
Magnolol and Luteolin Inhibition of α-Glucosidase Activity: Kinetics and Type of Interaction Detected by In Vitro and In Silico Studies

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
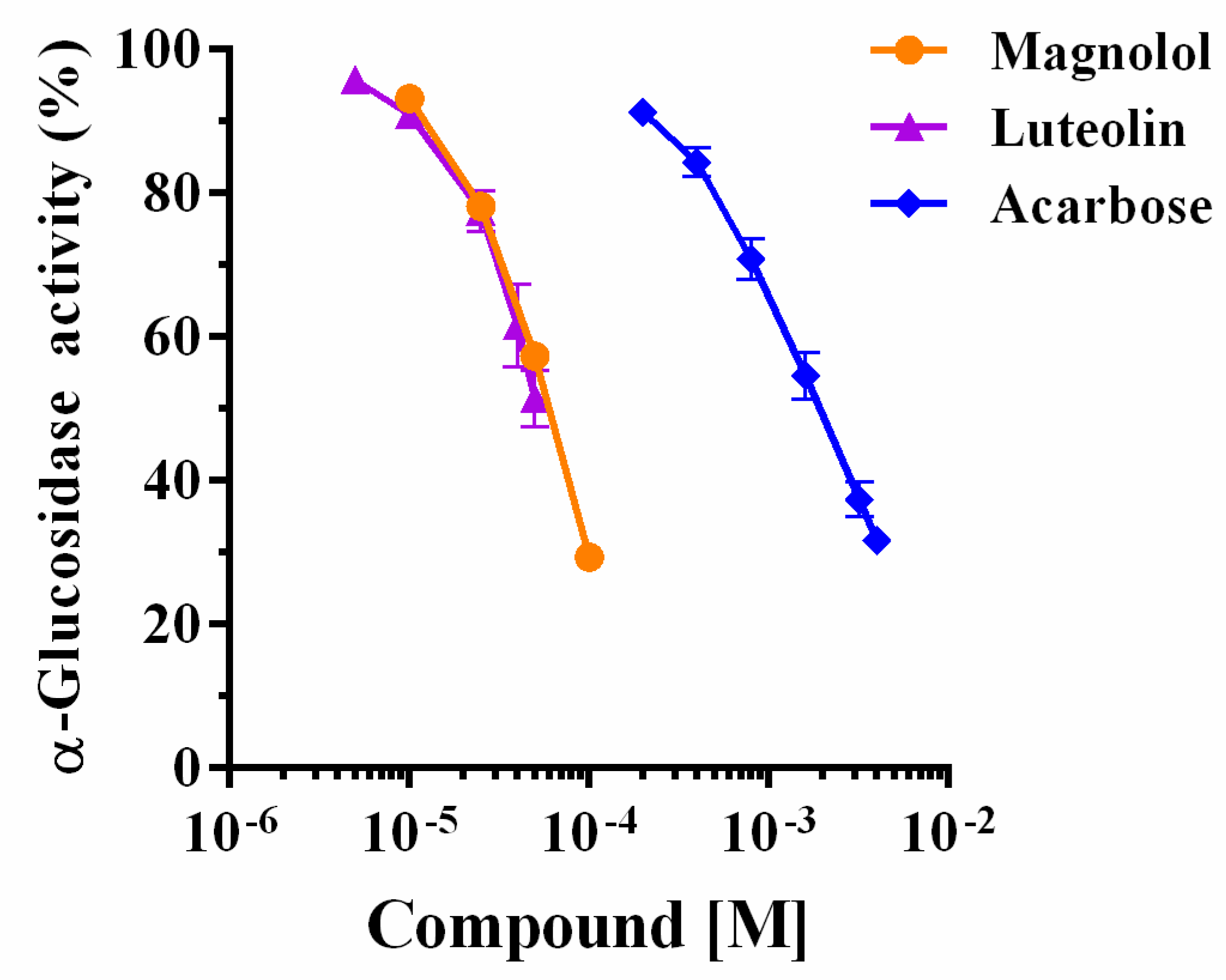
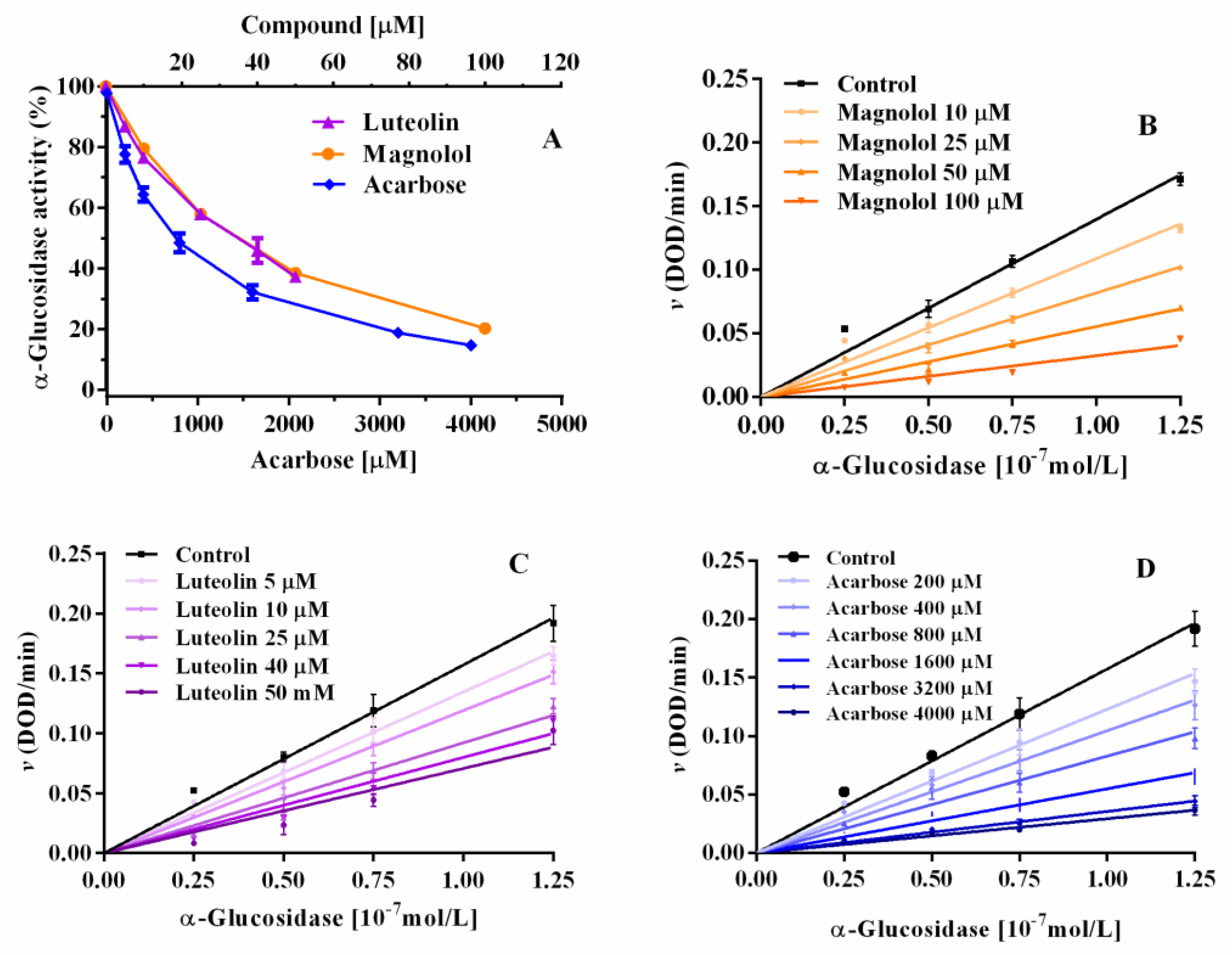
2.1. Yeast α-Glucosidase Inhibition
2.2. α-Glucosidase Inhibition: A Reversible Interaction
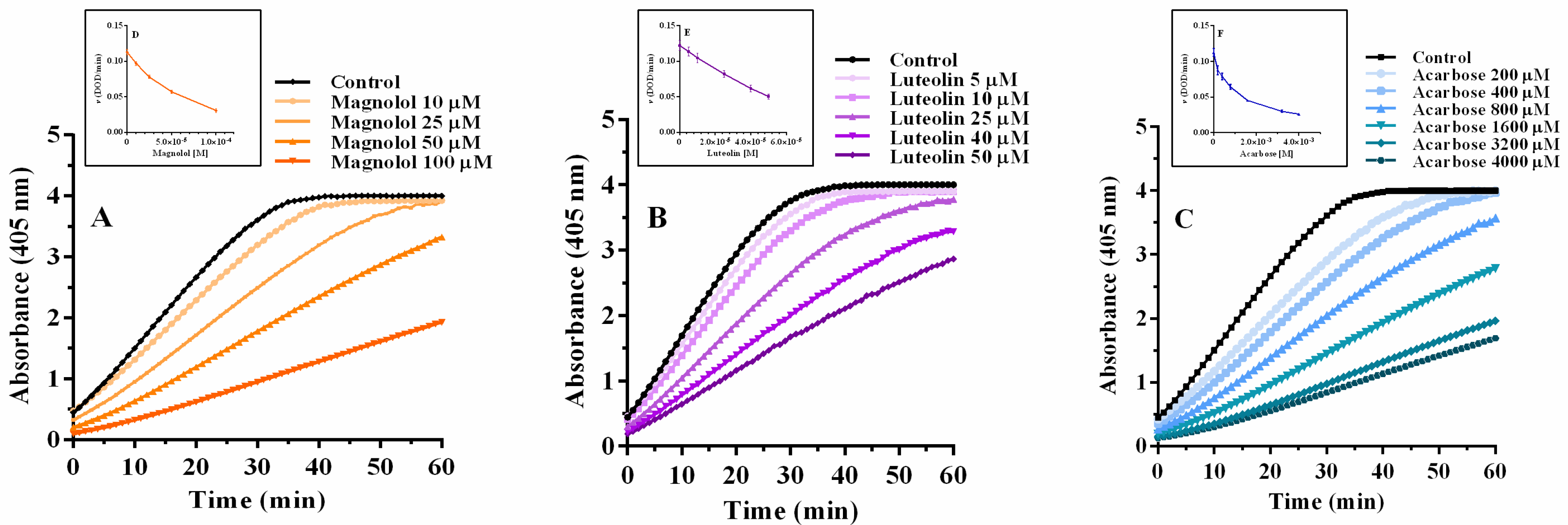
2.3. α-Glucosidase Inhibition: Inhibitory Kinetic Analysis
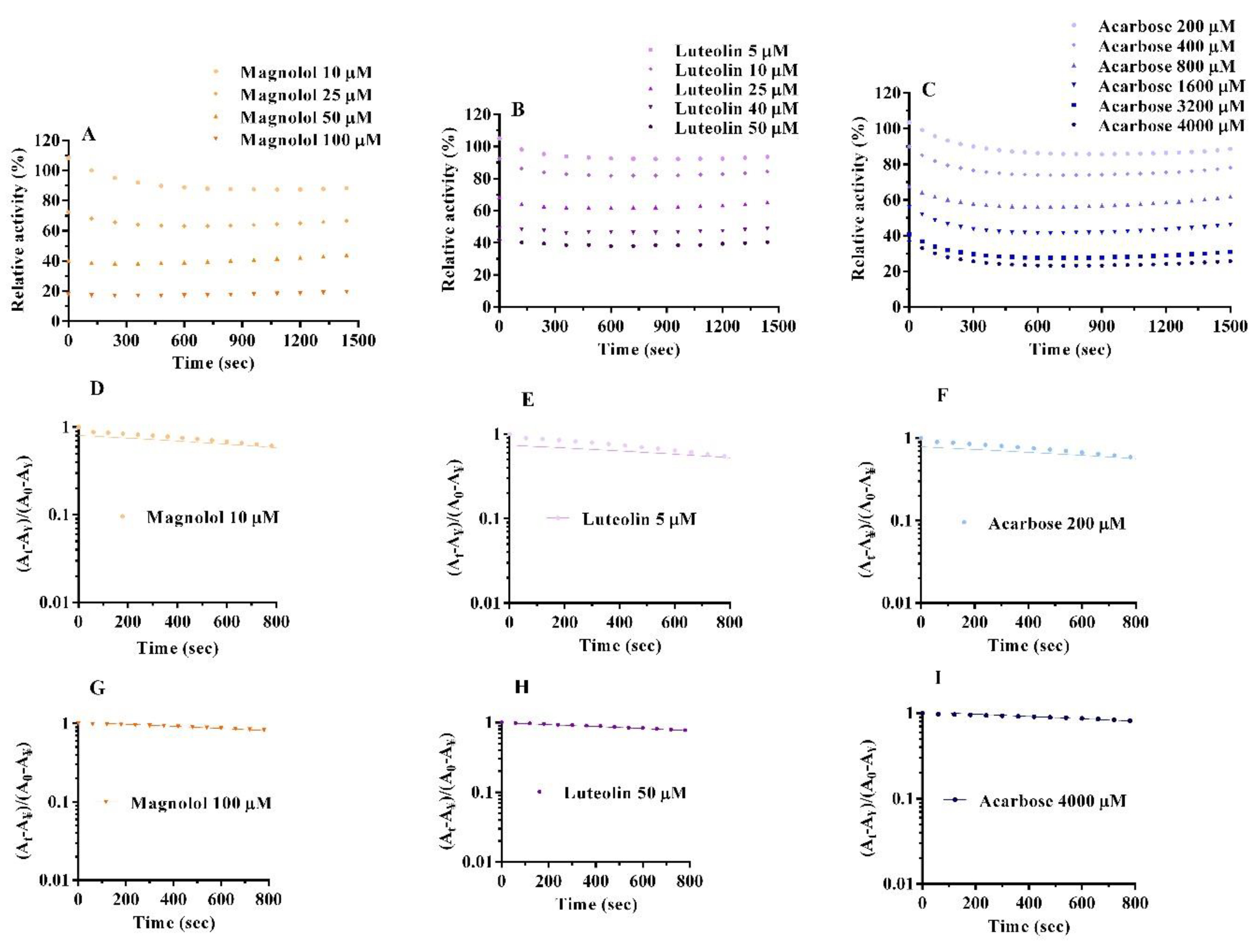
2.4. Inactivation Kinetics, Time Course and Thermodynamics
2.5. Interaction Characteristics between Inhibitors and α-Glucosidase
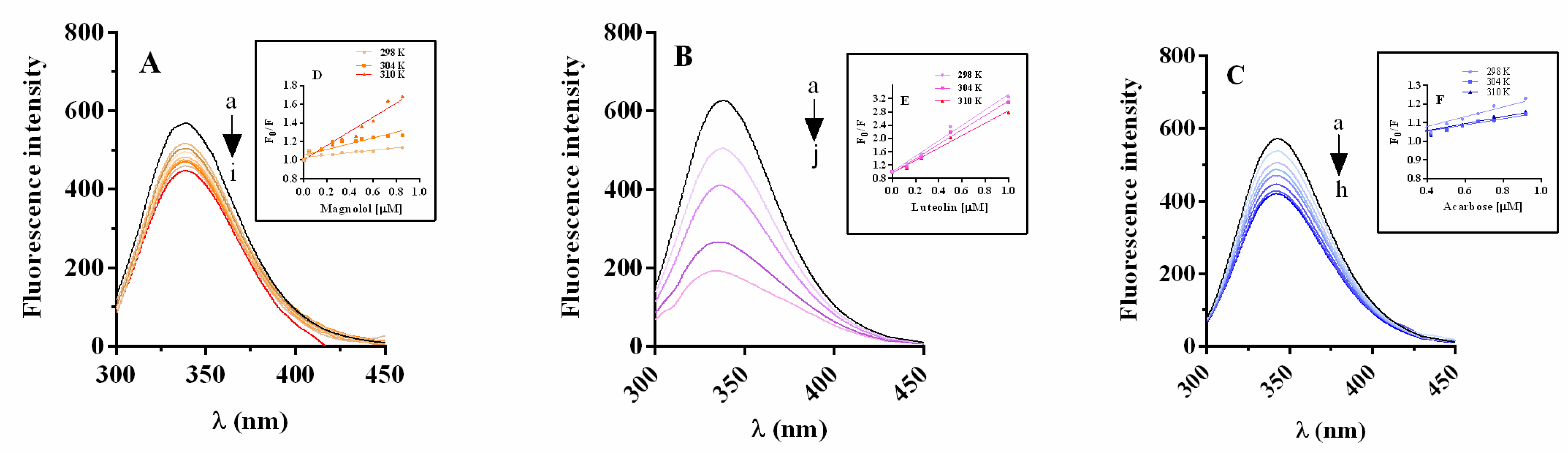
2.5.1. α-Glucosidase Fluorescence Quenching by Magnolol, Luteolin, and Acarbose
2.5.2. Thermodynamic Parameters and Nature of Binding Forces
2.5.3. Energy Transfer between Inhibitor and α-Glucosidase
2.6. Conformational Change of α-Glucosidase
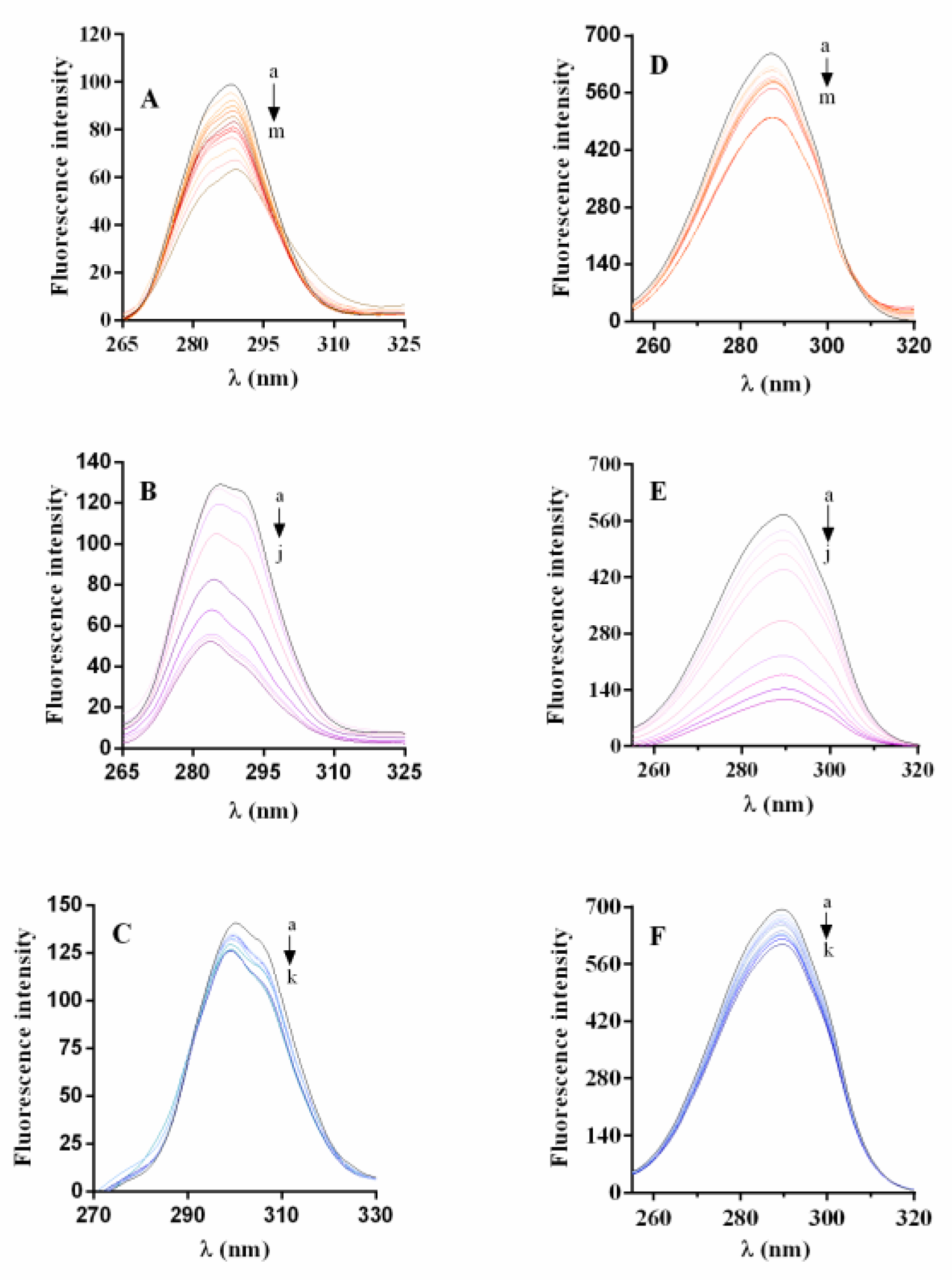
2.6.1. Synchronous Fluorescence Spectra
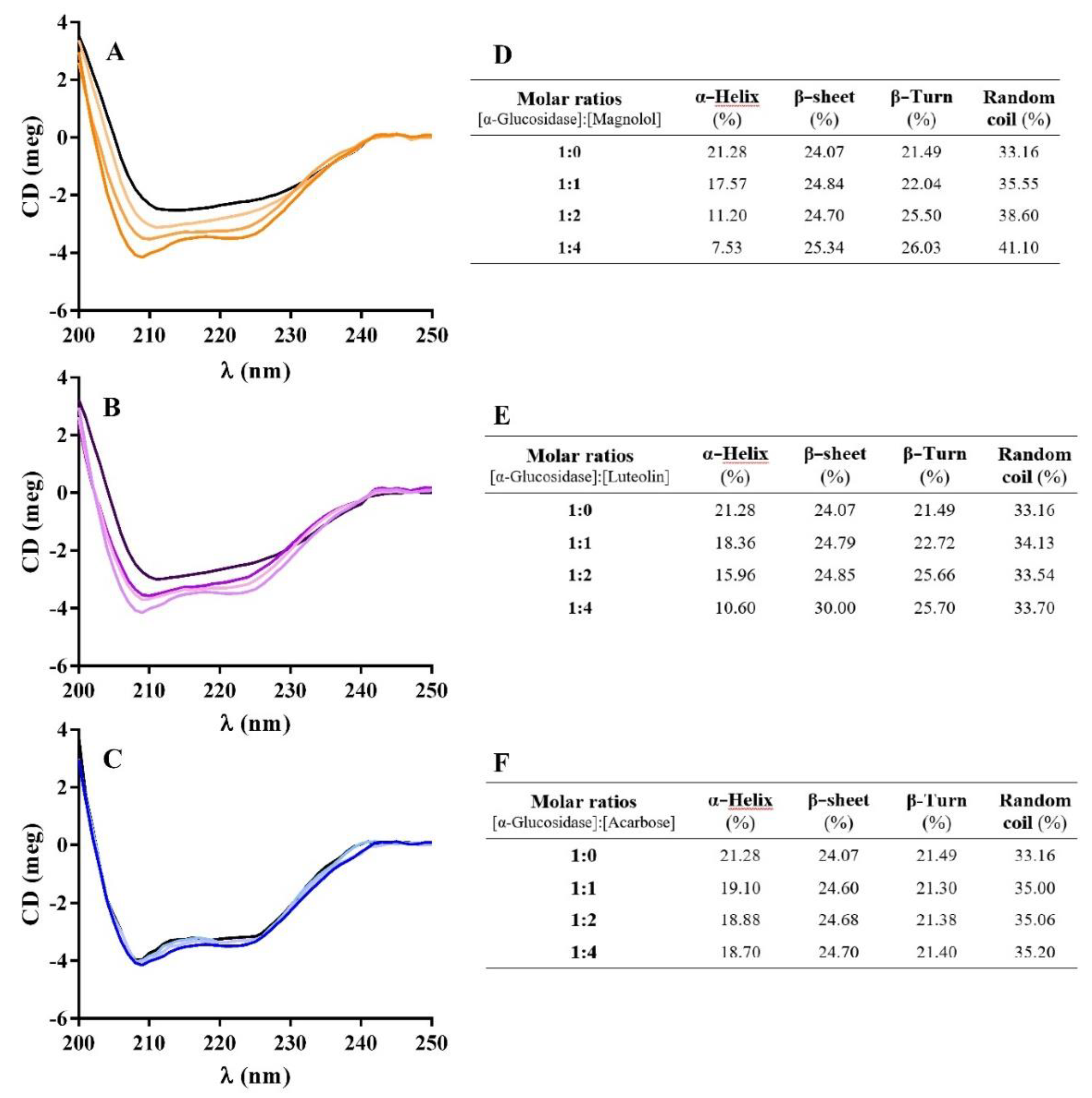
2.6.2. Circular Dichroism (CD) Measurements
2.7. α-Glucosidase Inhibition: Theoretical Homology Modeling
2.7.1. α-Glucosidase Inhibition: Binding Site Analysis and Molecular Docking Studies
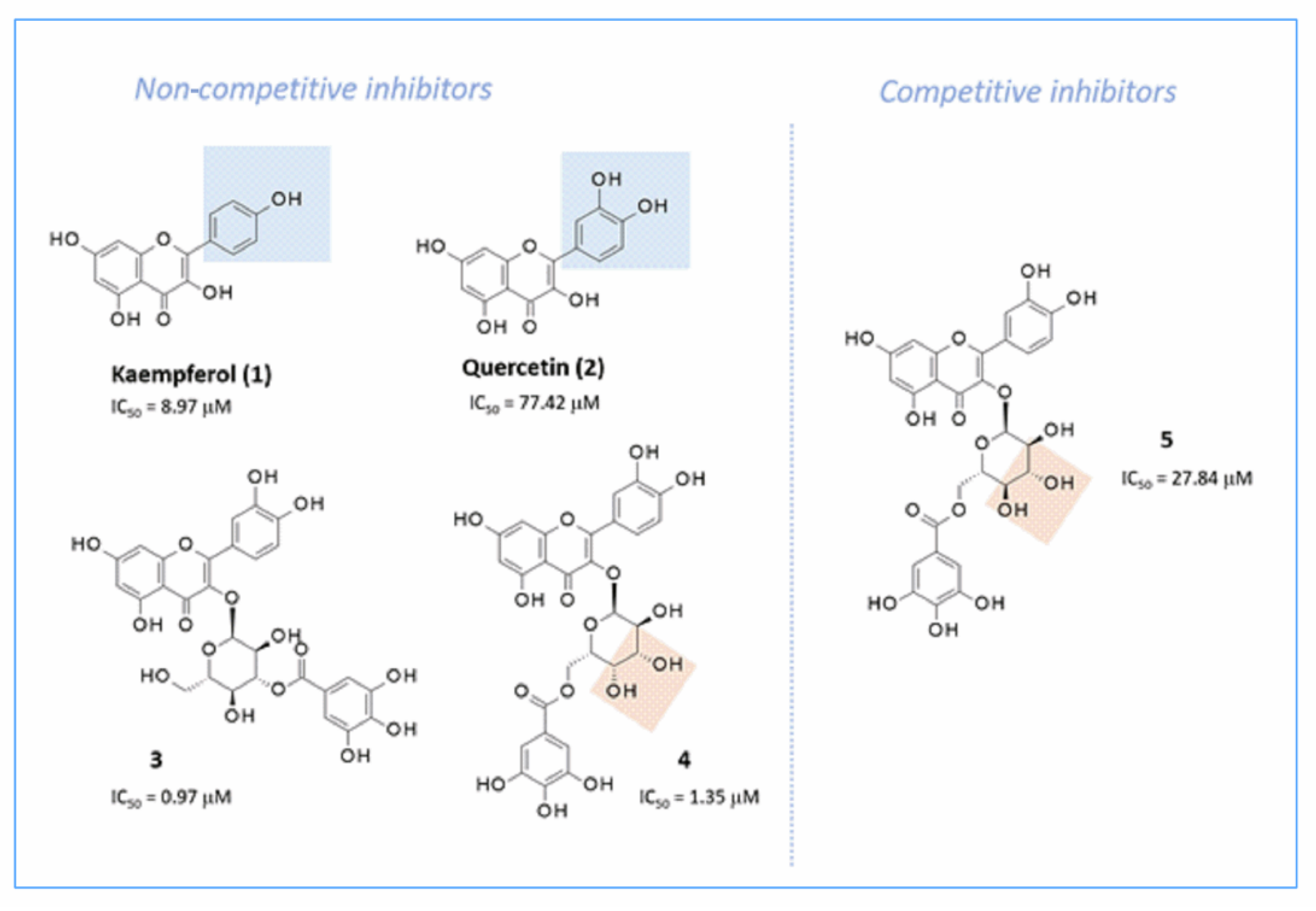
2.7.2. Docking Studies of Non-Sugar-Containing α-Glucosidase Inhibitors: Kaempferol and Quercetin
2.7.3. Docking Studies of Non-Sugar Containing α-Glucosidase Inhibitors: Magnolol and Luteolin
3. Materials and Methods
3.1. Reagents
3.2. Yeast α-Glucosidase Inhibitory Assay
3.3. Kinetic Analysis of Yeast α-Glucosidase Inhibition
3.4. Interaction Characteristics between Inhibitors and Yeast α-Glucosidase
3.4.1. Fluorescence Quenching Analysis
3.4.2. Thermodynamic Parameters and Nature of Binding Forces
3.4.3. Non-Radiation Energy Transfer
3.5. Conformational Changes of Yeast α-Glucosidase during Magnolol-Mediated Inhibition
3.5.1. Synchronous Fluorescence Spectra
3.5.2. Circular Dichroism Measurements
3.6. Homology Modeling and Molecular Docking Studies
