3.1. Chemistry–General Remarks
Melting points were determined on a Kofler melting point apparatus and were uncorrected. 1H NMR spectra were recorded on BRUKER AC 300 P (300 MHz) spectrometer, 13C NMR spectra on BRUKER AC 300 P (75 MHz) spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Data are given in the following order: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), and number of protons, and coupling constants J is given in Hertz. The mass spectra (HRMS) were taken, respectively, on a MS/MS ZABSpec Tof Micromass (EBE TOF geometry) at an ionizing potential of 8 eV and on a VARIAN MAT 311 at an ionizing potential of 70 eV in the “Centre Régional de Mesures Physiques de l’Ouest” (CRMPO, Rennes, France). Reactions under microwave irradiations were realized in the Synthewave® 402 apparatus (Merck Eurolab, Div. Prolabo, France) or in the Explorer® 24 CEM microwave reactor (CEM France, Saclay, France), as well as in the Anton Paar Monowave 300® microwave reactor (Anton Paar France, Les Ulis, France). Microwave irradiation reactions were realized in open cylindrical quartz reactor (Ø = 1.8 cm) with the Synthewave® 402 apparatus or in borosilicate glass vials of 10 mL equipped with snap caps (at the end of the irradiation, cooling reaction was realized by compressed air) with the Explorer® 24 or Monowave® 300 reactors. The microwave instrument consists of a continuous focused microwave power output from 0 to 300W for the Synthewave® 402 or the Explorer® 24 CEM apparatus and from 0 to 800W for the Anton Paar Monowave 300® apparatus. All the experiments in each microwave reactor were performed using the stirring option. The target temperature was reached with a ramp of 2–5 min, and the chosen microwave power stays constant to hold the mixture at this temperature. The reaction temperature is monitored using calibrated infrared sensor and the reaction time included the ramp period. The microwave irradiation parameters (power and temperature) were monitored by the ChemDriver software package for the Explorer® 24 CEM apparatus and by the Monowave software package for the Anton Paar Monowave 300® reactor. Solvents were evaporated with a BUCHI rotary evaporator. All reagents and solvents were purchased from Acros (Geel, Belgium), Sigma-Aldrich Chimie (Saint-Quentin-Fallavier, France), and Fluka France and were used without further purification.
3.1.1. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3a–l) in the Synthewave® 402 Microwave Reactor
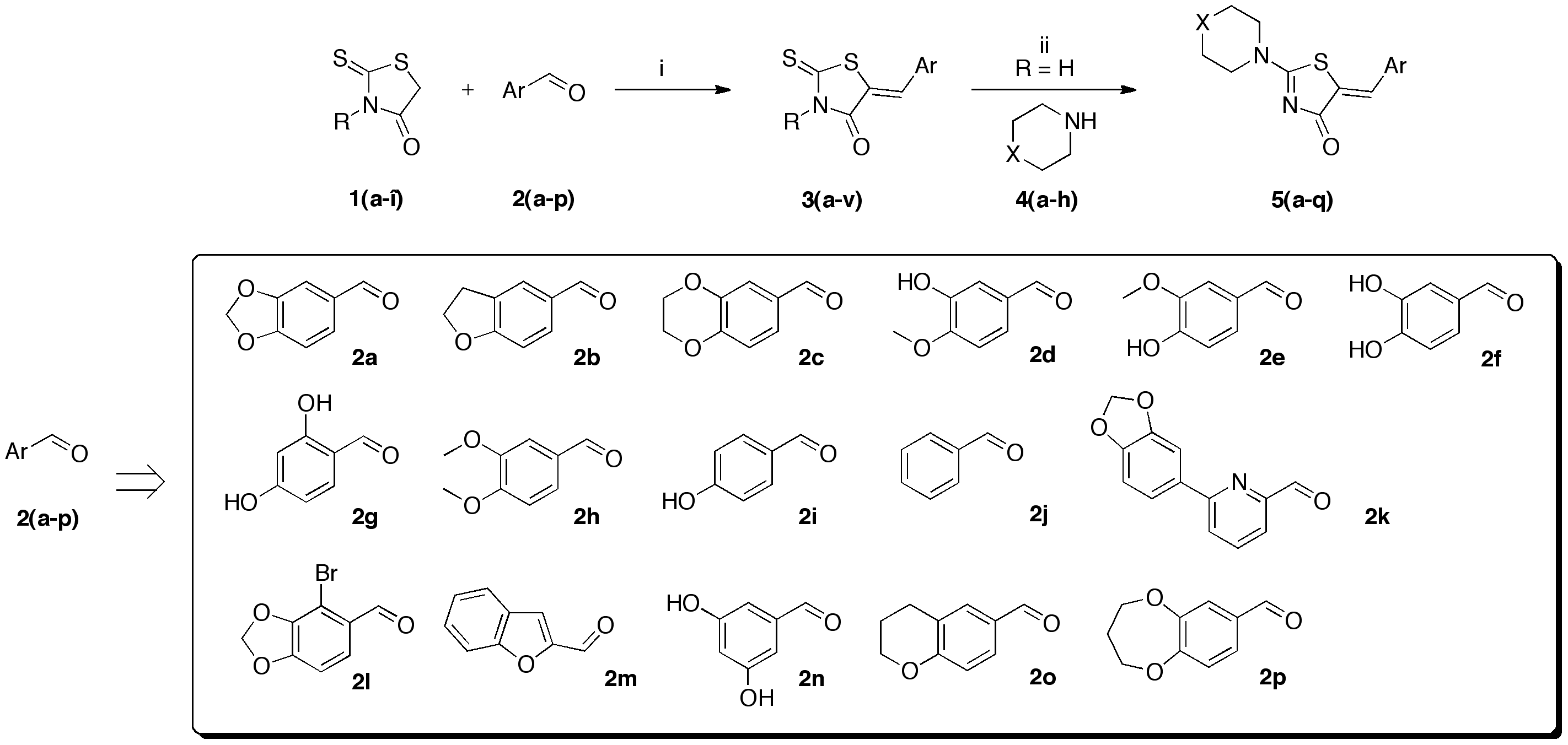
In a cylindrical quartz reactor (Ø = 1.8 cm), successively commercial rhodanine 1a (1 eq), aromatic aldehyde 2 (1 eq), and n-propylamine (2 eq) were placed. The reactor was then introduced into the Synthewave® 402 Prolabo microwave cavity (power: 300 W). The stirred mixture was irradiated at 80 °C (after a ramp of 2 min. from 20 to 80 °C) for 60 min (power level: 30%, 90 W). After microwave dielectric heating, the crude reaction mixture was allowed to cooling down at room temperature, and ethanol (20 mL) was added in the cylindrical quartz reactor. The resulting insoluble product 3 was filtered, recrystallized in EtOH, and dried under high vacuum (10−2 Torr) for 1 h.
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3a). According to the standard procedure, 3a was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), piperonaldehyde 2a (1.081 g, 7.2 mmol), and n-propylamine (1.17 mL, 839 mg, 14.2 mmol) in 79% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.13 (s, 2H, OCH2O); 7.11 (d, 3H, J = 8.7 Hz, H-2, H-5, H-6, Ar); 7.54 (s, 1H, =CH); 13.74 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.1 (OCH2O); 109.2 (C-2′); 109.4 (C-5′); 122.8 (C=, C-5); 126.6 (C-6′); 127.1 (CH=); 131.8 (C-7′); 148.2 (C-4′); 149.6 (C-3′); 169.3 (C=O, C-4); 195.3 (C=S, C-2). HRMS, m/z: 264.9864 found (calculated for C11H7NO3S2, M+. requires 264.9867).
(5Z)-5-(3-Hydroxy-4-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3b). According to the standard procedure, 3b was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 3-hydroxy-4-methoxybenzaldehyde 2d (1.095 g, 7.2 mmol), and n-propylamine (1.17 mL, 839 mg, 14.2 mmol) in 81% yield as light-yellow powder; mp = 222–224 °C. 1H NMR (DMSO-d6) δ: 3.82 (s, 3H, OCH3); 6.94 (d, 1H, J = 6 Hz, H-6, Ar); 7.04 (d, 1H, J = 7.9 Hz, H-5, Ar); 7.11 (s, 1H, H-2, Ar); 7.53 (s, 1H, =CH); 10.06 (br s, 1H, OH); 13.65 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 56.0 (OCH3); 114.7 (C-2′); 116.7 (C-5′); 121.5 (C=, C-5); 124.8 (C-6′); 125.5 (C-1′); 133.1 (=CH); 148.5 (C-4′); 150.4 (C-3′); 169.8 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 267.0023 found (calculated for C11H9NO3S2, M+. requires 267.0024).
(5Z)-5-(4-Hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3c). According to the standard procedure, 3c was synthesized from commercial rhodanine 1a (665 mg, 5 mmol), 4-hydroxy-3-methoxybenzaldehyde 2e (1.52 g, 10 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 80% yield as light-yellow powder; mp = 220–222 °C. 1H NMR (DMSO-d6) δ: 3.82 (s, 3H, OCH3); 7.00 (d, 1H, J = 8.9 Hz, H-6, Ar); 7.07 (d, 1H, J = 8.6 Hz, H-5, Ar); 7.13 (s, H-2, Ar); 7.48 (s, 1H, =CH); 9.57 (br s, 1H, OH); 13.62 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 56.1 (OCH3); 112.9 (C-2′); 116.5 (C-5′); 122.4 (C=, C-5); 124.8 (C-6′); 126.1 (C-1′); 132.7 (=CH); 147.5 (C-4′); 150.9 (C-3′); 169.9 (C=O, C-4); 196.0 (C=S, C-2). HRMS, m/z: 267.0022 found (calculated for C11H9NO3S2, M+. requires 267.0024).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3d). According to the standard procedure, 3d was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 3,4-dimethoxybenzaldehyde 2h (1.19 g, 7.2 mmol), and n-propylamine (1.18 mL, 851 mg, 14.4 mmol) in 83% yield as light-yellow powder; mp = 232–234 °C. 1H NMR (DMSO-d6) δ: 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 7.14 (s, 1H, H-2, Ar); 7.32 (d, 1H, J = 8.6 Hz, H-5, Ar); 7.44 (d, 1H, J = 6.3 Hz, H-6, Ar); 7.59 (s, 1H, =CH); 13.72 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 112.4 (C-2′); 112.7 (C-5′); 123.6 (C=, C-5); 124.8 (C-6′); 127.5 (C-1′); 128.7 (=CH); 150.4 (C-4′); 150.7 (C-3′); 169.5 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 281.0167 found (calculated for C12H11NO3S2, M+. requires 281.0184).
