior. Health Aff. Proj. Hope 2006, 25, 1371–1377. [Google Scholar] [CrossRef]
- Topol, E.J. Failing the Public Health—Rofecoxib, Merck, and the FDA. N. Engl. J. Med. 2004, 351, 1707–1709. [Google Scholar] [CrossRef] [Green Version]
- Prakash, S.; Valentine, V. Timeline: The Rise and Fall of Vioxx; National Public Radio: Washington, DC, USA, 2007. [Google Scholar]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors. JAMA 2001, 286, 954–959. [Google Scholar] [CrossRef]
- Topol, E.J.; Falk, G.W. A Coxib a Day Won’t Keep the Doctor Away. Lancet 2004, 364, 639–640. [Google Scholar] [CrossRef]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; et al. Cardiovascular Events Associated with Rofecoxib in a Colorectal Adenoma Chemoprevention Trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Ott, E.; Nussmeier, N.A.; Duke, P.C.; Feneck, R.O.; Alston, R.P.; Snabes, M.C.; Hubbard, R.C.; Hsu, P.H.; Saidman, L.J.; Mangano, D.T.; et al. Efficacy and Safety of the Cyclooxygenase 2 Inhibitors Parecoxib and Valdecoxib in Patients Undergoing Coronary Artery Bypass Surgery. J. Thorac. Cardiovasc. Surg. 2003, 125, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Nussmeier, N.A.; Whelton, A.A.; Brown, M.T.; Langford, R.M.; Hoeft, A.; Parlow, J.L.; Boyce, S.W.; Verburg, K.M. Complications of the COX-2 Inhibitors Parecoxib and Valdecoxib after Cardiac Surgery. N. Engl. J. Med. 2005, 352, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; McMurray, J.J.V.; Pfeffer, M.A.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.F.; Zauber, A.; Hawk, E.; Bertagnolli, M.; et al. Cardiovascular Risk Associated with Celecoxib in a Clinical Trial for Colorectal Adenoma Prevention. N. Engl. J. Med. 2005, 352, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Wadman, M. Merck Settles Vioxx Lawsuits for $4.85 Billion. Nature 2007. [Google Scholar] [CrossRef]
- Melnikova, I. Future of COX 2 Inhibitors. Nat. Rev. Drug Discov. 2005, 4, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Stempel, J. Pfizer in $486 Million Settlement of Celebrex, Bextra Litigation; Reuters: Toronto, OM, USA, 2016. [Google Scholar]
- Burton, B. Australian Drugs Regulator Cancels Registration of COX 2 Inhibitor. BMJ 2007, 335, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, B. COX 2 Inhibitor Rejected in North America but Retained in Europe. BMJ 2007, 335, 791. [Google Scholar] [CrossRef] [Green Version]
- EMA. European Medicines Agency Recommends Withdrawal of the Marketing Authorisations for Lumiracoxib-Containing Medicines. Available online: https://www.ema.europa.eu/en/news/european-medicines-agency-recommends-withdrawal-marketing-authorisations-lumiracoxib-containing (accessed on 20 February 2022).
- Konstantinopoulos, P.A.; Lehmann, D.F. The Cardiovascular Toxicity of Selective and Nonselective Cyclooxygenase Inhibitors: Comparisons, Contrasts, and Aspirin Confounding. J. Clin. Pharmacol. 2005, 45, 742–750. [Google Scholar] [CrossRef]
- Arunasree, K.M.; Roy, K.R.; Anilkumar, K.; Aparna, A.; Reddy, G.V.; Reddanna, P. Imatinib-Resistant K562 Cells Are More Sensitive to Celecoxib, a Selective COX-2 Inhibitor: Role of COX-2 and MDR-1. Leuk. Res. 2008, 32, 855–864. [Google Scholar] [CrossRef]
- Kalle, A.M.; Sachchidanand, S.; Pallu, R. Bcr-Abl-Independent Mechanism of Resistance to Imatinib in K562 Cells: Induction of Cyclooxygenase-2 (COX-2) by Histone Deacetylases (HDACs). Leuk. Res. 2010, 34, 1132–1138. [Google Scholar] [CrossRef]
- Patel, V.A.; Dunn, M.J.; Sorokin, A. Regulation of MDR-1 (P-Glycoprotein) by Cyclooxygenase-2. J. Biol. Chem. 2002, 277, 38915–38920. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.R.; Reddy, G.V.; Maitreyi, L.; Agarwal, S.; Achari, C.; Vali, S.; Reddanna, P. Celecoxib Inhibits MDR1 Expression through COX-2-Dependent Mechanism in Human Hepatocellular Carcinoma (HepG2) Cell Line. Cancer Chemother. Pharmacol. 2010, 65, 903–911. [Google Scholar] [CrossRef]