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martinez, M.; Santamarina, J.; Pavesi, A.; Musso, C.; Umpierrez, G.E. Glycemic variability and cardiovascular disease in patients with type 2 diabetes. BMJ Open Diabetes Res. Care 2021, 9, e002032. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Bhutani, J.; O’Keefe, J.H. Acarbose: Safe and effective for lowering postprandial hyperglycaemia and improving cardiovascular outcomes. Open Heart 2015, 2, e000327. [Google Scholar] [CrossRef] [Green Version]
- Jugran, A.K.; Rawat, S.; Devkota, H.P.; Bhatt, I.D.; Rawal, R.S. Diabetes and plant-derived natural products: From ethnopharmacological approaches to their potential for modern drug discovery and development. Phytother. Res. 2021, 35, 223–245. [Google Scholar] [CrossRef]
- Tadera, K.; Minami, Y.; Takamatsu, K.; Matsuoka, T. Inhibition of α-glucosidase and α-amylase by flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Yeh, W.J.; Hsia, S.M.; Lee, W.H.; Wu, C.H. Polyphenols with antiglycation activity and mechanisms of action: A review of recent findings. J. Food Drug Anal. 2017, 25, 84–92. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.F.T.; Sousa, J.L.C.; Tomé, S.M.; Ramos, M.J.; Silva, A.M.S.; Fernandes, P.A.; Fernandes, E. α-Glucosidase inhibition by flavonoids: An in vitro and in silico structure–activity relationship study. J. Enzym. Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Poivre, M.; Duez, P. Biological activity and toxicity of the Chinese herb Magnolia officinalis Rehder & E. Wilson (Houpo) and its constituents. J. Zhejiang Univ. Sci. B 2017, 18, 194–214. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Lee, Y.M.; Lee, C.K.; Jung, J.K.; Han, S.B.; Hong, J.T. Therapeutic applications of compounds in the Magnolia family. Pharmacol. Ther. 2011, 130, 157–176. [Google Scholar] [CrossRef]
- Suh, K.S.; Chon, S.; Jung, W.W.; Choi, E.M. Magnolol protects pancreatic β-cells against methylglyoxal-induced cellular dysfunction. Chem. Biol. Interact. 2017, 277, 101–109. [Google Scholar] [CrossRef]
- Wang, J.-J.; Zhao, R.; Liang, J.-C.; Chen, Y. The antidiabetic and hepatoprotective effects of magnolol on diabetic rats induced by high-fat diet and streptozotocin. Yao Xue Xue Bao Acta Pharm. Sin. 2014, 49, 476–481. [Google Scholar]