(5Z)-5-(4-Hydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3e). According to the standard procedure, 3e was synthesized from commercial rhodanine 1a (1.33 g, 10 mmol), 4-hydroxybenzaldehyde 2i (1.22 g, 10 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 80% yield as light-yellow powder; mp = 240–242 °C. 1H NMR (DMSO-d6) δ: 6.91 (d, 2H, J = 8.6 Hz, H-3, Ar); 7.47 (d, 2H, J = 8.6 Hz, H-2, Ar); 7.58 (S, 1H, =CH); 10.48 (br s, 1H, OH); 13.68 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 116.7 (C-3′); 123.9 (C-2′); 123.8 (C=, C-5); 129.1 (C-1′); 130.7 (=CH); 159.9 (C-4′); 169.7 (C=O, C-4); 195.7 (C=S, C-2). HRMS, m/z: 236.9918 found (calculated for C10H7NO2S2, M+. requires 236.99182).
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3f). According to the standard procedure, 3f was synthesized from commercial rhodanine 1a (958 mg, 7.2 mmol), 2,3-dihydro-1,4-benzodioxin-6-carboxaldehyde 2c (1.18 g, 7.2 mmol), and n-propylamine (1.18 mL, 851 mg, 14.4 mmol) in 89% yield as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 4.29 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 7.02 (d, 1H, J = 8.1 Hz, H-5, Ar); 7.08 (s, 1H, H-2, Ar); 7.10 (d, 1H, J = 8.2 Hz; H-6, Ar); 7.53 (s, 1H, =CH); 13.76 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 63.8 (OCH2CH2O); 64.4 (OCH2CH2O); 118.6 (C-5′); 119.7 (C-2′); 123.5 (=CH); 124.7 (C-6′); 126.7 (C-1′); 132.1 (C=, C-5); 144.2 (C-4′); 146.5 (C-3′); 170.0 (C=O, C-4); 195.9 (C=S, C-2). HRMS, m/z: 279.0016 found (calculated for C12H9NO3S2, M+. requires 279.0024).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3g). According to the standard procedure, 3g was synthesized from commercial rhodanine 1a (1.357 g, 10.2 mmol), 2,3-dihydro-1-benzofuran-5-carbaldehyde 2b (1.067 mg, 7.2 mmol), and n-propylamine (1.67 mL, 1.205 g, 20.4 mmol) in 79% yield as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 4.02 (t, 2H, J = 6.8 Hz; CH2CH2O); 6.02 (t, 2H, J = 6.9 Hz; CH2CH2O); 6.84 (d, 1H, J = 8 Hz, H-3, Ar); 7.12 (dd, 1H, J = 8, 1.5 Hz, H-2, Ar); 7.37 (d, 1H, J = 1.4 Hz, H-6, Ar); 8.16 (s, 1H, =CH); 13.66 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 29.2 (CH2CH2O); 71.7 (CH2CH2O); 107.0 (C-5′); 108.4 (C-2′); 123.5 (=CH); 124.7 (C-6′); 126.7 (C-1′); 132.1 (C=, C-5); 144.2 (C-4′); 148.6 (C-3′); 170.0 (C=O, C-4); 195.9 (C=S, C-2). HRMS, m/z: 263.0063 found (calculated for C12H9NO2S2, M+. requires 263.0075).
(5Z)-5-{[6-(1,3-Benzodioxol-5-yl)pyridin-2-yl]methylene}-2-thioxo-1,3-thiazolidin-4-one (3h). According to the standard procedure, 3h was synthesized from commercial rhodanine 1a (18 mg, 0.134 mmol), 6-(1,3-benzodioxol-5-yl)-2-pyridinecarbaldehyde 2k (30 mg, 0.134 mmol), and n-propylamine (22 mL, 15.8 mg, 0.268 mmol) in 79% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.05 (s, 2H, OCH2O); 6.94 (d, 1H, J = 8 Hz, H-3′); 7.55 (dd, 1H, J = 8, 1.5 Hz, H-2′); 7.59 (d, 1H, J = 8 Hz, H-3); 7.66 (dd, 1H, J = 8, 1.5 Hz, H-5); 7.77 (t, 1H, J = 8 Hz, H-4); 7.93 (dd, 1H, J = 1.4 Hz, H-6′); 8.46 (s, 1H, =CH); 12.46 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 107.0 (C-5″); 107.5 (C-6′); 108.4 (C-2″); 114.0 (C-4′); 116.0 (C=, C-5); 118.0 (C-5″); 124.5 (C-6″); 13.,6 (C-1″); 131.9 (=CH); 132.1 (C-3″); 132.4 (C-2′); 148.6 (C-3″); 150.0 (C-4″); 169.9 (C=O, C-4); 192.4 (C=S, C-2). HRMS, m/z: 342.0131 found (calculated for C16H10N2O3S2, M+. requires 342.0133).
(5Z)-5-[(7-Bromo-1,3-benzodioxol-5-yl)methylene]-2-thioxo-1,3-thiazolidin-4-one (3i). According to the standard procedure, 3i was synthesized from commercial rhodanine 1a (1.31 g, 9.8 mmol), 7-bromo-1,3-benzodioxole-5-carbaldehyde 2l (2.24 g, 9.8 mmol), and n-propylamine (1.6 mL, 1.15 g, 19.6 mmol) in 70% yield as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 6.18 (s, 2H, OCH2O); 6.37 (s, 1H, H-6); 7.39 (s, 1H, H-2); 7.88 (s, 1H, =CH); 12.10 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 100.7 (C-Br); 102.7 (OCH2O); 109.4 (C-2′); 110.7 (=CH); 127.7 (C-6′); 128.1 (C-1′); 128.7 (C=, C-5); 146.8 (C-4′); 148.7 (C-3′); 166.4 (C=O, C-4); 192.6 (C=S, C-2). HRMS, m/z: 342.8923 found (calculated for C11H1679BrNO3S2, M+. requires 342.8972).
(5Z)-5-(3,4-Dihydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3j). According to the standard procedure, 3j was synthesized from commercial rhodanine 1a (1.385 g, 10.4 mmol), 3,4-dihydroxybenzaldehyde 2f (1.44 g, 10.4 mmol), and n-propylamine (1.7 mL, 1.23 g, 20.8 mmol) in 79% yield as light-yellow powder; mp = 210–212 °C. 1H NMR (DMSO-d6) δ: 6.82 (s, 1H, H-2); 6.89 (d, 1H, J = 8.6 Hz, H-6); 7.05 (d, 1H, J = 8.2 Hz, H-5); 7.47 (s, 1H, =CH); 9.54 (br s, 1H, OH); 9.96 (br s, 1H, OH); 13.66 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 115.5 (C-2′); 117.5 (C-5′); 123.2 (C=, C-5); 128.51 (C-6′); 129.8 (C-1′); 132.7 (=CH); 148.7 (C-4′); 149.4 (C-3′); 169.6 (C=O, C-4); 195.8 (C=S, C-2). HRMS, m/z: 252.9862 found (calculated for C10H7NO3S2, M+. requires 252.9867).
(5Z)-5-(3,5-Dihydroxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one (3k). According to the standard procedure, 3k was synthesized from commercial rhodanine 1a (1.385 g, 10.4 mmol), 3,5-dihydroxybenzaldehyde 2n (1.44 g, 10.4 mmol), and n-propylamine (1.7 mL, 1.23 g, 20.8 mmol) in 79% yield as light-yellow powder; mp = 230–232 °C. 1H NMR (DMSO-d6) δ: 6.62 (s, 1H, H-3); 6.65 (d, 1H, J = 8.7 Hz, H-5); 7.46 (d, 1H, J = 5.8 Hz, H-6); 7.94 (s, 1H, =CH); 10.23 (br s, 1H, OH); 10.54 (br s, 1H, OH); 13.72 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 104.9 (C-3′); 110.1 (C-5′); 114.1 (C-6′); 122.4 (C=, C-5); 130.5 (C-1′); 130.8 (=CH); 159.8 (C-2′); 161.1 (C-4′); 169.9 (C=O, C-4); 196.1 (C=S, C-2). HRMS, m/z: 252.9859 found (calculated for C10H7NO3S2, M+. requires 252.9867).