- Kalle, A.M.; Rizvi, A. Inhibition of Bacterial Multidrug Resistance by Celecoxib, a Cyclooxygenase-2 Inhibitor. Antimicrob. Agents Chemother. 2011, 55, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Varma, G.Y.N.; Kummari, G.; Paik, P.; Kalle, A.M. Celecoxib Potentiates Antibiotic Uptake by Altering Membrane Potential and Permeability in Staphylococcus Aureus. J. Antimicrob. Chemother. 2019, 74, 3462–3472. [Google Scholar] [CrossRef]
- Tzeng, S.-R.; Huang, Y.-W.; Zhang, Y.-Q.; Yang, C.-Y.; Chien, H.-S.; Chen, Y.-R.; Yu, S.-L.; Chen, C.S.; Chiu, H.-C. A Celecoxib Derivative Eradicates Antibiotic-Resistant Staphylococcus Aureus and Biofilms by Targeting YidC2 Translocase. Int. J. Mol. Sci. 2020, 21, 9312. [Google Scholar] [CrossRef]
- Thangamani, S.; Younis, W.; Seleem, M.N. Repurposing Celecoxib as a Topical Antimicrobial Agent. Front. Microbiol. 2015, 6, 750. [Google Scholar] [CrossRef] [Green Version]
- Salunke, S.B.; Azad, A.K.; Kapuriya, N.P.; Balada-Llasat, J.-M.; Pancholi, P.; Schlesinger, L.S.; Chen, C.-S. Design and Synthesis of Novel Anti-Tuberculosis Agents from the Celecoxib Pharmacophore. Bioorg. Med. Chem. 2015, 23, 1935–1943. [Google Scholar] [CrossRef]
- Naftalin, C.M.; Verma, R.; Gurumurthy, M.; Hee, K.H.; Lu, Q.; Yeo, B.C.M.; Tan, K.H.; Lin, W.; Yu, B.; Seng, K.Y.; et al. Adjunctive Use of Celecoxib with Anti-Tuberculosis Drugs: Evaluation in a Whole-Blood Bactericidal Activity Model. Sci. Rep. 2018, 8, 13491. [Google Scholar] [CrossRef]
- Chiu, H.-C.; Yang, J.; Soni, S.; Kulp, S.K.; Gunn, J.S.; Schlesinger, L.S.; Chen, C.-S. Pharmacological Exploitation of an Off-Target Antibacterial Effect of the Cyclooxygenase-2 Inhibitor Celecoxib against Francisella Tularensis. Antimicrob. Agents Chemother. 2009, 53, 2998–3002. [Google Scholar] [CrossRef] [Green Version]
- Hoang, K.V.; Adcox, H.E.; Fitch, J.R.; Gordon, D.M.; Curry, H.M.; Schlesinger, L.S.; White, P.; Gunn, J.S. AR-13, a Celecoxib Derivative, Directly Kills Francisella In Vitro and Aids Clearance and Mouse Survival In Vivo. Front. Microbiol. 2017, 8, 1695. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Mei, Y.; Li, Q.; Zhang, M.; Tang, H.; Liu, T.; Feng, F.; Fu, Q.; Jiang, Y.; Ye, Q. Repurposing Celecoxib Analogues as Leads for Antibiotics. Future Med. Chem. 2021, 13, 959–974. [Google Scholar] [CrossRef]
- Annamanedi, M.; Varma, G.Y.N.; Anuradha, K.; Kalle, A.M. Celecoxib Enhances the Efficacy of Low-Dose Antibiotic Treatment against Polymicrobial Sepsis in Mice and Clinical Isolates of ESKAPE Pathogens. Front. Microbiol. 2017, 8, 805. [Google Scholar] [CrossRef]
- Borah, P.; Deb, P.K.; Deka, S.; Venugopala, K.N.; Singh, V.; Mailavaram, R.P.; Kalia, K.; Tekade, R.K. Current Scenario and Future Prospect in the Management of COVID-19. Curr. Med. Chem. 2021, 28, 284–307. [Google Scholar] [CrossRef]
- Borah, P.; Deb, P.K.; Chandrasekaran, B.; Goyal, M.; Bansal, M.; Hussain, S.; Pottathil, S.; Venugopala, K.N.; Al-Shar, N.A.; Deka, S. Neurological Consequences of SARS-CoV-2 Infection and Concurrence of Treatment-Induced Neuropsychiatric Adverse Events in COVID-19 Patients: Navigating the Uncharted. Front. Mol. Biosci. 2021, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Chen, Y.; You, K.; Tan, S.; Wu, F.; Tao, J.; Chen, X.; Zhang, J.; Xiong, Y.; Yuan, F.; et al. Celebrex Adjuvant Therapy on Coronavirus Disease 2019: An Experimental Study. Front. Pharmacol. 2020, 11, 561674. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The Effect of Celecoxib, a Cyclooxygenase-2 Inhibitor, in Familial Adenomatous Polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yue, P.; Zhou, Z.; Khuri, F.R.; Sun, S.-Y. Death Receptor Regulation and Celecoxib-Induced Apoptosis in Human Lung Cancer Cells. J. Natl. Cancer Inst. 2004, 96, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Schellhorn, M.; Haustein, M.; Frank, M.; Linnebacher, M.; Hinz, B. Celecoxib Increases Lung Cancer Cell Lysis by Lymphokine-Activated Killer Cells via Upregulation of ICAM-1. Oncotarget 2015, 6, 39342–39356. