
State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy

 , ,
, , 
Abstract
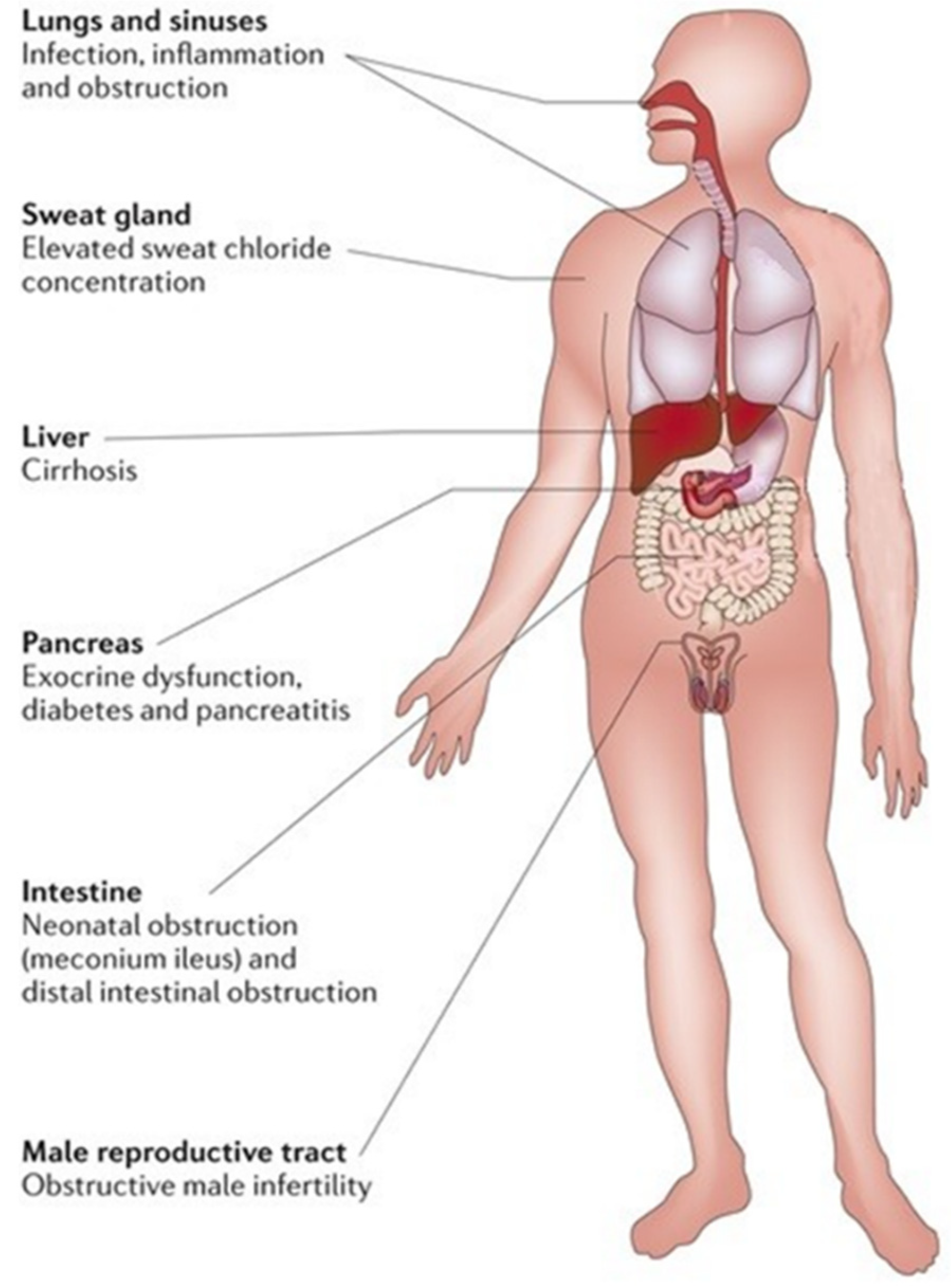
:1. Introduction
2. CTFR Potentiators
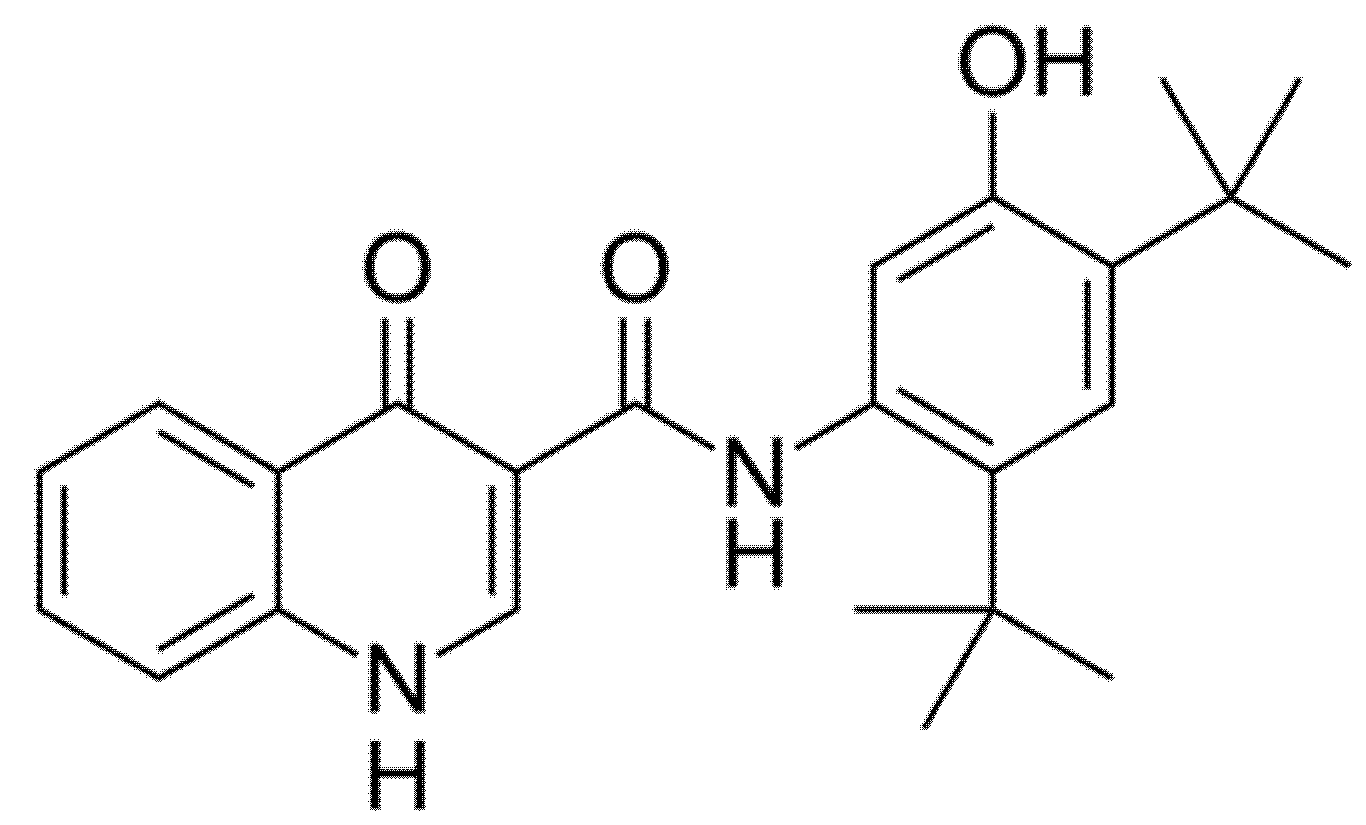
2.1. Ivacaftor (Formerly VX-770; KALYDECO®)