(5Z)-5-(1-Benzofuran-2-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one (3l). According to the standard procedure, 3l was synthesized from commercial rhodanine 1a (1.33 g, 10 mmol), 1-benzofuran-2-carbaldehyde 2m (1.52 g, 10.4 mmol), and n-propylamine (1.64 mL, 1.18 g, 20 mmol) in 62% yield as light-yellow powder; mp = 246–250 °C. 1H NMR (DMSO-d6) δ: 6.98 (s, 1H, C-3); 7.11–7.28 (m, 2H, H-5, H-6); 7.47 (d, 1H, J = 7.7 Hz, H-7); 7.83 (d, 1H, J = 7.5 Hz, H-4); 8.51 (s, 1H, H-2); 8.98 (s, 1H, =CH); 12.10 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 107.2 (=CH); 108.8 (C-3′); 112.6 (C-7′); 118.6 (C-4′); 121.2 (C-5′); 122.1 (C=, C-5); 123.1 (C-6′); 127.6 (C-3a′); 129.7 (C-2′); 136.4 (C-7a′); 164.3 (C=O, C-4); 194.1 (C=S, C-2). HRMS, m/z: 260.9842 found (calculated for C12H7NO2S2, M+. requires 260.9918).
7-Bromo-1,3-benzodioxole-5-carbaldehyde (2l). In a 25 mL round-bottomed flask provided with a magnetic stirrer and condenser, a mixture of commercial 3-bromo-4,5-dihydroxybenzaldehyde (217 mg, 1 mmol), potassium fluoride (529 mg, 9.1 mmol, 9.1 eq), and dibromomethane (70 mL, 173 mg, 1 mmol) in 3 mL of dimethylformamide was stirred vigorously under a stream of nitrogen for 4 h at 140 °C in an oil bath. After cooling down to room temperature, 3 mL of deionized water was added in one portion to the crude reaction mixture. The resulting solution was submitted to extraction with Et2O (3 × 5 mL), and, after decantation, the organic layer was dried over MgSO4. After filtration, the solvent of the filtrate was eliminated in a rotary evaporator under reduced pressure; then, the crude residue was dried under high vacuum (10−2 Torr) at 25 °C for 2 h. The desired 7-bromo-1,3-benzodioxole-5-carbaldehyde 2l was obtained as a light-yellow powder in 51% yield (117 mg) and was sufficiently pure to be used further without purification; mp = 123–125 °C. 1H NMR (DMSO-d6) δ: 6.17 (s, 2H, OCH2O); 7.28 (d, 1H, J = 1.4 Hz, H-2, Ar); 7.55 (d, 1H, J = 1.3 Hz, H-6); 9.78 (s, 1H, CHO). 13C NMR (DMSO-d6) δ: 100.9 (C-Br); 102.6 (OCH2O); 106.2 (C-2); 131.0 (C-6); 132.8 (C-1); 148.9 (C-4); 151.2 (C-3); 189.1 (C=O). HRMS, m/z: 227.9426 found (calculated for C8H5O379Br, M+. requires 227.9422).
3.1.2. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3m,n) in the Monowave® 300 Microwave Reactor
In a 10 mL glass tube were placed successively commercial rhodanine 1a (1 eq), aromatic aldehyde 2o,p (1.1–1.2 eq), sodium acetate (26.4 mg, 0.31 mmol, 0.1 eq), and glacial acetic acid (1.2 eq). The glass tube was sealed with a snap cap and placed in the Monowave 300® Anton-Paar microwave cavity (power = 850 W). The mixture was irradiated at 140 °C for 20–30 min under vigorous magnetic stirring. After microwave dielectric heating, the crude reaction mixture was allowed to cool down at room temperature. To this crude mixture, 4 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired compound 3 was collected by filtration, and the crude solid was washed with 5 mL of deionized water and dried under high vacuum (10−2 Torr) at 25 °C for 2 h.
(5Z)-5-(Chroman-6-yl)methylene-2-thioxo-1,3-thiazolidin-4-one (3m). According to the standard procedure, 3m was synthesized after a reaction time of 20 min from commercial rhodanine 1a (200 mg, 1.5 mmol), 6-chromanecarbaldehyde 2o (292 mg, 1.8 mmol), sodium acetate (147 mg, 1.8 mmol), and glacial acetic acid (0.6 mL, 108 mg, 1.8 mmol) in 93% yield as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 1.92 (m, 2H, CH2); 2.77 (t, 2H, J = 6.3 Hz, CH2); 4.19 (m, 2H, CH2); 6.83 (d, 1H, J = 8.4 Hz, Ar); 7.27 (m, 2H, Ar); 7.47 (s, 1H, CH=); 12.06 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 21.3 (CH2); 24.1 (CH2); 66.6 (CH2); 117.5 (=CH); 121.9 (Ar); 123.6 (C=, C-5); 124.9 (Ar); 130.2 (Ar); 131.9 (Ar); 132.8 (Ar); 157.0 (Ar); 169.7 (C=O, C-4); 195.6 (C=S, C-2). HRMS, m/z: 300.0129 found (calculated for C15H11NO2NaS2, [M+Na]+. requires 300.0129).
(5Z)-5-(3,4-Dihydro-2H-1,5-benzodioxepine-7-yl)methylene-2-thioxo-1,3-thiazolidin-4-one (3n). According to the standard procedure, 3n was synthesized after a reaction time of 20 min from commercial rhodanine 1a (200 mg, 1.5 mmol), 3,4-dihydro-2H-1,5-benzodioxepine-7-carbaldehyde 2p (321 mg, 1.8 mmol), sodium acetate (147 mg, 1.8 mmol), and glacial acetic acid (0.6 mL, 108 mg, 1.8 mmol) in 94% yield as light-yellow powder; mp = 220–222 °C. 1H NMR (DMSO-d6) δ: 2.13 (m, 2H, CH2); 4.20 (m, 4H, 2xCH2); 7.05 (m, 1H, Ar); 7.15 (m, 2H, Ar); 7.50 (s, 1H, CH=); 13.68 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 30.7 (CH2); 70.4 (CH2); 70.5 (CH2); 122.4 (CH=) 123.7 (Ar); 123.8 (C=, C-5); 125.9 (Ar); 128.1 (Ar); 131.0 (Ar); 150.9 (Ar); 153.0 (Ar); 169.3 (C=O, C-4); 195.4 (C=S, C-2). HRMS, m/z: 316.0079 found (calculated for C18H13NO2NaS2, [M+Na]+. requires 316.0078).
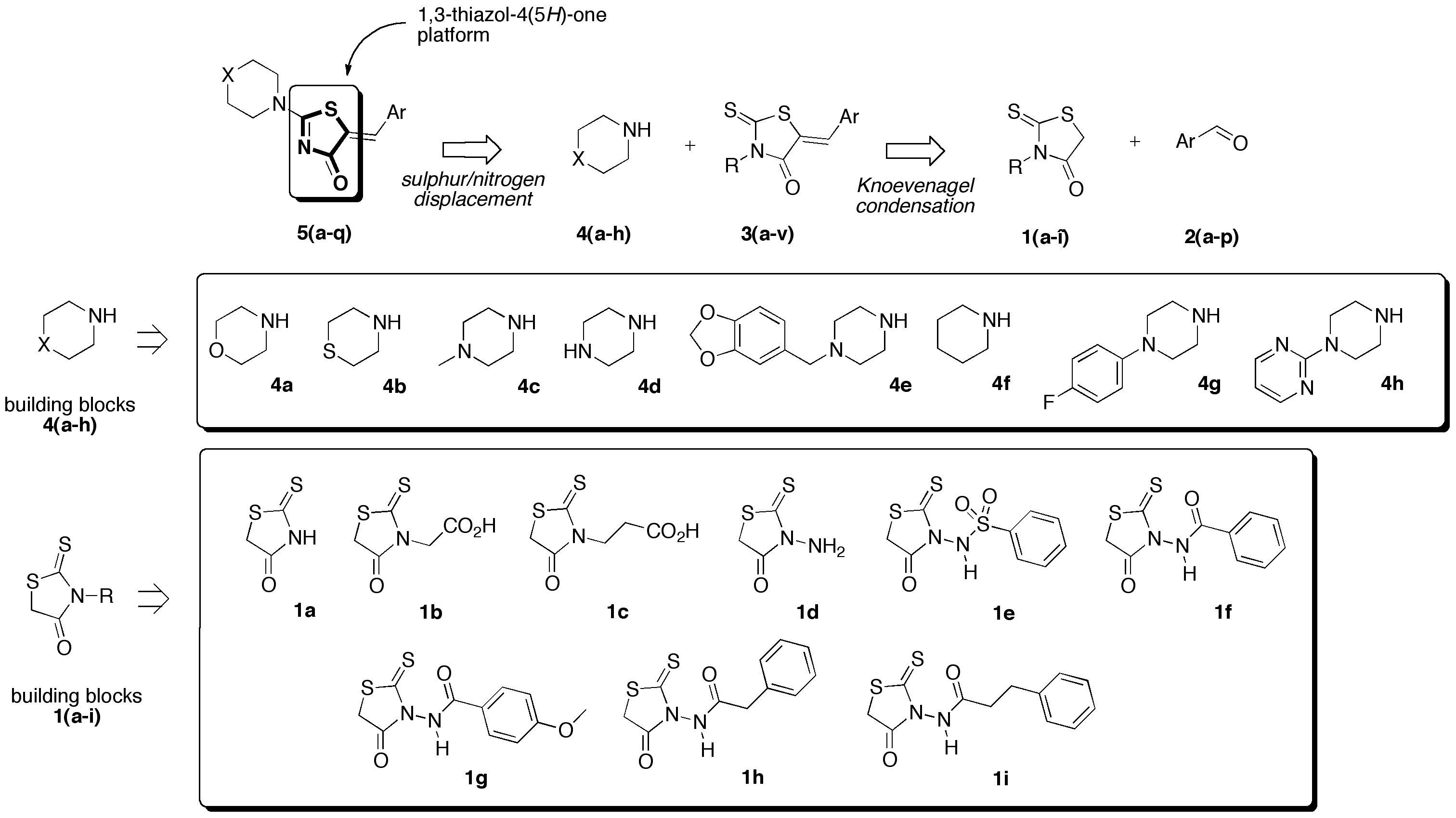
(5Z)-3-Amino-5-benzo[1,3]dioxol-5-ylmethylene-2-thioxo-thiazolidin-4-one (3o). In a 25 mL round-bottomed flask provided with a magnetic stirrer and condenser, a mixture of commercial 3-amino rhodanine 1d (197 mg, 1.33 mmol), piperonaldehyde 2a (200 g, 1.33 mmol), triethylamine (19 mL, 13 mg, 0.13 mmol, 0.1 eq), and glacial acetic acid (8 μL, 8 mg, 0.13 mmol, 0.1 eq) in 4.3 mL of ethyl acetate was stirred vigorously under a stream of argon for 3.5 h at 85 °C in an oil bath. After cooling down to room temperature, the insoluble product of the orange reaction mixture was filtered, washed with absolute ethanol (2 × 3 mL), and was dried under high vacuum (10−2 Torr) for 1 h. The desired 3-amino-5-benzo[1,3]dioxol-5-ylmethylene-2-thioxo-thiazolidin-4-one 3o was obtained as a light-yellow powder in 26% yield (97 mg); mp = 224–226 °C. 1H NMR (DMSO-d6) δ: 5.92 (s, 2H, NH2); 6.15 (s, 2H, H-1); 7.12 (d, 1H, J = 8.1Hz, H-7); 7.19–7.25 (m, 2H, H-3, H-4); 7.79 (s, 1H, H-5); 12.71 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.1 (C-1′); 109.3 (C-4′); 109.6 (C-3′); 117.5 (C=, C-5); 127.0 (C-6′); 127.2 (C-5′); 133.5 (CH=); 148.3 (C-2′); 149.9 (C-2′); 163.7 (C=O, C-4); 187.1 (C=S, C-2). HRMS, m/z: 302.9872 found (calculated for C11H8N2O3NaS2, [M+Na]+. requires 302.9874).