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 Promotes Human Cholangiocarcinoma Growth: Evidence for Cyclooxygenase-2-Independent Mechanism in Celecoxib-Mediated Induction of P21waf1/Cip1 and P27kip1 and Cell Cycle Arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Wang, L.; He, Y.; Xiong, H.Q.; Abbruzzese, J.L.; Xie, K. Celecoxib Inhibits Vascular Endothelial Growth Factor Expression in and Reduces Angiogenesis and Metastasis of Human Pancreatic Cancer via Suppression of Sp1 Transcription Factor Activity. Cancer Res. 2004, 64, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Perroud, H.A.; Rico, M.J.; Alasino, C.M.; Queralt, F.; Mainetti, L.E.; Pezzotto, S.M.; Rozados, V.R.; Scharovsky, O.G. Safety and Therapeutic Effect of Metronomic Chemotherapy with Cyclophosphamide and Celecoxib in Advanced Breast Cancer Patients. Future Oncol. Lond. Engl. 2013, 9, 451–462. [Google Scholar] [CrossRef]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase II Study of the Combination Carboplatin plus Celecoxib in Heavily Pre-Treated Recurrent Ovarian Cancer Patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef]
- Penas-Prado, M.; Hess, K.R.; Fisch, M.J.; Lagrone, L.W.; Groves, M.D.; Levin, V.A.; De Groot, J.F.; Puduvalli, V.K.; Colman, H.; Volas-Redd, G.; et al. Randomized Phase II Adjuvant Factorial Study of Dose-Dense Temozolomide Alone and in Combination with Isotretinoin, Celecoxib, and/or Thalidomide for Glioblastoma. Neuro Oncol. 2015, 17, 266–273. [Google Scholar] [CrossRef]
- Hou, L.-C.; Huang, F.; Xu, H.-B. Does Celecoxib Improve the Efficacy of Chemotherapy for Advanced Non-Small Cell Lung Cancer? Br. J. Clin. Pharmacol. 2016, 81, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-W.; Chen, B.; Zhang, J.; Qi, Y.-P.; Liang, J.-H.; Zhong, J.-H.; Xiang, B.-D. Novel Combination of Celecoxib and Metformin Improves the Antitumor Effect by Inhibiting the Growth of Hepatocellular Carcinoma. J. Cancer 2020, 11, 6437–6444. [Google Scholar] [CrossRef]
- Veltman, J.D.; Lambers, M.E.H.; van Nimwegen, M.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G.J.V.; Hegmans, J.P.J.J. COX-2 Inhibition Improves Immunotherapy and Is Associated with Decreased Numbers of Myeloid-Derived Suppressor Cells in Mesothelioma. Celecoxib Influences MDSC Function. BMC Cancer 2010, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Schwarz, M.J.; Dehning, S.; Douhe, A.; Cerovecki, A.; Goldstein-Müller, B.; Spellmann, I.; Hetzel, G.; Maino, K.; Kleindienst, N.; et al. The Cyclooxygenase-2 Inhibitor Celecoxib Has Therapeutic Effects in Major Depression: Results of a Double-Blind, Randomized, Placebo Controlled, Add-on Pilot Study to Reboxetine. Mol. Psychiatry 2006, 11, 680–684. [Google Scholar] [CrossRef]
- Majd, M.; Hashemian, F.; Hosseini, S.M.; Vahdat Shariatpanahi, M.; Sharifi, A. A Randomized, Double-Blind, Placebo-Controlled Trial of Celecoxib Augmentation of Sertraline in Treatment of Drug-Naive Depressed Women: A Pilot Study. Iran. J. Pharm. Res. IJPR 2015, 14, 891–899. [Google Scholar]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of Anti-Inflammatory Treatment on Depression, Depressive Symptoms, and Adverse Effects: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Elnazer, H.Y.; Sampson, A.P.; Baldwin, D.S. Effects of Celecoxib Augmentation of Antidepressant or Anxiolytic Treatment on Affective Symptoms and Inflammatory Markers in Patients with Anxiety Disorders: Exploratory Study. Int. Clin. Psychopharmacol. 2021, 36, 126–132. [Google Scholar] [CrossRef]
- Baune, B.T.; Sampson, E.; Louise, J.; Hori, H.; Schubert, K.O.; Clark, S.R.; Mills, N.; Fourrier, C. No Evidence for Clinical Efficacy of Adjunctive Celecoxib with Vortioxetine in the Treatment of Depression: A 6-Week Double-Blind Placebo Controlled Randomized Trial. Eur. Neuropsychopharmacol. 2021, 53, 34. [Google Scholar] [CrossRef]
- Konheim, Y.L.; Wolford, J.K. Association of a Promoter Variant in the Inducible Cyclooxygenase-2 Gene (PTGS2) with Type 2 Diabetes Mellitus in Pima Indians. Hum. Genet. 2003, 113, 377–381. [Google Scholar] [CrossRef]
- Helmersson, J.; Vessby, B.; Larsson, A.; Basu, S. Association of Type 2 Diabetes with Cyclooxygenase-Mediated Inflammation and Oxidative Stress in an Elderly Population. Circulation 2004, 109, 1729–1734. [Google Scholar] [CrossRef]