2.2. D9-Ivacaftor (Formerly VX-561 or CTP-656)
2.3. Icenticaftor (Formerly QBW-251)
2.4. ABBV-3067 (Formerly GLPG3067)
2.5. ABBV-974 (Formerly GLPG1837)
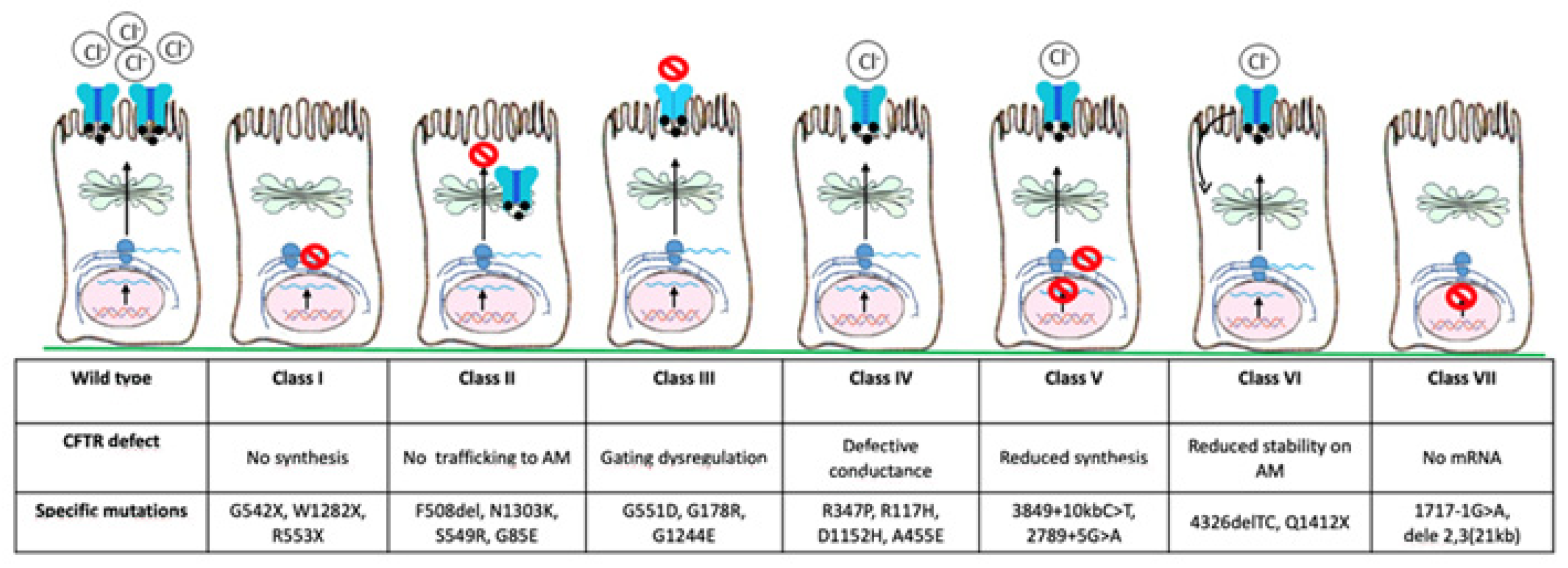
3. CTFR Correctors