- Alonso-Castro, A.J.; Zapata-Bustos, R.; Domínguez, F.; García-Carrancá, A.; Salazar-Olivo, L.A. Magnolia dealbata Zucc and its active principles honokiol and magnolol stimulate glucose uptake in murine and human adipocytes using the insulin-signaling pathway. Phytomedicine 2011, 18, 926–933. [Google Scholar] [CrossRef]
- Sohn, E.J.; Kim, C.S.; Kim, Y.S.; Jung, D.H.; Jang, D.S.; Lee, Y.M.; Kim, J.S. Effects of magnolol (5,5′-diallyl-2,2′-dihydroxybiphenyl) on diabetic nephropathy in type 2 diabetic Goto-Kakizaki rats. Life Sci. 2007, 80, 468–475. [Google Scholar] [CrossRef]
- Aziz, N.; Kim, M.Y.; Cho, J.Y. Anti-inflammatory effects of luteolin: A review of in vitro, in vivo, and in silico studies. J. Ethnopharmacol. 2018, 225, 342–358. [Google Scholar] [CrossRef]
- Manzoor, M.F.; Ahmad, N.; Ahmed, Z.; Siddique, R.; Zeng, X.A.; Rahaman, A.; Muhammad Aadil, R.; Wahab, A. Novel extraction techniques and pharmaceutical activities of luteolin and its derivatives. J. Food Biochem. 2019, 43, 1–19. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M.; et al. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; He, Z. Dietary luteolin: A narrative review focusing on its pharmacokinetic properties and effects on glycolipid metabolism. J. Agric. Food Chem. 2021, 69, 1441–1454. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, G.; Pan, J.; Wang, Y. α-Glucosidase inhibition by luteolin: Kinetics, interaction and molecular docking. Int. J. Biol. Macromol. 2014, 64, 213–223. [Google Scholar] [CrossRef]
- Li, H.; Song, F.; Xing, J.; Tsao, R.; Liu, Z.; Liu, S. Screening and structural characterization of α-glucosidase inhibitors from hawthorn leaf flavonoids extract by ultrafiltration LC-DAD-MSn and SORI-CID FTICR MS. J. Am. Soc. Mass Spectrom. 2009, 20, 1496–1503. [Google Scholar] [CrossRef] [Green Version]
- Şöhretoğlu, D.; Sari, S.; Barut, B.; Özel, A. Discovery of potent α-glucosidase inhibitor flavonols: Insights into mechanism of action through inhibition kinetics and docking simulations. Bioorganic Chem. 2018, 79, 257–264. [Google Scholar] [CrossRef]