- Hsieh, P.-S.; Tsai, H.-C.; Kuo, C.-H.; Chan, J.Y.-H.; Shyu, J.-F.; Cheng, W.-T.; Liu, T.-T. Selective COX2 Inhibition Improves Whole Body and Muscular Insulin Resistance in Fructose-Fed Rats. Eur. J. Clin. Investig. 2008, 38, 812–819. [Google Scholar] [CrossRef]
- Hsieh, P.-S.; Jin, J.-S.; Chiang, C.-F.; Chan, P.-C.; Chen, C.-H.; Shih, K.-C. COX-2-Mediated Inflammation in Fat Is Crucial for Obesity-Linked Insulin Resistance and Fatty Liver. Obesity 2009, 17, 1150–1157. [Google Scholar] [CrossRef]
- El-bahrawy, H.; Hegazy, S.; Farrag, W.; Werida, R.H. Targeting Inflammation Using Celecoxib with Glimepiride in the Treatment of Obese Type 2 Diabetic Egyptian Patients. Int. J. Diabetes Dev. Ctries. 2015, 37, 97–102. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, Y.J.; Li, Y.; Xie, Y. Celecoxib Ameliorates Non-Alcoholic Steatohepatitis in Type 2 Diabetic Rats via Suppression of the Non-Canonical Wnt Signaling Pathway Expression. PLoS ONE 2014, 9, e83819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, L. Celecoxib Alleviates Memory Deficits by Downregulation of COX-2 Expression and Upregulation of the BDNF-TrkB Signaling Pathway in a Diabetic Rat Model. J. Mol. Neurosci. 2017, 62, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Zhao, L.; Ke, T.; Wang, X.; Cao, L.; Liu, S.; He, J.; Rong, W. Celecoxib Ameliorates Diabetic Neuropathy by Decreasing Apoptosis and Oxidative Stress in Dorsal Root Ganglion Neurons via the MiR-155/COX-2 Axis. Exp. Ther. Med. 2021, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pernaute, R.; Ferree, A.; Cooper, O.; Yu, M.; Brownell, A.-L.; Isacson, O. Selective COX-2 Inhibition Prevents Progressive Dopamine Neuron Degeneration in a Rat Model of Parkinson’s Disease. J. Neuroinflamm. 2004, 1, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizaki, A.; Tien, L.-T.; Pang, Y.; Cai, Z.; Tanaka, S.; Numazawa, S.; Bhatt, A.J.; Fan, L.-W. Celecoxib Reduces Brain Dopaminergic Neuronaldysfunction, and Improves Sensorimotor Behavioral Performance in Neonatal Rats Exposed to Systemic Lipopolysaccharide. J. Neuroinflamm. 2013, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Sarbishegi, M.; Mahmoudzadeh-Sagheb, H.; Heidari, Z.; Baharvand, F. The Protective Effect of Celecoxib on CA1 Hippocampal Neurons and Oxidative Stress in a Rat Model of Parkinson’s Disease. Acta Med. Iran. 2019, 57, 94–102. [Google Scholar] [CrossRef]
- Dassati, S.; Schweigreiter, R.; Buechner, S.; Waldner, A. Celecoxib Promotes Survival and Upregulates the Expression of Neuroprotective Marker Genes in Two Different in Vitro Models of Parkinson’s Disease. Neuropharmacology 2021, 194, 108378. [Google Scholar] [CrossRef]
- de Craen, A.J.M.; Gussekloo, J.; Vrijsen, B.; Westendorp, R.G.J. Meta-Analysis of Nonsteroidal Antiinflammatory Drug Use and Risk of Dementia. Am. J. Epidemiol. 2005, 161, 114–120. [Google Scholar] [CrossRef]
- ADAPT Research Group; Lyketsos, C.G.; Breitner, J.C.S.; Green, R.C.; Martin, B.K.; Meinert, C.; Piantadosi, S.; Sabbagh, M. Naproxen and Celecoxib Do Not Prevent AD in Early Results from a Randomized Controlled Trial. Neurology 2007, 68, 1800–1808. [Google Scholar] [CrossRef]
- Soininen, H.; West, C.; Robbins, J.; Niculescu, L. Long-Term Efficacy and Safety of Celecoxib in Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2007, 23, 8–21. [Google Scholar] [CrossRef]
- Zadori, Z.S.; Lazar, B.; Brenner, G.; Laszlo, S.; Bato, E.; Ostorhazi, E.; Juhasz, J.; Szabo, D.; Ferdinandy, P.; Gyires, K. Rofecoxib, in Contrast to Celecoxib, Does Not Have Direct Antimicrobial Effect and Does Not Cause Small Intestinal Dysbiosis in the Rat. Proc. Annu. Meet. Jpn. Pharmacol. Soc. 2018, PO2-6-23. [Google Scholar] [CrossRef]
- Zhu, F.S.; Chen, X.M.; Huang, Z.G.; Wang, Z.R.; Zhang, D.W.; Zhang, X. Rofecoxib Augments Anticancer Effects by Reversing Intrinsic Multidrug Resistance Gene Expression in BGC-823 Gastric Cancer Cells. J. Dig. Dis. 2010, 11, 34–42. [Google Scholar] [CrossRef]
- Buecher, B.; Bouancheau, D.; Broquet, A.; Bezieau, S.; Denis, M.G.; Bonnet, C.; Heymann, M.-F.; Jarry, A.; Galmiche, J.-P.; Blottière, H.M. Growth Inhibitory Effect of Celecoxib and Rofecoxib on Human Colorectal Carcinoma Cell Lines. Anticancer Res. 2005, 25, 225–233. [Google Scholar]