3.1. Lumacaftor (Formerly VX-809)
3.2. Tezacaftor (Formerly VX-661)
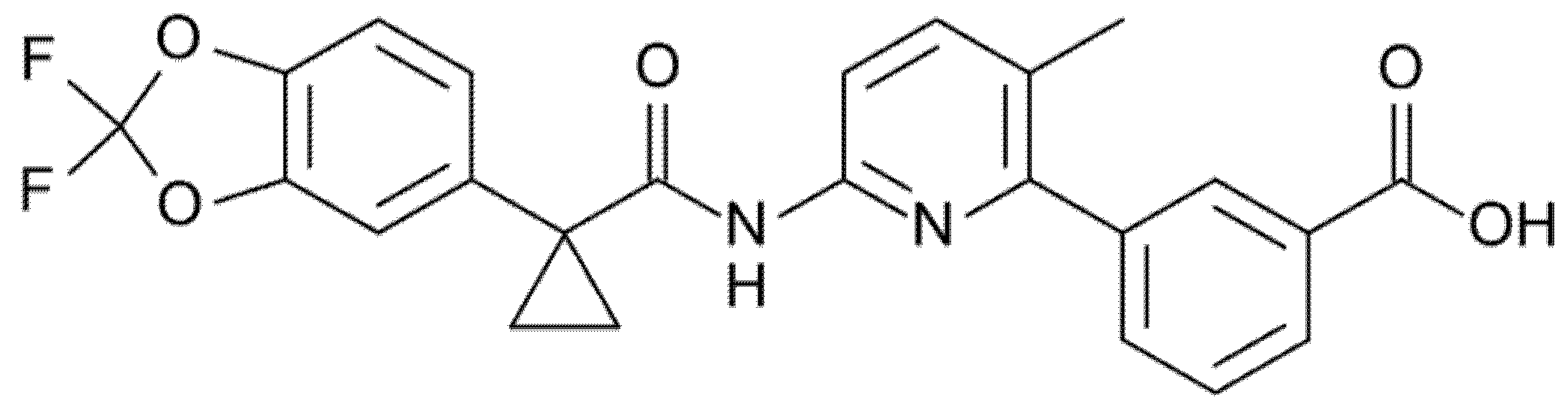
3.3. Galicaftor (Formerly GLPG2222 or ABBV-2222)
3.4. VX-152, VX-440, VX-445, and VX-659
3.5. FDL 169
4. Double Combination CFTR Modulator Therapy