- Li, X.; Lü, Z.R.; Wang, W.; Han, X.P.; Yang, J.M.; Park, Y.D.; Zhou, H.M.; Sheng, Q.; Lee, J. Effect of Ba2+ on the activity and structure of α-glucosidase: Inhibition kinetics and molecular dynamics simulation. Process Biochem. 2015, 50, 582–588. [Google Scholar] [CrossRef]
- Gupta, M.N. Enzyme function in organic solvents. Eur. J. Biochem. 1992, 203, 25–32. [Google Scholar] [CrossRef]
- Nakamura, A.; Haga, K.; Yamane, K. Three histidine residues in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: Effects of the replacement on pH dependence and transition-state stabilization. Biochemistry 1993, 32, 6624–6631. [Google Scholar] [CrossRef]
- Luo, T.; Lee, J.; Lü, Z.R.; Mu, H.; Yue, L.M.; Park, Y.D.; Ye, Z.M. Effect of cadmium ion on α-glucosidase: An inhibition kinetics and molecular dynamics simulation integration study. Protein J. 2016, 35, 218–224. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, G.; Pan, J.; Gong, D. Novel insights into the inhibitory mechanism of kaempferol on xanthine oxidase. J. Agric. Food Chem. 2015, 63, 526–534. [Google Scholar] [CrossRef]
- Aghili, Z.; Taheri, S.; Zeinabad, H.A.; Pishkar, L.; Saboury, A.A.; Rahimi, A.; Falahati, M. Investigating the interaction of Fe nanoparticles with lysozyme by biophysical and molecular docking studies. PLoS ONE 2016, 11, 1–21. [Google Scholar] [CrossRef]
- Gehlen, M.H. The centenary of the Stern-Volmer equation of fluorescence quenching: From the single line plot to the SV quenching map. J. Photochem. Photobiol. C Photochem. Rev. 2020, 42, 100338. [Google Scholar] [CrossRef]
- Ding, H.; Hu, X.; Xu, X.; Zhang, G.; Gong, D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int. J. Biol. Macromol. 2018, 107, 1844–1855. [Google Scholar] [CrossRef]
- Shuai, L.; Chen, Z.; Fei, P.; Wang, Q.; Yang, T. Spectroscopic analysis on the interaction of ferulic acid and tetramethylpyrazine with trypsin. Luminescence 2014, 29, 79–86. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of macromolecular association reactions: Analysis of forces contributing to stabilization. Biophys. J. 1980, 32, 79–81. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J. Principles of Fluorescence Spectroscopy; Lakowicz, J.R., Ed.; Springer: Boston, MA, USA, 2013; ISBN 9781475787702/1475787707. [Google Scholar]