- Hohenforst-Schmidt, W.; Domvri, K.; Zogas, N.; Zarogoulidis, P.; Petanidis, S.; Kioseoglou, E.; Zachariadis, G.; Kakolyris, S.; Porpodis, K.; Gaga, M.; et al. COX-2 Inhibitors, a Potential Synergistic Effect with Antineoplastic Drugs in Lung Cancer. Oncomedicine 2017, 2, 28–36. [Google Scholar] [CrossRef]
- Al-Nimer, M.S.M.; Al-Deen, S.M.; Lateef, Z.W.A. Rofecoxib Prevents CtdsDNA against Damage Induced by Copper Sulfate and Ultraviolet B Radiation in vitro Study. J. Basic Clin. Pharm. 2011, 2, 21–25. [Google Scholar]
- Zhang, Y.; Hoda, M.N.; Zheng, X.; Li, W.; Luo, P.; Maddipati, K.R.; Seki, T.; Ergul, A.; Wang, M.-H. Combined Therapy with COX-2 Inhibitor and 20-HETE Inhibitor Reduces Colon Tumor Growth and the Adverse Effects of Ischemic Stroke Associated with COX-2 Inhibition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R693–R703. [Google Scholar] [CrossRef] [Green Version]
- Gridelli, C.; Gallo, C.; Ceribelli, A.; Gebbia, V.; Gamucci, T.; Ciardiello, F.; Carozza, F.; Favaretto, A.; Daniele, B.; Galetta, D.; et al. Factorial Phase III Randomised Trial of Rofecoxib and Prolonged Constant Infusion of Gemcitabine in Advanced Non-Small-Cell Lung Cancer: The GEmcitabine-COxib in NSCLC (GECO) Study. Lancet Oncol. 2007, 8, 500–512. [Google Scholar] [CrossRef]
- Midgley, R.S.; McConkey, C.C.; Johnstone, E.C.; Dunn, J.A.; Smith, J.L.; Grumett, S.A.; Julier, P.; Iveson, C.; Yanagisawa, Y.; Warren, B.; et al. Phase III Randomized Trial Assessing Rofecoxib in the Adjuvant Setting of Colorectal Cancer: Final Results of the VICTOR Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4575–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzharevski, A.; Paskaš, S.; Sárosi, M.-B.; Laube, M.; Lönnecke, P.; Neumann, W.; Murganić, B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; et al. Carboranyl Derivatives of Rofecoxib with Cytostatic Activity against Human Melanoma and Colon Cancer Cells. Sci. Rep. 2020, 10, 4827. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Mikelis, C.M.; Dixit, A.; Arutla, V.; Srivenugopal, K.S. Conception, Synthesis, and Characterization of a Rofecoxib-Combretastatin Hybrid Drug with Potent Cyclooxygenase-2 (COX-2) Inhibiting and Microtubule Disrupting Activities in Colon Cancer Cell Culture and Xenograft Models. Oncotarget 2018, 9, 26109–26129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.H.; Bigbee, M.J.; Boynton, G.E.; Wakeham, C.M.; Rosenheim, H.M.; Staral, C.J.; Morrissey, J.L.; Hund, A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer’s Disease and Parkinson’s Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals 2010, 3, 1812–1841. [Google Scholar] [CrossRef]
- Hewett, S.J.; Silakova, J.M.; Hewett, J.A. Oral Treatment with Rofecoxib Reduces Hippocampal Excitotoxic Neurodegeneration. J. Pharmacol. Exp. Ther. 2006, 319, 1219–1224. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J.; et al. Effects of Rofecoxib or Naproxen vs Placebo on Alzheimer Disease Progression: A Randomized Controlled Trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.S. Can Rofecoxib Delay a Diagnosis of Alzheimer’s Disease in Patients with Mild Cognitive Impairment? Nat. Clin. Pract. Neurol. 2005, 1, 20–21. [Google Scholar] [CrossRef]
- Al-Nimer, M.S.M. Effects of Meloxicam and Rofecoxib on Psychomotor Performance: A Randomized, Double-Blind, Placebo-Controlled Cross-over Study. Indian J. Pharmacol. 2007, 39, 291. [Google Scholar] [CrossRef]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A Randomized, Double-Blind, Study of Rofecoxib in Patients with Mild Cognitive Impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.S.; Thal, L.J.; Ferris, S.H.; Assaid, C.; Nessly, M.L.; Giuliani, M.J.; Lines, C.R.; Norman, B.A.; Potter, W.Z. Rofecoxib in Patients with Mild Cognitive Impairment: Further Analyses of Data from a Randomized, Double-Blind, Trial. Curr. Alzheimer Res. 2008, 5, 73–82. [Google Scholar] [CrossRef]
- Tremeau Pharmaceuticals. Pipeline. Available online: https://tremeau.com/pipeline (accessed on 18 February 2022).