3.1.3. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3p,q) and (3s,t) in the Explorer® 24 Microwave Reactor
In a 10 mL glass tube were placed successively the 3-N-amino rhodanine derivative 1 (1 eq), piperonaldehyde 2a (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq). The glass tube was placed in the Explorer® 24 CEM microwave cavity (power = 300 W). The mixture was irradiated at 120 or 150 °C (with a power of 100 or 200 W) for 20 min under vigorous magnetic stirring. After microwave dielectric heating, the insoluble compound 3 was separated from the crude reaction mixture by filtration. To the collected crude compound 3, absolute ethanol or deionized water was added, and the resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide (3p). According to the standard procedure, 3p was synthesized at 150 °C with a power of 200 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide 1f (222 mg, 1 mmol), piperonaldehyde 2a (150 mg, 1 mmol), piperidine (10 μL, 9 mg, 0.1 mmol, 0.1 eq), and glacial acetic acid (6 μL, 6 mg, 0.1 mmol, 0.1 eq) in 27% yield as light-yellow powder; mp = 182–184 °C. 1H NMR (DMSO-d6) δ: 6.64 (s, 2H, H-1); 7.56 (d, J = 8.1 Hz, 1H, Ar); 7.66 (d, J = 1.7 Hz, 1H, Ar); 7.77 (dd, J = 1.3, 8.2 Hz, 1H, Ar); 8.03 (m, 2H, Ar); 8.29 (s, 1H, CH=); 8.50 (m, 1H, Ar); 8.53 (m, 1H, Ar); 12.03 (br s, 1H, H-6′). 13C NMR (DMSO-d6) δ: 102.3 (OCH2O); 109.4 (Ar); 109.80 (Ar); 116.4 (C=, C-5); 126.9 (C=6); 127.6 (Ar); 127.8 (Ar); 128.8 (Ar); 132.9 (Ar); 135.2 (CH=); 148.4 (Cipso, Cortho, Ar); 150.3 (C-2′); 163.4 (C=O); 164.5 (C=O, C-4); 190.3 (C=S, C-2). HRMS, m/z: 385.0307 found (calculated for C18H13N2O4S2, [M+H]+. requires 385.0317).
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-2-phenyl-acetamide (3q). According to the standard procedure, 3q was synthesized at 120 °C with a power of 100 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-2-phenyl-acetamide 1h (50 mg, 0.19 mmol), piperonaldehyde 2a (28 mg, 0.19 mmol), sodium acetate (15 mg, 0.19 mmol, 0.1 eq), and glacial acetic acid (70 μL, 74 mg, 1.24 mmol, 6.6 eq) in 98% yield as light-yellow powder; mp = 216–218 °C. 1H NMR (DMSO-d6) δ: 3.71 (s, 2H, CH2Ph); 6.16 (s, 2H, OCH2O); 7.13 (d, 1H, J = 8.1 Hz, Ar); 7.21 (m, 1H, Ar); 7.25 (m, 1H, Ar); 7.28 (m, 1H, Ar); 7.32–7.34 (m, 4H, Ar); 7.84 (s, 1H, CH=); 11.44 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 39.5 (CH2); 102.3 (OCH2O); 109.4 (Ar); 109.7 (Ar); 116.6 (C=, C-5); 126.7 (Ar); 126.9 (Cipso, Ar); 127.4 (Ar); 128.3 (Ar); 129.1 (Ar); 134.6 (Cipso, Ar); 134.9 (Ar); 148.4 (CAr-O); 150.2 (CAr-O); 163.2 (C=O); 168.6 (C=O, C-4); 190.2 (C=S, C-2). HRMS, m/z: 421.0294 found (calculated for C19H14N2O4NaS2, [M+Na]+. requires 421.0293).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-3-[2-(4-methoxy-phenyl)-2-oxo-ethyl]-2-thioxo-thiazolidin-4-one (3s). According to the standard procedure, 3s was synthesized at 120 °C with a power of 100 W from 4-methoxy-N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzamide 1g (50 mg, 0.18 mmol), piperonaldehyde 2a (27 mg, 0.18 mmol), sodium acetate (15 mg, 0.18 mmol, 0.1 eq), and glacial acetic acid (70 μL, 70 mg, 1.17 mmol, 6.6 eq) in 83% yield (61 mg) as light-yellow powder; mp = 226–228 °C. 1H NMR (DMSO-d6) δ: 3.85 (s, 3H, MeO); 6.18 (s, 2H, OCH2O); 7.11 (m, 2H, Ar); 7.15 (d, 1H, J = 8.2 Hz, Ar); 7.26 (d, 1H, J = 1.7 Hz, Ar); 7.31 (dd, 1H, J = 1.5, 1.6, 8.4 Hz, Ar); 7.90 (s, 1H, CH=); 7.93 (m, 1H, Ar); 7.96 (m, 1H, Ar); 11.61 (s, 1H, Ar). 13C NMR (DMSO-d6) δ: 55.5 (CH3O); 102.3 (OCH2O); 109.4 (Ar); 109.8 (Ar); 114.0 (Ar); 116.5 (C=, C-5); 122.8 (Cipso, Ar); 126.9 (Cipso, Ar); 127.6 (Ar); 129.9 (Ar); 135.1 (CH=); 148.4 (O-CAr); 150.3 (O-CAr); 162.8 (Cipso, Ar); 163.5 (C=O); 163.9 (C=O, C-4); 190.5 (C=S, C-2). HRMS, m/z: 437.0237 found (calculated for C19H14N2O5NaS2, [M+Na]+. requires 437.0242).
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-benzenesulfonamide (3t). According to the standard procedure, 3t was synthesized at 150 °C with a power of 200 W from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-benzenesulfonamide 1e (100 mg, 0.35 mmol), piperonaldehyde 2a (52 mg, 0.35 mmol), piperidine (3 μL, 3 mg, 0.034 mmol, 0.1 eq), and glacial acetic acid (2 μL, 2 mg, 0.034 mmol, 0.1 eq) in 64% yield (94 mg) as light-yellow powder; mp = 214–216 °C. 1H NMR (DMSO-d6) δ: 6.16 (s, 2H, OCH2O); 7.12 (d, 1H, J = 8.1 Hz, Ar); 7.19 (d, 1H, J = 1.6 Hz, Ar); 7.24 (dd, 1H, J = 1.7, 8.2 Hz, Ar); 7.59 (m, 2H, Ar); 7.69 (m, 1H, Ar); 7.79 (s, 1H, CH=); 7.86 (m, 2H, Ar); 11.57 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 102.3 (OCH2O); 109.4 (Ar); 109.7 (Ar); 115.9 (C=, C-5); 126.8 (Cipso, Ar); 127.3 (Ar); 127.5 (Ar); 129.1 (Ar); 133.5 (Ar); 134.9 (CH=); 140.7 (Cipso, Ar); 148.3 (O-CAr); 150.2 (O-CAr); 163.4 (C=O, C-4); 189.6 (C=S, C-2). HRMS, m/z: 442.9785 found (calculated for C17H12N2O5NaS3, [M+Na]+. requires 442.9806) and 301.9780 found (calculated for C11H7N2O3NaS2, [M-SO2Ph + Na]+. requires 301.9796).