- Li, Y.Q.; Zhou, F.C.; Gao, F.; Bian, J.S.; Shan, F. Comparative evaluation of quercetin, isoquercetin and rutin as inhibitors of α-glucosidase. J. Agric. Food Chem. 2009, 57, 11463–11468. [Google Scholar] [CrossRef]
- Varlan, A.; Hillebrand, M. Bovine and human serum albumin interactions with 3-carboxyphenoxathiin studied by fluorescence and circular dichroism spectroscopy. Molecules 2010, 15, 3905–3919. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Zhang, G.; Liao, Y.; Gong, D. Inhibitory mechanism of morin on α-glucosidase and its anti-glycation properties. Food Funct. 2016, 7, 3953–3963. [Google Scholar] [CrossRef]
- Pacheco, M.E.; Bruzzone, L. Synchronous fluorescence spectrometry: Conformational investigation or inner filter effect? J. Lumin. 2013, 137, 138–142. [Google Scholar] [CrossRef]
- Wani, T.A.; Bakheit, A.H.; Ansari, M.N.; Al-Majed, A.R.A.; Al-Qahtani, B.M.; Zargar, S. Spectroscopic and molecular modeling studies of binding interaction between bovine serum albumin and roflumilast. Drug Des. Dev. Ther. 2018, 12, 2627–2634. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, G.; Liao, Y.; Gong, D. Inhibitory kinetics and mechanism of kaempferol on α-glucosidase. Food Chem. 2016, 190, 207–215. [Google Scholar] [CrossRef]
- Ma, H.; Wang, L.; Niesen, D.B.; Cai, A.; Cho, B.P.; Tan, W.; Gu, Q.; Xu, J.; Seeram, N.P. Structure activity related, mechanistic, and modeling studies of gallotannins containing a glucitol-core and α-glucosidase. RSC Adv. 2015, 5, 107904–107915. [Google Scholar] [CrossRef]
- Han, L.; Fang, C.; Zhu, R.; Peng, Q.; Li, D.; Wang, M. Inhibitory effect of phloretin on α-glucosidase: Kinetics, interaction mechanism and molecular docking. Int. J. Biol. Macromol. 2017, 95, 520–527. [Google Scholar] [CrossRef]
- Jia, Y.; Ma, Y.; Cheng, G.; Zhang, Y.; Cai, S. Comparative study of dietary flavonoids with different structures as α-glucosidase inhibitors and insulin sensitizers. J. Agric. Food Chem. 2019, 67, 10521–10533. [Google Scholar] [CrossRef]