- Erdélyi, P.; Fodor, T.; Varga, Á.K.; Czugler, M.; Gere, A.; Fischer, J. Chemical and Biological Investigation of N-Hydroxy-Valdecoxib: An Active Metabolite of Valdecoxib. Bioorg. Med. Chem. 2008, 16, 5322–5330. [Google Scholar] [CrossRef]
- Schröder, H.; Höllt, V.; Becker, A. Parecoxib and Its Metabolite Valdecoxib Directly Interact with Cannabinoid Binding Sites in CB1-Expressing HEK 293 Cells and Rat Brain Tissue. Neurochem. Int. 2011, 58, 9–13. [Google Scholar] [CrossRef]
- Ahmad, M.; Zhang, Y.; Liu, H.; Rose, M.E.; Graham, S.H. Prolonged Opportunity for Neuroprotection in Experimental Stroke with Selective Blockade of Cyclooxygenase-2 Activity. Brain Res. 2009, 1279, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Jing, X.; Zou, X.; Wu, J. Role of cyclooxygenase 2 and its inhibitor valdecoxib in liver fibrosis. Zhonghua Yi Xue Za Zhi 2014, 94, 784–787. [Google Scholar]
- Man-Li, Z.; Hui-Ci, W.; Jin-Jin, L.I.U.; Ding, Y.U.; Zi-Wei, W.; Jia-Xin, W.U.; Jun-Xia, L.I. Valdecoxib Induces Apoptosis of Human Breast Cancer MCF-7 Cells by Increasing the Level of ROS. Basic Clin. Med. 2015, 35, 491. [Google Scholar]
- Reis, A.; Birnbaum, F.; Hansen, L.L.; Reinhard, T. Successful Treatment of Cystoid Macular Edema with Valdecoxib. J. Cataract Refract. Surg. 2007, 33, 682–685. [Google Scholar] [CrossRef]
- Benson, S.E.; Ratcliffe, S.; Van Raders, P.; Schlottmann, P.G.; Khan, I.; Newsom, R.; Langford, R.M.; Charteris, D.G. A Randomized Comparison of Parecoxib/Valdecoxib and Placebo for the Prevention of Cystoid Macular Edema after Scleral Buckling Surgery. Retina 2009, 29, 387–394. [Google Scholar] [CrossRef]
- Mishra, D.; Ghosh, G.; Kumar, P.S.; Panda, P.K. Anticancer Activity of Selective Cyclooxygenase-2 Inhibitor with Conventional NSAIDs. Asian J. Chem. 2011, 23, 427–430. [Google Scholar]
- Jain, N.K.; Baghel, K.S. Selective Cyclooxygenase-2 Inhibitor Etoricoxib Attenuated Hypoxic Cancer Milieu Induced M2-Polarization of Macrophages and Acquisition of Pro-Angiogenic and Pro-Invasive Attributes. Res. J. Pharm. Technol. 2019, 12, 5871–5877. [Google Scholar] [CrossRef]
- Md, S.; Alhakamy, N.A.; Alharbi, W.S.; Ahmad, J.; Shaik, R.A.; Ibrahim, I.M.; Ali, J. Development and Evaluation of Repurposed Etoricoxib Loaded Nanoemulsion for Improving Anticancer Activities against Lung Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13284. [Google Scholar] [CrossRef]
- Tripathi, S.; Dwarkanath, B.; Chandna, S.; Saluja, D.; Chopra, M. Cyclooxygenase-2-Dependent and -Independent Effects of Etoricoxib in Human Cervical Cancer Cell Lines. Cancer Res. 2010, 70 (Suppl. S8), 5671. [Google Scholar] [CrossRef]
- Narayanan, B.L.; Venkatesan, N.; Subburaju, T.; Fathah, A. Chemopreventive Role of Combination of Etoricoxib and Atorvastatin on Colon Cancer Induced by 1, 2-Dimethyl Hydrazine on Rats. Int. J. Pharm. Pharm. Sci. 2015, 7, 299–303. [Google Scholar]
- Ahlers, I.; Kubatka, P.; Ahlersová, E.; Bojková, B.; Kassayová, M.; Friedmanová, L.; Kisková, J.; Ďatelinka, I.; Starostová, M. Etoricoxib in the Prevention of Rat Mammary Carcinogenesis. Acta Vet. Brno 2007, 76, 613–618. [Google Scholar] [CrossRef]
- Ali, G.; Omar, H.; Hersi, F.; Abo-Youssef, A.; Ahmed, O.; Mohamed, W. The Protective Role of Etoricoxib Against Diethylnitrosamine/2-Acetylaminofluorene- Induced Hepatocarcinogenesis in Wistar Rats: The Impact of NF-ΚB/COX-2/PGE2 Signaling. Curr. Mol. Pharmacol. 2022, 15, 252–262. [Google Scholar] [CrossRef]
- Liao, H.; Ou, S.; Dong, X.; Liu, J.; Xiao, C. Association of Etoricoxib Treatment and Incident Hypoxia in Patients with Aortic Dissection Undergoing Endovascular Aortic Repair. Biomed. Pharmacother. 2021, 139, 111625. [Google Scholar] [CrossRef]
- Kabir, F.; Nahar, K.; Rahman, M.M.; Mamun, F.; Lasker, S.; Khan, F.; Yasmin, T.; Akter, K.A.; Subhan, N.; Alam, M.A. Etoricoxib Treatment Prevented Body Weight Gain and Ameliorated Oxidative Stress in the Liver of High-Fat Diet-Fed Rats. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 33–47. [Google Scholar] [CrossRef]