3.1.4. Standard Procedure for the Preparation of 5-Arylidene-2-thioxo-1,3-thiazolidin-4-one (3r) and (3u,v) in the Monowave® 300 Microwave Reactor
In a 10 mL glass tube, the 3-N-amino rhodanine derivative 1 (1 eq), piperonaldehyde 2a (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq) were successively placed. The glass tube was placed in the Monowave® 300 Anton Paar microwave cavity (power = 850 W). The mixture was irradiated at 120 or 150 °C for 20 min under vigorous magnetic stirring. After microwave dielectric heating, the reaction was allowed to cool down to room temperature. To this crude mixture, 2.5 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired insoluble compound 3 was collected by filtration, and 2 mL of absolute ethanol was added to the collected compound 3. The resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was finally collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
(5Z)-N-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-3-phenyl-propionamide (3r). According to the standard procedure, 3r was synthesized at 120 °C from N-(4-oxo-2-thioxo-thiazolidin-3-yl)-3-phenyl-propionamide 1i (50 mg, 0.18 mmol), piperonaldehyde 2a (27 mg, 0.18 mmol), sodium acetate (15 mg, 0.18 mmol, 0.1 eq), and glacial acetic acid (68 μL, 396 mg, 6.6 mmol, 6.6 eq) in 68% yield (50 mg) as light-yellow powder; mp = 226–228 °C. 1H NMR (DMSO-d6) δ: 2.65 (m, 2H, CH2CO); 2.91 (m, 2H, CH2Ph); 6.16 (s, 2H, OCH2O); 7.12–7.32 (m, 8H, Ar); 7.85 (s, 1H, CH=); 11.23 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 30.4 (CH2Ar); 34.5 (CH2CO); 102.3 (OCH2O); 109.4 (Ar); 109.8 (Ar); 116.7 (C=, C-5); 126.1 (Ar); 126.9 (Cipso, Ar); 127.4 (Ar); 128.3–128.4 (Ar); 134.8 (CH=); 140.5 (Cipso, Ar); 148.4 (O-CAr); 150.2 (O-CAr); 163.3 (C=O); 170.0 (C=O, C-4); 190.2 (C=S, C-2). HRMS, m/z: 435.0447 found (calculated for C20H16N2O4NaS2, [M+Na]+. requires 435.0449).
(5Z)-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-acetic acid (3u). According to the standard procedure, 3u was synthesized at 120 °C from 2-(4-oxo-2-thioxo-thiazolidin-3-yl)acetic acid 1b (255 mg, 1.33 mmol), piperonaldehyde 2a (200 mg, 1.33 mmol), piperidine (13 μL, 11 mg, 0.13 mmol, 0.1 eq), and glacial acetic acid (8 μL, 7.8 mg, 0.13 mmol, 0.1 eq) in 72% yield (308 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 4.81 (s, 2H, CH2COH); 6.26 (s, 2H, OCH2O); 7.23 (d, 1H, J = 8.1 Hz, Ar); 7.31–7.35 (m, 2H, Ar); 7.92 (s, 1H, CH=); 12.37 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 45.5 (CH2CO2H); 102.0 (OCH2O); 109.1 (Ar); 109.4 (Ar); 118.9 (C=, C-5); 126.7 (Cipso, Ar); 127.0 (Ar); 133.9 (CH=); 148.1 (O-CAr); 149.9 (O-CAr); 166.1 (C=O); 167.1 (C=O, C-4); 192.7 (C=S, C-2). HRMS, m/z: 322.9922 found (calculated for C13H9NO5S2, [M]+. requires 322.9922) and 305.9897 found (calculated for C13H8NO4S2, [M − OH]+ requires 305.9895).
(5Z)-3-(5-Benzo[1,3]dioxol-5-ylmethylene-4-oxo-2-thioxo-thiazolidin-3-yl)-propionic acid (3v). According to the standard procedure, 3v was synthesized at 150°C from 3-(4-oxo-2-thioxo-thiazolidin-3-yl)-propionic acid 1c (50 mg, 0.24 mmol), piperonaldehyde 2a (37 mg, 0.24 mmol), piperidine (2 μL, 2 mg, 0.024 mmol, 0.1 eq), and glacial acetic acid (1.4 μL, 1.4 mg, 0.024 mmol, 0.1 eq) in 62% yield (50 mg) as light-yellow powder; mp = 218–220 °C. 1H NMR (DMSO-d6) δ: 2.58 (m, 2H, CH2CO2H); 4.21 (t, 2H, J = 6.9Hz, NCH2); 6.15 (s, 2H, OCH2O); 7.10–7.24 (m, 3H, Ar); 7.75 (br s, 1H, CH=); 11.95 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 31.0 (CH2CO2H); 102.2 (OCH2O); 109.3–109.6 (Ar); 119.7 (Cipso, Ar); 126.8 (Ar); 127.2 (C=, C-5); 133.2 (CH=); 148.3 (O-CAr); 149.9 (O-CAr); 166.7 (C=O); 171.8 (C=O, C-4); 193.0 (C=S, C-2). HRMS, m/z: 336.0005 found (calculated for C14H10NO5S2, [M]+. requires 336.0004).
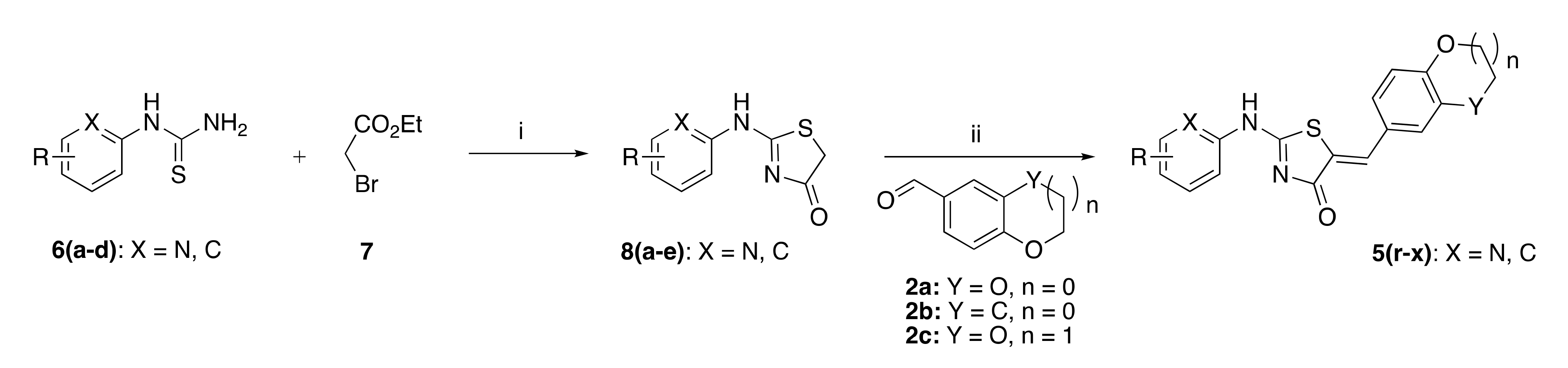
3.1.5. Standard Procedure for the Preparation of (5Z) 2-Amino-5-arylidene-1,3-thiazol-4(5H)-4-one (5a–q) by Sulphur-Nitrogen Displacement in the Synthewave® 402 Microwave Reactor
In a cylindrical quartz reactor (Ø = 1.8 cm), the (5Z)-5-arylidene-2-thioxo-1,3-thiazolidin-4-one 3 (0.5 mmol, 1 eq), and the secondary cyclic amine 4 (1.5 mmol, 3 eq) were successively placed. The reactor was then introduced into the Synthewave® 402 Prolabo microwave cavity (power = 300 W). The stirred mixture was irradiated (after a ramp of 3 min from 20 to the appropriate reaction temperature) at 80–120 °C (power: 80–150 W) for 20–120 min. After microwave dielectric heating, the crude reaction mixture was allowed to cooling down at room temperature, and ethanol (20 mL) was added in the cylindrical quartz reactor. The resulting insoluble product 3 was filtered and recrystallized in absolute EtOH and then dried under high vacuum (10−2 Torr) for 1 h.
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5a). According to the standard procedure, 5a was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 96% yield (153 mg) as light-yellow powder; mp = 250–252 °C. 1H NMR (DMSO-d6) δ: 3.63 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2OCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.1 (CH2NCH2); 74.4 (CH2OCH2); 102.2 (OCH2O); 109.1 (C-3′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 318.0688 found (calculated for C15H14N2O4S, [M]+. requires 318.0674).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5b). According to the standard procedure, 5b was synthesized at 80 °C (power: 80 W) after a reaction time of 25 min from (5Z)-5-(3,4-dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3d (141 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 64% yield (107 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.73 (m, 4H, CH2NCH2); 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 66.3 (CH2NCH2); 74.40 (CH2OCH2); 102.2 (OCH2O); 109.1 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 334.0970 found (calculated for C16H18N2O4S, [M]+. requires 334.0987).
(5Z)-5-(4-Hydroxy-3-methoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5c). According to the standard procedure, 5c was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(4-hydroxy-3-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3b (134 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 88% yield (141 mg) as light-yellow powder; mp = 251–253 °C. 1H NMR (DMSO-d6) δ: 3.46 (m, 4H, CH2NCH2); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7,15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=); 9.57 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 58.1 (OCH3); 66.1 (CH2NCH2); 74.5 (CH2OCH2); 109.1 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 150.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 320.0836 found (calculated for C15H16N2O4S, [M]+. requires 320.0831).