- Ahmad, M.U.; Rafiq, M.; Zahra, B.; Islam, M.; Ashraf, M.; al-Rashida, M.; Khan, A.; Hussain, J.; Shafiq, Z.; Al-Harrasi, A. Synthesis of benzimidazole based hydrazones as non-sugar based α-glucosidase inhibitors: Structure activity relation and molecular docking. Drug Dev. Res. 2021, 1–11. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Steric hindrance by 2 amino acid residues determines the substrate specificity of isomaltase from Saccharomyces cerevisiae. J. Biosci. Bioeng. 2011, 112, 545–550. [Google Scholar] [CrossRef]
- Şöhretoğlu, D.; Sari, S. Flavonoids as α-glucosidase inhibitors: Mechanistic approaches merged with enzyme kinetics and molecular modelling. Phytochem. Rev. 2020, 19, 1081–1092. [Google Scholar] [CrossRef]
- Cichero, E.; Menozzi, G.; Guariento, S.; Fossa, P. Ligand-based homology modelling of the human CB2 receptor SR144528 antagonist binding site: A computational approach to explore the 1,5-diaryl pyrazole scaffold. Med. Chem. Commun. 2015, 6, 1978–1986. [Google Scholar] [CrossRef]
- Iqbal, S.; Rehman, N.U.; Kortz, U.; Iqbal, J. Development of a fast and efficient CE enzyme assay for the characterization and inhibition studies of α-glucosidase inhibitors. J. Sep. Sci. 2013, 36, 3623–3628. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Green, R.J.; Frazier, R.A. Interaction of flavonoids with bovine serum albumin: A fluorescence quenching study. J. Agric. Food Chem. 2005, 53, 158–163. [Google Scholar] [CrossRef]
- Li, Y.; Gao, F.; Gao, F.; Shan, F.; Bian, J.; Zhao, C. Study on the interaction between 3 flavonoid compounds and α-amylase by fluorescence spectroscopy and enzymatic kinetics. J. Food Sci. 2009, 74, C199–C203. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Zhang, C.; Ma, L.; Wei, T.; Zhao, Y.; Peng, X. Comparative study of inhibition mechanisms of structurally different flavonoid compounds on α-glucosidase and synergistic effect with acarbose. Food Chem. 2021, 347, 129056. [Google Scholar] [CrossRef]
- Miles, A.J.; Ramalli, S.G.; Wallace, B.A. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31, 37–46. Available online: http://dichroweb.cryst.bbk.ac.uk/html/home.shtml (accessed on 1 December 2021). [CrossRef]
- MOE: Chemical Computing Group Inc. Montreal. H3A 2R7 Canada. Available online: http://www.chemcomp.com (accessed on 1 December 2021).