- Jenum, S.; Tonby, K.; Rueegg, C.S.; Rühwald, M.; Kristiansen, M.P.; Bang, P.; Olsen, I.C.; Sellæg, K.; Røstad, K.; Mustafa, T.; et al. A Phase I/II Randomized Trial of H56:IC31 Vaccination and Adjunctive Cyclooxygenase-2-Inhibitor Treatment in Tuberculosis Patients. Nat. Commun. 2021, 12, 6774. [Google Scholar] [CrossRef]
- Sil, S.; Ghosh, T. Cox-2 Plays a Vital Role in the Impaired Anxiety Like Behavior in Colchicine Induced Rat Model of Alzheimer Disease. Behav. Neurol. 2016, 2016, 1501527. [Google Scholar] [CrossRef] [Green Version]
- Citraro, R.; Leo, A.; Marra, R.; De Sarro, G.; Russo, E. Antiepileptogenic Effects of the Selective COX-2 Inhibitor Etoricoxib, on the Development of Spontaneous Absence Seizures in WAG/Rij Rats. Brain Res. Bull. 2015, 113, 1–7. [Google Scholar] [CrossRef]
- Hung, Y.-M.; Lin, L.; Chen, C.-M.; Chiou, J.-Y.; Wang, Y.-H.; Wang, P.Y.-P.; Wei, J.C.-C. The Effect of Anti-Rheumatic Medications for Coronary Artery Diseases Risk in Patients with Rheumatoid Arthritis Might Be Changed over Time: A Nationwide Population-Based Cohort Study. PLoS ONE 2017, 12, e0179081. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.-H.; Peng, Y.-S.; Ting, H.-F.; Peijer Hsieh, J.; Huang, Y.-K.; Wang, Y.-H.; Chiou, J.-Y.; Wei, J.C.-C. Etoricoxib and Diclofenac Might Reduce the Risk of Dementia in Patients with Osteoarthritis: A Nation-Wide, Population-Based Retrospective Cohort Study. Dement. Geriatr. Cogn. Disord. 2018, 45, 262–271. [Google Scholar] [CrossRef]
- Attia, J.Z.; Mansour, H.S. Perioperative Duloxetine and Etoricoxibto Improve Postoperative Pain after Lumbar Laminectomy: A Randomized, Double-Blind, Controlled Study. BMC Anesthesiol. 2017, 17, 162. [Google Scholar] [CrossRef]
- Hao, J.; Li, Q.; Xu, S.; Shen, Y.; Sun, G. Effect of Lumiracoxib on Proliferation and Apoptosis of Human Nonsmall Cell Lung Cancer Cells in Vitro. Chin. Med. J. 2008, 121, 602–607. [Google Scholar] [CrossRef]
- Ji-Qing, H.; Gen-Yun, S.; Yu-Xian, S. Mechanisms of Lumiracoxib on Apoptosis in Human Lung Cancer Cells. Chin. J. Clin. Pharmacol. Ther. 2012, 17, 47. [Google Scholar]
- Hu, W.; Criswell, M.H.; Ottlecz, A.; Cornell, T.L.; Danis, R.P.; Lambrou, G.N.; Ciulla, T.A. Oral Administration of Lumiracoxib Reduces Choroidal Neovascular Membrane Development in the Rat Laser-Trauma Model. Retina 2005, 25, 1054–1064. [Google Scholar] [CrossRef]
- Mehra, M.R.; Ruschitzka, F.; Patel, A.N. Retraction-Hydroxychloroquine or Chloroquine with or without a Macrolide for Treatment of COVID-19: A Multinational Registry Analysis. Lancet Lond. Engl. 2020, 395, 1820. [Google Scholar] [CrossRef]
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Retraction: Cardiovascular Disease, Drug Therapy, and Mortality in COVID-19. N. Engl. J. Med. 2020, 382, 2582. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Intervention (Drug) | CT Phase and Identifier | Sponsor(s) | Objective of the Study |
|---|---|---|---|
| Celecoxib | Phase 4 NCT04814355 | Stony Brook University/Brain & Behavior Research Foundation | To assess the effect of celecoxib on neuroinflammation associated with major depressive disorder |
| Phase 2 NCT03896113 | Cliniques universitaires Saint-Luc- Université Catholique de Louvai | To evaluate the influence of prior administration of celecoxib in endometrial cancer | |
| Phase 1 NCT04120636 | Targeted Therapy Technologies, LLC | To determine the effect of sequestered transscleral celecoxib delivery in macular oedema and inflammatory eye disorders | |
| Phase 2 NCT04673578 | University of British Columbia | To assess the efficacy of adjunctive celecoxib to treatment-as-usual in obsessive-compulsive disorder | |
| Phase 4 NCT04147013 | Lawson Health Research Institute | To determine the effect of celecoxib on postoperative narcotic use, aspirin-exacerbated respiratory disease (AERD), and chronic rhinosinusitis | |
| Phase 4 NCT03645187 | Tanta University | To evaluate the efficacy of adjunctive celecoxib therapy to cancer chemotherapy in metastatic colorectal cancer patients | |