(5Z)-5-(3-Hydroxy-4-methoxybenzylidene)-2-(morpholin-1-yl)-1,3-thiazol-4(5H)-one (5d). According to the standard procedure, 5d was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3c (134 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 86% yield (138 mg) as light-yellow powder; mp = 256–258 °C. 1H NMR (DMSO-d6) δ: 3.45 (m, 4H, CH2NCH2); 3.91 (s, 3H, OCH3); 4.01 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=); 9.54 (br s, 1H, OH). 13C NMR (DMSO-d6) δ: 58.1 (OCH3); 66.1 (CH2NCH2); 74.4 (CH2OCH2); 109.1 (C-2′); 109.4 (C-5′); 113.5 (CH=); 125.3 (C-6′); 126.9 (C-1′); 130.3 (C=, C-4); 148.8 (C-4′); 149.2 (C-3′); 175.1 (C=O, C-4). HRMS, m/z: 320.0834 found (calculated for C15H16N2O4S, [M]+. requires 320.0831).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(piperazin-1-yl)-1,3-thiazol-4(5H)-one (5e). According to the standard procedure, 5e was synthesized at 80 °C (power: 80 W) after a reaction time of 60 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial piperazine 4d (129 mg, 1.5 mmol) in 78% yield (124 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.75 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NHCH2); 6.10 (s, 2H, OCH2O); 7.06 (d, 1H, J = 8.1 Hz, H-2′); 7.17 (d, 1H, J = 8 Hz, H-5′); 7.15 (d, 1H, J = 7.9 Hz, H-6′); 7.56 (s, 1H, CH=); 12.58 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 66.2 (CH2NCH2); 74.4 (CH2NHCH2); 102.2 (OCH2O); 109.3 (C-2′); 109.1 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 317.0831 found (calculated for C15H15N3O3S, [M]+. requires 317.0834).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(4-N-methylpiperazin-1-yl)-1,3-thiazol-4(5H)-one (5f). According to the standard procedure, 5f was synthesized at 80 °C (power: 80 W) after a reaction time of 60 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial N-methylpiperazine 4d (166 μL, 150 mg, 1.5 mmol) in 84% yield (139 mg) as light-yellow powder; mp = 202–204 °C. 1H NMR (DMSO-d6) δ: 3.03 (S, 3H, NCH3); 3.73 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NMeCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-6′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.57 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 36.1 (NCH3); 65.1 (CH2NCH2); 74.2 (CH2NMeCH2); 102.2 (OCH2O); 109.4 (C-5′); 113.9 (CH=); 125.4 (C-6′); 126.2 (C-1′); 130.6 (C=, C-4); 148.1 (C-4′); 149.6 (C-3′); 173.6 (C=O, C-4). HRMS, m/z: 331.0962 found (calculated for C16H17N3O3S, [M]+. requires 331.0991).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(thiomorpholin-1-yl)-1,3-thiazol-4(5H)-one (5g). According to the standard procedure, 5g was synthesized at 80 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 80% yield (134 mg) as light-yellow powder; mp = 204–208 °C. 1H NMR (DMSO-d6) δ: 3.79 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2SCH2); 6.10 (s, 2H, OCH2O); 7.01 (d, 1H, J = 8.1 Hz, H-2′); 7.16 (d, 1H, J = 8 Hz, H-6′); 7.17 (d, 1H, J = 7.8 Hz, H-6′); 7.57 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 67.0 (CH2NCH2); 74.4 (CH2SCH2); 102.7 (OCH2O); 109.5 (C-5′); 114.1 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.8 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 334.0441 found (calculated for C15H14N2O3S2, [M]+. requires 334.0449).
(5Z)-5-(3,4-Dimethoxybenzylidene)-2-(thiomorpholin-1-yl)-1,3-thiazol-4(5H)-one (5h). According to the standard procedure, 5h was synthesized at 80 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(3,4-dimethoxybenzylidene)-2-thioxo-1,3-thiazolidin-4-one 3d (141 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 78% yield (137 mg) as light-yellow powder; mp = 240–242 °C. 1H NMR (DMSO-d6) δ: 3.73 (m, 4H, CH2NCH2); 3.74 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 3.90 (m, 2H, CH2SCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 57.0 (OCH3); 57.1 (OCH3); 66.0 (CH2NCH2); 74.4 (CH2SCH2); 109.1 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.2 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 350.0749 found (calculated for C16H18N2O3S2, [M]+. requires 350.0759).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one (5i). According to the standard procedure, 5i was synthesized at 80 °C (power: 80 W) after a reaction time of 20 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 80% yield (127 mg) as light-yellow powder; mp = 201–203 °C. 1H NMR (DMSO-d6) δ: 3.79 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2SCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′; 7.17 (d, 1H, J = 7.8 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 60.0 (CH2CH2CH2); 65.9 (CH2CH2CH2); 74.4 (CH2NCH2); 102.3 (OCH2O); 109.1 (C-5′); 109.4 (C-5′); 113.7 (CH=); 125.1 (C-6′); 126.6 (C-1′); 130.5 (C=, C-4); 148.8 (C-4′); 149.4 (C-3′); 174.7 (C=O, C-4). HRMS, m/z: 316.0860 found (calculated for C16H16N2O3S, [M]+. requires 316.0882).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[1-(1,3-benzodioxol-5-ylmethyl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5j). According to the standard procedure, 5j was synthesized at 100 °C (power: 80 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 1-(1,3-benzodioxol-5-ylmethyl)piperazine 4e (330 mg, 1.5 mmol) in 75% yield (169 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.24 (s, 2H, CH2N); 3.73 (m, 4H, CH2NCH2); 3.92 (m, 4H, CH2NCH2); 6.12 (s, 2H, OCH2O); 6.15 (s, 2H, OCH2O); 7.06 (d, 1H, J = 8.1 Hz, H-2′); 7.09 (d, 1H, J = 8.1 Hz, H-2″); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 8 Hz, H-3″); 7.18 (d, 1H, J = 7.9 Hz, H-6′); 7.19 (d, 1H, J = 7.9 Hz, H-2″); 7.59 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.1 (CH2NCH2); 74.6 (CH2NCH2); 102.4 (OCH2O); 103.2 (OCH2O); 109.1 (C-2′); 109.2 (Ar); 109.4 (C-5′); 110.1 (Ar); 113.9 (CH=); 125.3 (C-6′); 125.6 (Ar); 126.9 (C-1′); 127.3 (Cipso Ar); 130.6 (C=, C-4); 148.6 (C-4′); 148.8 (Cipso Ar); 149.2 (C-3′); 149.8 (Ar); 174.9 (C=O, C-4). HRMS, m/z: 451.1191 found (calculated for C21H21N3O5S, [M]+. requires 451.1202).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[1-(4-fluorophenyl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5k). According to the standard procedure, 5k was synthesized at 120 °C (power: 150 W) after a reaction time of 30 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 1-(4-fluorophenyl)piperazine 4g (270 mg, 1.5 mmol) in 72% yield (148 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 4H, CH2NCH2); 3.90 (m, 4H, CH2NCH2); 6.10 (s, 2H, OCH2O); 7.04 (d, 1H, J = 8 Hz, H-2′); 7.12 (d, 2H, J = 8.1 Hz, H-2″); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 7.9 Hz, H-6); 7.18 (d, 1H, J = 7.9 Hz, H-3″); 7.56 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 65.9 (CH2NCH2); 74.4 (CH2NCH2); 102.7 (OCH2O); 109.4 (C-2′); 109.2 (Cortho Ar); 109.4 (C-5′); 110.1 (Cmeta Ar); 113.7 (CH=); 125.1 (C-6′); 125.6 (C-4″); 126.9 (C-1′); 127.3 (Ar); 130.6 (C=, C-4); 148.4 (C-4′); 148.7 (Cipso Ar); 149.5 (C-3′); 174.5 (C=O, C-4). HRMS, m/z: 411.1021 found (calculated for C21H18N3O3S19F, [M]+. requires 411.1053).
(5Z)-5-(1,3-Benzodioxol-5-ylmethylene)-2-[4-(pyrimidin-2-yl)piperazin-1-yl]-1,3-thiazol-4(5H)-one (5l). According to the standard procedure, 5l was synthesized at 100 °C (power: 150 W) after a reaction time of 25 min from (5Z)-5-(1,3-benzodioxol-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3a (132 mg, 0.5 mmol) and commercial 2-(1-piperazin-1-yl)pyrimidine 4h (212 μL, 246 mg, 1.5 mmol) in 80% yield (158 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.75 (m, 4H, CH2NCH2); 3.87 (m, 4H, CH2NCH2); 6.14 (s, 2H, OCH2O); 7.06 (d, 1H, J = 7.9 Hz, H-2′); 7.13 (d, 1H, J = 8 Hz, H-5′); 7.16 (d, 1H, J = 7.9 Hz, H-6′); 7.18 (m, 3H, Ar); 7.61 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 66.0 (CH2NCH2); 74.2 (CH2NCH2); 102.5 (OCH2O); 109.2 (C-2′); 109.4 (C-5′); 110.1 (Ar); 113.9 (CH=); 125.2 (C-6′); 125.6 (Cipso Ar); 126.9 (C-1′); 130.5 (C=, C-4); 148.6 (C-4′); 148.8 (Cipso Ar); 149.2 (C-3′); 174.7 (C=O, C-4). HRMS, m/z: 395.1006 found (calculated for C19H17N5O3S, [M]+. requires 395.1052).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(morpholin-4-yl)-1,3-thiazol-4(5H)-one(5m). According to the standard procedure, 5m was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial morpholine 4a (131 μL, 131 mg, 1.5 mmol) in 80% yield (127 mg) as light-yellow powder; mp = 222–224 °C. 1H NMR (DMSO-d6) δ: 3.70 (t, 2H, J = 6.8 Hz; CH2CH2O); 3.73 (m, 4H, CH2NCH2); 3.90 (t, 2H, J = 6.9 Hz; CH2CH2O); 5.20 (m, 2H, CH2OCH2); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 29.2 (CH2CH2O); 65.9 (CH2NCH2); 71.7 (CH2CH2O); 74.4 (CH2OCH2); 109.1 (C-2′); 109.5 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 316.0864 found (calculated for C16H16N2O3S, [M]+. requires 316.0882).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(thiomorpholin-4-yl)-1,3-thiazol-4(5H)-one(5n). According to the standard procedure, 5n was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 84% yield (140 mg) as light-yellow powder; mp = 230–232 °C. 1H NMR (DMSO-d6) δ: 3.73 (t, 2H, J = 6.8 Hz; CH2CH2O); 4.02 (m, 4H, CH2NCH2); 4.09 (m, 4H, CH2SCH2); 5.22 (t, 2H, J = 6.9 Hz; CH2CH2O); 7.02 (d, 1H, J = 8.1 Hz, H-2′); 7.13 (d, 1H, J = 8 Hz, H-5′); 7.19 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 29.3 (CH2CH2O); 66.1 (CH2NCH2); 71.7 (CH2SCH2); 74.4 (CH2CH2O); 108.9 (C-2′); 109.4 (C-5′); 113.7 (CH=); 125.2 (C-6′); 126.9 (C-1′); 130.7 (C=, C-4); 148.4 (C-4′); 149.2 (C-3′); 174.6 (C=O, C-4). HRMS, m/z: 332.0643 found (calculated for C16H16N2O2S2, [M]+. requires 332.0653).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one(5o). According to the standard procedure, 5o was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-benzofuran-5-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3g (131 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 86% yield (135 mg) as light-yellow powder; mp = 252–254 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 6H, CH2); 3.73 (t, 2H, J = 6.8 Hz; CH2CH2O); 4.02 (m, 4H, CH2NCH2); 5.22 (t, 2H, J = 6.9 Hz; CH2CH2O); 7.06 (d, 1H, J = 8 Hz, H-2′); 7.15 (d, 1H, J = 8.2 Hz, H-5′); 7.17 (d, 1H, J = 7.9 Hz, H-6′); 7.56 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 24.1 (CH2CH2CH2); 29.1 (CH2CH2CH2); 31.2 (CH2CH2O); 66.2 (CH2NCH2); 74.3 (CH2CH2O); 109.1 (C-2′); 109.3 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.8 (C-1′); 130.6 (C=, C-4); 148.6 (C-4′); 149.3 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 314.1075 found (calculated for C17H18N2O2S, [M]+. requires 314.1089).