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. Proc. Annu. Hawaii Int. Conf. Syst. Sci. 1995, 5, 256–264. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Liessi, N.; Tasso, B.; Salis, A.; Tonelli, M.; Millo, E.; Pedemonte, N.; Fossa, P.; Cichero, E. Molecular docking and QSAR studies as computational tools exploring the rescue ability of F508DEL CFTR correctors. Int. J. Mol. Sci. 2020, 21, 8084. [Google Scholar] [CrossRef]
- Soga, S.; Shirai, H.; Koborv, M.; Hirayama, N. Use of amino acid composition to predict ligand-binding sites. J. Chem. Inf. Modeling 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Francesconi, V.; Cichero, E.; Kanov, E.V.; Laurini, E.; Pricl, S.; Gainetdinov, R.R.; Tonelli, M. Novel 1-amidino-4-phenylpiperazines as potent agonists at human taar1 receptor: Rational design, synthesis, biological evaluation and molecular docking studies. Pharmaceuticals 2020, 13, 391. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
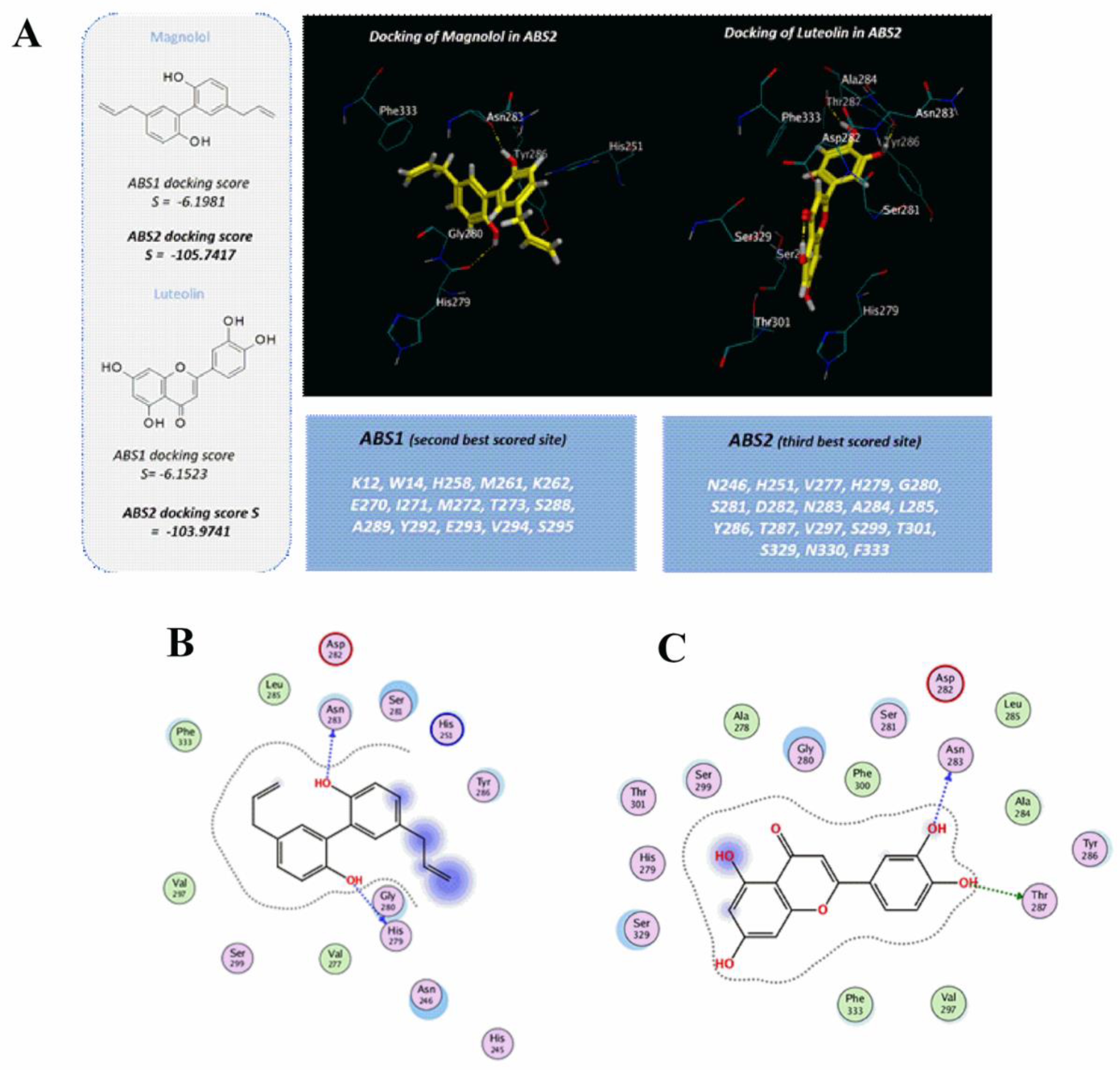{kind=link}
| α-Glucosidase10−6 M | Magnolol IC50 | Luteolin IC50 | Acarbose IC50 | |||
|---|---|---|---|---|---|---|
| 10−5 M | −Log IC50 | 10−5 M | −Log IC50 | 10−5 M | −Log IC50 | |
| 0.025 | 2.83 ± 1.07 a | 4.55 ± 0.03 | 1.40 ± 1.05 a | 4.85 ± 0.02 | 80.54 ± 1.02 b | 3.09 ± 0.01 |
| 0.05 | 2.85 ± 1.10 a | 4.55 ± 0.04 | 2.16 ± 1.17 a | 4.66 ± 0.07 | 86.80 ± 1.05 b | 3.06 ± 0.02 |
| 0.075 | 3.26 ± 1.05 a | 4.49 ± 0.02 | 3.23 ± 1.17 a | 4.49 ± 0.07 | 81.54 ± 1.12 b | 3.09 ± 0.05 |
| 0.125 | 3.55 ± 1.02 a | 4.26 ± 0.01 | 5.94 ± 1.23 a | 4.23 ± 0.09 | 80.70 ± 1.10 b | 3.09 ± 0.04 |
| Compound | Concentration (µM) | Km (mM) | Kcat (sec−1) | Kcat/Km (sec−1 mM−1) | Ki (µM) | Ki’ (µM) | Inhibitor Potency |
|---|---|---|---|---|---|---|---|
| Magnolol | 0 | 0.48 ± 0.054 | 3.89 ± 0.14 | 8.18 ± 2.62 | 78.3 | 132.4 | 4.5 |
| 10 | 0.53 ± 0.08 | 3.59 ± 0.17 | 6.78 ± 2.17 | ||||
| 25 | 0.59 ± 0.08 | 3.29 ± 0.15 | 5.54 ± 1.82 | ||||
| 50 | 0.54 ± 0.08 | 2.86 ± 0.13 | 5.52 ± 1.55 | ||||
| 100 | 0.58±0.24 | 2.22 ± 0.29 | 3.87 ± 1.26 | ||||
| Luteolin | 0 | 0.32 ± 0.04 | 3.52 ± 0.14 | 11.01 ± 0.43 | 34.2 | 35.4 | 10.4 |
| 5 | 0.35 ± 0.05 | 2.95 ± 0.12 | 8.43 ± 0.32 | ||||
| 10 | 0.37 ± 0.05 | 2.65 ± 0.13 | 7.17 ± 0.34 | ||||
| 25 | 0.36 ± 0.07 | 2.02 ± 0.12 | 5.60 ± 0.34 | ||||
| 40 | 0.36 ± 0.07 | 1.78 ± 0.12 | 4.94 ± 0.33 | ||||
| 50 | 0.37 ± 0.05 | 1.41 ± 0.11 | 3.81 ± 0.31 | ||||
| Acarbose | 0 | 0.30 ± 0.08 | 2.92 ± 0.23 | 9.75 ± 2.83 | 356.3 | _ | 1 |
| 200 | 0.91 ± 0.23 | 2.83 ± 0.33 | 3.11 ± 1.42 | ||||
| 400 | 1.31 ± 0.34 | 2.87 ± 0.39 | 2.19 ± 1.14 | ||||
| 800 | 1.96 ± 0.52 | 2.77 ± 0.44 | 1.41 ± 0.84 | ||||
| 1600 | 3.24 ± 0.64 | 2.66 ± 0.36 | 0.82 ± 0.56 | ||||
| 3200 | 5.91 ± 1.81 | 2.79 ± 0.67 | 0.47 ± 0.37 | ||||
| 4000 | 8.30 ± 3.32 | 3.12 ± 1.04 | 0.38 ± 0.36 |
| Compound | Concentration (µM) | K (10−4 s−1) | ᴧᴧG° (kJ mol−1 s−1) |
|---|---|---|---|
| Magnolol | 10 | 2.78 ± 0.05 | 21.10 |
| 25 | 2.81 ± 0.03 | 21.08 | |
| 50 | 2.81 ± 0.01 | 21.07 | |
| 100 | 2.85 ± 0.08 | 21.04 | |
| Luteolin | 5 | 2.67 ± 0.08 | 21.21 |
| 10 | 2.78 ± 0.08 | 21.10 | |
| 25 | 2.81 ± 0.03 | 21.07 | |
| 40 | 2.82 ± 0.04 | 21.06 | |
| 50 | 2.84 ± 0.01 | 21.05 | |
| Acarbose | 200 | 2.76 ± 0.07 | 21.12 |
| 400 | 2.81 ± 0.07 | 21.07 | |
| 800 | 2.83 ± 0.05 | 21.06 | |
| 1600 | 2.84 ± 0.03 | 21.05 | |
| 3200 | 2.85 ± 0.01 | 21.04 | |
| 4000 | 2.85 ± 0.09 | 21.03 |