| Phase 2 NCT03498326 | Zhejiang University | To determine the efficacy of a combination of celecoxib and gemcitabine in the treatment of R0 resection pancreatic cancer | |
| Phase 2 NCT04786548 | New York State Psychiatric Institute | To examine the effectiveness of celecoxib in combination with ongoing medication in the treatment of obsessive-compulsive disorder (OCD) | |
| Phase 2 NCT04162873 | University of Utah | To assess the efficacy of celecoxib adjunct to standard-of-care therapy in the treatment of patients with advanced head and neck cancer | |
| Phase 1 NCT02885974 | Baylor College of Medicine | To determine the efficacy of celecoxib combined with cisplatin and gemcitabine in the neoadjuvant treatment of localized muscle-invasive bladder cancer | |
| Phase 2 NCT04093323 | Roswell Park Cancer Institute | To study the combination of the polarized dendritic cell (aDC1) vaccine, celecoxib, interferon α-2, and rintatolimod in the treatment of patients with refractory HLA-A2(+) melanoma. | |
| Phase 1/2 NCT03686657 | ARKAY Therapeutics | To determine the effect of RK-01 co-administered with celecoxib, valsartan, and metformin-HCl XR on insulin resistance | |
| Phase 1/2 NCT03926338 | Sun Yat-sen University | To determine the effect of celecoxib combined with anti-PD-1 monoclonal antibody (mAb) in the treatment of dMMR/MSI-H phenotype resectable colorectal cancer | |
| Phase 2 NCT03026140 | The Netherlands Cancer Institute | To assess the effectiveness of celecoxib, nivolumab, and ipilimumab in early-stage colon cancer | |
| Phase 1 NCT04081389 | Roswell Park Cancer Institute | To determine the effect of chemokine modulation therapy (including celecoxib, recombinant interferon α-2, and rintatolimod) and standard chemotherapy administered prior to surgery in treating subjects with early-stage triple-negative breast cancer | |
| Phase 2 NCT01356290 | Medical University of Vienna | To assess the effect of biweekly bevacizumab (i.v.) in combination with celecoxib, thalidomide, fenofibrate, etoposide, and cyclophosphamide in the treatment of recurrent, progressive medulloblastoma, and ependymoma | |
| Phase 2/3 NCT00268476 | Medical Research Council | To assess the multiple therapeutic strategies (including a celecoxib arm) in the treatment of metastatic hormone-naïve prostate cancer | |
| Rofecoxib | Phase 3 NCT04684511 | Tremeau Pharmaceuticals | To determine the safety and efficacy of rofecoxib (TRM-201) in subjects with haemophilic arthropathy |
| Etoricoxib | Phase 1 NCT04830579 | Pharmtechnology LLC | To determine the bioequivalence of two formulations of etoricoxib by Pharmtechnology and Merck |
| Early Phase 1 NCT05142098 | Dow University of Health Sciences | To compare the anti-inflammatory effect of etoricoxib and pre-emptive dexamethasone following impacted third molar surgery | |
| Phase 3 NCT04968158 | Laboratorios Silanes S.A. de C.V. | To compare and determine the safety and efficacy of a combination of etoricoxib and tramadol compared with a combination of acetaminophen and tramadol in the treatment of acute low back pain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Malah, A.A.; Gineinah, M.M.; Deb, P.K.; Khayyat, A.N.; Bansal, M.; Venugopala, K.N.; Aljahdali, A.S. Selective COX-2 Inhibitors: Road from Success to Controversy and the Quest for Repurposing. Pharmaceuticals 2022, 15, 827. https://doi.org/10.3390/ph15070827
El-Malah AA, Gineinah MM, Deb PK, Khayyat AN, Bansal M, Venugopala KN, Aljahdali AS. Selective COX-2 Inhibitors: Road from Success to Controversy and the Quest for Repurposing. Pharmaceuticals. 2022; 15(7):827. https://doi.org/10.3390/ph15070827
Chicago/Turabian StyleEl-Malah, Afaf A., Magdy M. Gineinah, Pran Kishore Deb, Ahdab N. Khayyat, Monika Bansal, Katharigatta N. Venugopala, and Anfal S. Aljahdali. 2022. "Selective COX-2 Inhibitors: Road from Success to Controversy and the Quest for Repurposing" Pharmaceuticals 15, no. 7: 827. https://doi.org/10.3390/ph15070827