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-(piperidin-1-yl)-1,3-thiazol-4(5H)-one (5p). According to the standard procedure, 5p was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3f (140 mg, 0.5 mmol) and commercial piperidine 4f (149 μL, 128 mg, 1.5 mmol) in 88% yield (145 mg) as light-yellow powder; mp = 257–259 °C. 1H NMR (DMSO-d6) δ: 2.93 (m, 6H, CH2); 3.72 (m, 4H, CH2NCH2); 4.28 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 7.04 (d, 1H, J = 8.1 Hz, H-2′); 7,14 (d, 1H, J = 8 Hz, H-5′); 7.18 (d, 1H, J = 7.9 Hz, H-6′); 7.60 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 24.0 (CH2CH2CH2); 29.0 (CH2CH2CH2); 63.1 (CH2NCH2); 64.8 (OCH2CH2O); 66.4 (OCH2CH2O); 109.2 (C-2′); 109.4 (C-5′); 113.7 (CH=); 125.2 (C-6′); 126.9 (C-1′); 130.7 (C=, C-4); 148.7 (C-4′); 149.2 (C-3′); 175.1 (C=O, C-4). HRMS, m/z: 330.1081 found (calculated for C17H18N2O3S, [M]+. requires 330.1038).
(5Z)-5-(2,3-Dihydro-1,4-benzodioxin-6-ylmethylene)-2-(thiomorpholin-4-yl)-1,3-thiazol-4(5H)-one (5q). According to the standard procedure, 5q was synthesized at 80 °C (power: 90 W) after a reaction time of 20 min from (5Z)-5-(2,3-dihydro-1,4-benzodioxin-6-ylmethylene)-2-thioxo-1,3-thiazolidin-4-one 3f (140 mg, 0.5 mmol) and commercial thiomorpholine 4b (150 μL, 154 mg, 1.5 mmol) in 80% yield (139 mg) as light-yellow powder; mp > 260 °C. 1H NMR (DMSO-d6) δ: 3.71 (m, 4H, CH2NCH2); 3.90 (m, 2H, CH2SCH2); 4.27 (t, 2H, OCH2CH2O); 4.30 (t, 2H, OCH2CH2O); 5.10 (m, 2H, CH2OCH2); 7.01 (d, 1H, J = 8.2 Hz, H-2′); 7.15 (d, 1H, J = 8 Hz, H-5′); 7.19 (d, 1H, J = 7.9 Hz, H-6′); 7.58 (s, 1H, CH=). 13C NMR (DMSO-d6) δ: 64.8 (OCH2CH2O); 66.2 (CH2NCH2O); 66.6 (OCH2CH2O); 74.40 (CH2SCH2); 102.2 (OCH2O); 109.0 (C-2′); 109.4 (C-5′); 113.9 (CH=); 125.1 (C-6′); 126.9 (C-1′); 130.8 (C=, C-4); 148.6 (C-4′); 149.4 (C-3′); 174.9 (C=O, C-4). HRMS, m/z: 348.0603 found (calculated for C16H16N2O3S2, [M]+. requires 348.0602).
3.1.6. Standard Procedure for the Preparation of (5Z) 2-N-Heteroarylamino-5-arylidene-1,3-thiazol-4(5H)-one Derivatives (5r–x) by Solution Phase Condensation under Microwave in the Explorer® 24 Reactor
In a 10 mL glass tube, the 2-N-heteroarylamino-thiazolidin-4-one 8 (1 eq), aromatic aldehyde 2 (1 eq), piperidine (0.1 eq) or sodium acetate (0.1 eq), and glacial acetic acid (0.1–6.6 eq) were successively placed. The glass tube was placed in the Explorer® 24 CEM microwave cavity (power = 300 W). The mixture was irradiated at 120 or 150 °C (with a power of 100 or 200 W) for 20 or 40 min under vigorous magnetic stirring. After microwave dielectric heating, the reaction was allowed to cool down to room temperature. To this crude mixture, 1–2 mL of deionized water was added, and the resulting suspension was submitted to ultrasound in a Branson 1510 apparatus at 25 °C for 30 min. Then, the desired insoluble compound 3 was collected by filtration, and 2 mL of absolute ethanol was added to this collected compound 5. The resulting suspension was stirred vigorously under magnetic stirring for 18 h. The desired compound 3 was finally collected by filtration and was dried under high vacuum (10−2 Torr) at 25 °C for 1 h.
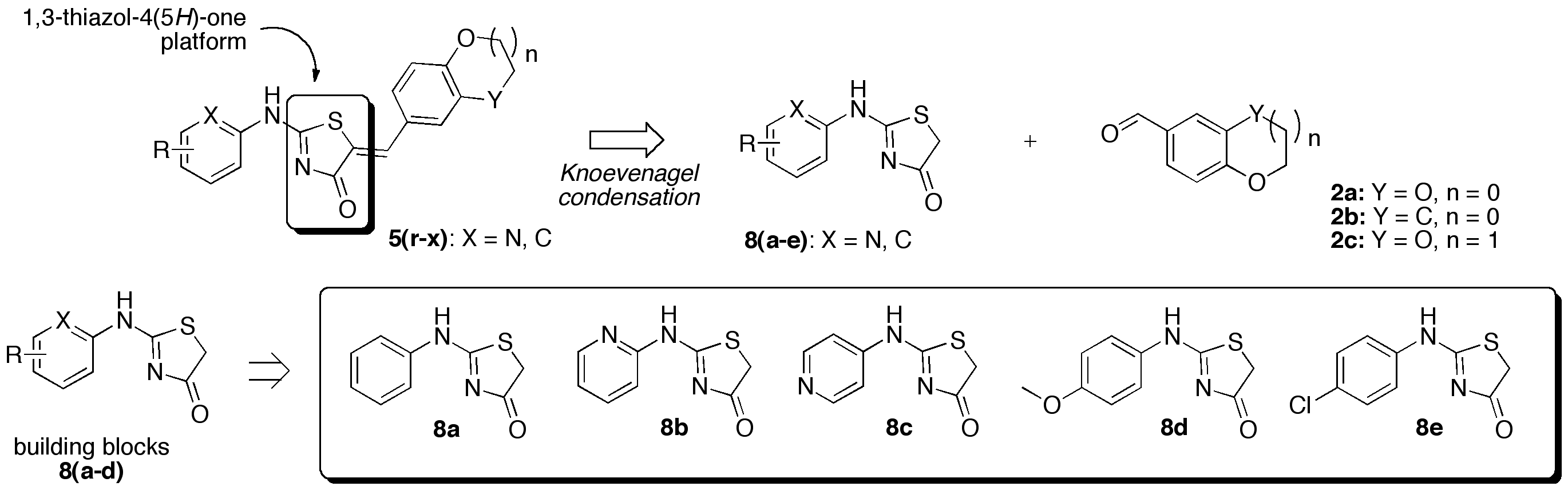
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-phenylamino-1,3-thiazol-4(5H)-one (5r). According to the standard procedure, 5r was synthesized at 150 °C with a power of 200 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (192 mg, 1 mmol), piperonaldehyde 2a (150 mg, 1 mmol), piperidine (10 μL, 9 mg, 0.1 mmol, 0.1 eq), and glacial acetic acid (6 μL, 6 mg, 0.1 mmol, 0.1 eq) in 34% yield (100 mg) as reddish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.08 (s, 2H, OCH2O); 7.03–7.22 (m, 5H, Ar + CH=); 7.42 (q, J = 7.4Hz, 2H, Ar); 7.56–7.78 (m, 2H, Ar); 12.21 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 108.7 (C-4′); 109.0 (C-7′); 120.4 (C-6′); 121.0 (Cipso, C-5′); 121.4 (C-2″); 124.8 (C-2″); 125.2 (C-4″); 127.5 (C=, C-5); 129.1 (C-3″); 129.4 (C-3″); 130.6 (CH=); 147.9 (Cipso, C-1″); 148.1 (Cipso, C-3a′); 148.7 (Cipso, C-7a′); 170.4 (C=N, C-2); 180.5 (C=O, C-4). HRMS, m/z: 325.0646 found (calculated for C17H13N2O3S, [M+H]+. requires 325.0647).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(pyridin-2-yl)amino-1,3-thiazol-4(5H)-one (5s). According to the standard procedure, 5s was synthesized at 120 °C with a power of 100 W for 20 min from 2-(pyridin-2-yl)amino-1,3-thiazolidin-4-one 8b (50 mg, 0.26 mmol), piperonaldehyde 2a (39 mg, 0.26 mmol), sodium acetate (21 mg, 0.26 mmol, 1 eq), and glacial acetic acid (0.1 mL, 102 mg, 1.7 mmol, 6.6 eq) in 90% yield (76 mg) as green-yellowish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.13 (s, 2H, OCH2O); 7.12 (d, J = 7.9 Hz, 1H, CH=); 7.17–7.23 (m, 4H, H-7′, H-4′, H-3″, H-5″); 7.58 (m, 1H, H-6′); 7.85 (t, J = 7.6 Hz, 1H, H-4″); 8.52 (d, J = 4.1 Hz, 1H, H-6″); 12.39 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.9 (OCH2O); 109.0 (C-4′); 120.1 (Ar); 127.8 (C=, C-5); 130.2 (Ar); 131.1 (CH=); 133.1 (Ar); 138.9 (Ar); 145.6 (Cipso, C-2″); 148.0 (Cipso, C-3a′); 148.8 (Cipso, C-7a′); 163.6 (C=N, C-2); 187.4 (C=O, C-4). HRMS, m/z: 348.0418 found (calculated for C16H11N3O3SNa, [M+Na]+. requires 348.0419).