| Compound | T (K) | Ksv (105 M−1) | Kq (1013 M−1 s−1) |
|---|---|---|---|
| Magnolol | 298 | 3.89 ± 0.01 | 3.90 ± 0.007 |
| 304 | 4.22 ± 0.01 | 4.22 ± 0.011 | |
| 310 | 5.30 ± 0.02 | 5.30 ± 0.022 | |
| Luteolin | 298 | 23.22 ± 0.16 | 23.22 ± 0.159 |
| 304 | 21.73 ± 0.14 | 21.73 ± 0.141 | |
| 310 | 18.68 ± 0.12 | 18.68 ± 0.120 | |
| Acarbose | 298 | 3.75 ± 0.04 | 3.75 ± 0.041 |
| 304 | 1.89 ± 0.01 | 1.89 ± 0.014 | |
| 310 | 1.80 ± 0.02 | 1.80 ± 0.020 |
| System | T (K) | Ka (105 M−1) | n | ΔG° (kJ mol−1) | ∆S° (J mol−1K−1) | ΔH° (kJ mol−1) |
|---|---|---|---|---|---|---|
| Magnolol-α-glucosidase | 298 | 2.97 ± 0.02 | 0.39 | −2.92 | 121.83 | 39.23 |
| 304 | 4.54 ± 0.02 | 0.42 | −2.19 | |||
| 310 | 5.48 ± 0.02 | 0.65 | −1.46 | |||
| Luteolin-α-glucosidase | 298 | 24.86 ± 0.05 | 1.10 | −2.29 | −22.95 | −9.13 |
| 304 | 23.99 ± 0.07 | 1.28 | −2.15 | |||
| 310 | 21.54 ± 0.10 | 1.34 | −2.01 | |||
| Acarbose- α-glucosidase | 298 | 3.91 ± 0.06 | 1.77 | 2.60 | −161.47 | −45.51 |
| 304 | 1.94 ± 0.04 | 1.38 | 3.57 | |||
| 310 | 1.93 ± 0.06 | 1.36 | 4.54 |
| Subscript | Description |
|---|---|
| hb | Interactions between hydrogen bond donor–acceptor pairs. An optimistic view is taken; for example, two hydroxyl groups are assumed to interact in the most favorable way. |
| ion | Ionic interactions. A Coulomb-like term is used to evaluate the interactions between charged groups. This can contribute to or detract from the binding affinity. |
| mlig | Metal ligation. Interactions between nitrogens/sulfurs and transition metals are assumed to be metal-ligation interactions. |
| hh | Hydrophobic interactions, for example, between alkane carbons. These interactions are generally favorable. |
| hp | Interactions between hydrophobic and polar atoms. These interactions are generally unfavorable. |
| aa | An interaction between two atoms. This interaction is weak and generally favorable. |
| S | The Final Score, Which Is the Score of the Last Stage of Refinement. |
|---|---|
| E_conf | The energy of the conformer. If there is a refinement stage, this is the energy calculated at the end of the refinement |
| E_place | Score from the placement stage |
| E_score1 E_score2 | Score from rescoring stages 1 and 2 |
| E_refine | Score from the refinement stage, calculated to be the sum of the van der Waals electrostatics and solvation energies, under the Generalized Born solvation model (GB/VI) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djeujo, F.M.; Ragazzi, E.; Urettini, M.; Sauro, B.; Cichero, E.; Tonelli, M.; Froldi, G. Magnolol and Luteolin Inhibition of α-Glucosidase Activity: Kinetics and Type of Interaction Detected by In Vitro and In Silico Studies. Pharmaceuticals 2022, 15, 205. https://doi.org/10.3390/ph15020205
Djeujo FM, Ragazzi E, Urettini M, Sauro B, Cichero E, Tonelli M, Froldi G. Magnolol and Luteolin Inhibition of α-Glucosidase Activity: Kinetics and Type of Interaction Detected by In Vitro and In Silico Studies. Pharmaceuticals. 2022; 15(2):205. https://doi.org/10.3390/ph15020205
Chicago/Turabian StyleDjeujo, Francine Medjiofack, Eugenio Ragazzi, Miriana Urettini, Beatrice Sauro, Elena Cichero, Michele Tonelli, and Guglielmina Froldi. 2022. "Magnolol and Luteolin Inhibition of α-Glucosidase Activity: Kinetics and Type of Interaction Detected by In Vitro and In Silico Studies" Pharmaceuticals 15, no. 2: 205. https://doi.org/10.3390/ph15020205