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(pyridin-4-yl)amino-1,3-thiazol-4(5H)-one (5t). According to the standard procedure, 5t was synthesized at 120 °C with a power of 100 W for 40 min from 2-(pyridin-4-yl)amino-1,3-thiazolidin-4-one 8c (150 mg, 0.78 mmol), piperonaldehyde 2a (117 mg, 0.78 mmol), sodium acetate (64 mg, 0.78 mmol, 1 eq), and glacial acetic acid (295 μL, 307 mg, 5.11 mmol, 6.6 eq) in 43% yield (109 mg) as orange-yellowish powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.11 (s, 2H, OCH2O); 7.11 (m, 2H, H-3″); 7.13 (s, 1H, H-7′); 7.72–7.67 (m, 3H, CH=, H-6′, H-4′); 8.72 (m, 2H, H-2″). 13C NMR (DMSO-d6) δ: 102.4 (OCH2O); 109.6–109.6 (C-7′, C-3″); 117.9 (C-4′); 126.1 (C-6′); 127.6 (C-5′); 132.4 (CH=); 146.3–146.3 (C-2″); 149.2–148.1 (C-3a′, C-7a′); 170.4 (C=N, C-2); 180.5 (C=O, C-4). HRMS, m/z: 370.0243 found (calculated for C16H10N3O3Na2S, [M-H+2Na]+. requires 370.0238).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(4-methoxy-phenylamino)-1,3-thiazol-4(5H)-one (5u). According to the standard procedure, 5u was synthesized at 120 °C with a power of 100 W for 20 min from 2-(4-methoxyphenyl)amino-1,3-thiazolidin-4-one 8d (50 mg, 0.22 mmol), piperonaldehyde 2a (34 mg, 0.22 mmol), sodium acetate (18 mg, 0.22 mmol, 1 eq), and glacial acetic acid (80 μL, 87 mg, 1.45 mmol, 6.6 eq) in 90% yield (72 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 3.76 (s, 3H, CH3O); 6.13 (s, 2H, OCH2O); 7.04–7.16 (m, 5H, CH=, Ar); 7.55–7.70 (m, 3H, Ar); 11.56 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 55.3 (CH3O); 101.8 (OCH2O); 108.7 (C-5′); 109.0 (C-4′); 114.2 (C-3″); 114.6 (C-3″); 122.0 (C-7′); 123.1 (C=, C-5); 124.8 (C-2″); 125.1 (C-2″); 127.7 (Cipso, C-1″); 128.2 (C-4″); 129.4 (C-6′); 130.0 (CH=); 147.9 (C-3a′); 148.1 (C-7a′); 148.7 (C=O, C-4); 156.5 (C=N, C-2). HRMS, m/z: 377.0574 found (calculated for C18H14N2O4NaS, [M+Na]+. requires 377.0572).
(5Z)-5-Benzo[1,3]dioxol-5-ylmethylene-2-(4-chloro-phenylamino)-1,3-thiazol-4(5H)-one (5v). According to the standard procedure, 5v was synthesized at 120 °C with a power of 100 W for 20 min from 2-(4-chlorophenyl)amino-1,3-thiazolidin-4-one 8e (50 mg, 0.22 mmol), piperonaldehyde 2a (34 mg, 0.22 mmol), sodium acetate (18 mg, 0.22 mmol, 1 eq), and glacial acetic acid (80 μL, 87 mg, 1.45 mmol, 6.6 eq) in 79% yield (62 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 6.08 (s, 2H, OCH2O); 7.04–7.17 (m, 4H, Ar); 7.43–7.51 (m, 2H, Ar); 7.57–7.68 (m, 2H, Ar); 7.81 (d, J = 7.2 Hz, 1H, CH=); 11.65 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 101.8 (OCH2O); 108.8 (C-4″); 109.1 (C-5′); 109.3 (C-4′); 122.0 (C-7′); 123.1 (C-2″); 124.9 (C-2″); 125.3 (C-5′); 127.4 (C=, C-5); 129.0 (C-1″); 129.4 (C-4″); 129.8 (C-6′); 131.0 (CH=); 148.0 (C-3a′); 148.1 (C-7a′); 148.8 (C=O, C-4); 156.7 (C=N, C-2). HRMS, m/z: 381.0079 found (calculated for C17H11N2O335ClSNa, [M+Na]+. requires 381.0077).
(5Z)-5-(2,3-Dihydro-benzofuran-5-ylmethylene)-2-phenylamino-1,3-thiazol-4(5H)-one (5w). According to the standard procedure, 5w was synthesized at 150 °C with a power of 200 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (100 mg, 0.22 mmol), 2,3-dihydro-1-benzofuran-5-carbaldehyde 2b (77 mg, 0.52 mmol), piperidine (5 mL, 4 mg, 0.052 mmol, 0.1 eq), and glacial acetic acid (3 μL, 3 mg, 0.052 mmol, 0.1 eq) in 64% yield (107 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 3.21 (m, 2H, CH2); 4.54–4.65 (m, 2H, CH2O); 6.90 (m, 1H, H-2″); 7.03 (m, 1H, H-4″); 7.19 (t, J = 7.4 Hz, 1H, H-7′); 7.27–7.66 (m, 5H, H-4′, H-6′, H-3″); 7.77 (d, J = 7.7 Hz, 1H, CH=); 12.25 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 28.5 (CH2); 71.8 (OCH2); 108.7 (C-7′); 120.4 (C-2″); 121.4 (C-2″); 124.8 (C-4″); 125.9 (C-3a′); 126.5 (C-6′); 128.9 (C=, C-5); 129.0 (C-1″); 129.2 (C-4′); 129.4 (C-3″); 129.9 (C-3″); 131.0 (CH=); 148.0 (C-3a′); 148.1 (C-7a′); 161.4 (C=O, C-4); 206.7 (C=N, C-2). HRMS, m/z: 345.0676 found (calculated for C18H14N2O2SNa, [M+Na]+. requires 345.0674).
(5Z)-5-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethylene)-2-phenylamino-1,3-thiazol-4(5H)-one (5x). According to the standard procedure, 5x was synthesized at 120 °C with a power of 100 W for 20 min from 2-phenylamino-1,3-thiazolidin-4-one 8a (100 mg, 0.52 mmol), 2,3-dihydro-1,4-benzodioxine-6-carbaldehyde 2c (85 mg, 0.52 mmol), sodium acetate (43 mg, 0.52 mmol, 1 eq), and glacial acetic acid (0.2 μL, 205 mg, 3.42 mmol, 6.6 eq) in 98% yield (175 mg) as light-yellow powder; mp > 250 °C. 1H NMR (DMSO-d6) δ: 4.24–4.33 (m, 4H, OCH2CH2O); 6.93–7.06 (m, 2H, H-2″); 7.12 (m, 1H, H-8′); 7.20 (t, J = 7.4 Hz, 1H, H-4″); 7.42 (m, 2H, H-3″); 7.52 (m, 1H, H-5′); 7.62 (m, 1H, H-7′); 7.77 (m, 1H, CH=); 12.26 (br s, 1H, NH). 13C NMR (DMSO-d6) δ: 63.9–64.4 (CH2CH2O); 117.7 (C-5′); 117.8 (C-8′); 120.5 (C-2″); 121.0 (C-1′); 121.4 (C-2″); 123.3 (C-7′); 124.9 (C-4″); 126.7 (C=, C-5); 127.1 (C-1″); 129.1 (C-3″); 129.4 (C-3″); 130.4 (CH=); 143.5 (C-4a′); 145.1 (C-8a′); 170.5 (C=O, C-4); 117.3 (C=N, C-2). HRMS, m/z: 361.0623 found (calculated for C18H14N2O3SNa, [M+Na]+. requires 361.0623).


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















































