4.1. Lumacaftor + Ivacaftor (ORKAMBI®)
4.2. Tezacaftor + Ivacaftor (SYMDEKO®/SYMKEVI®)
5. New CFTR Modulators and Triple Combination Therapy
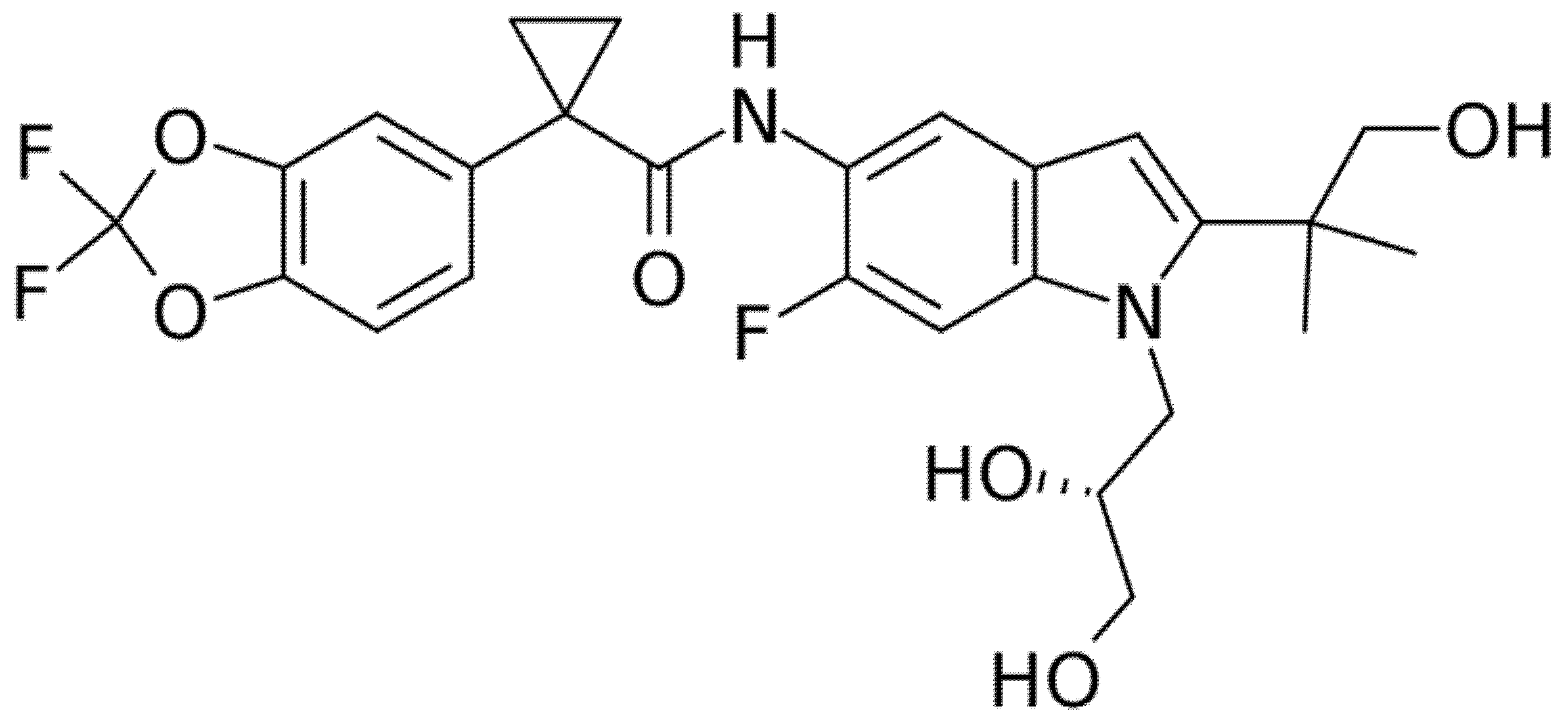
5.1. Elexacaftor (Formerly VX-445)
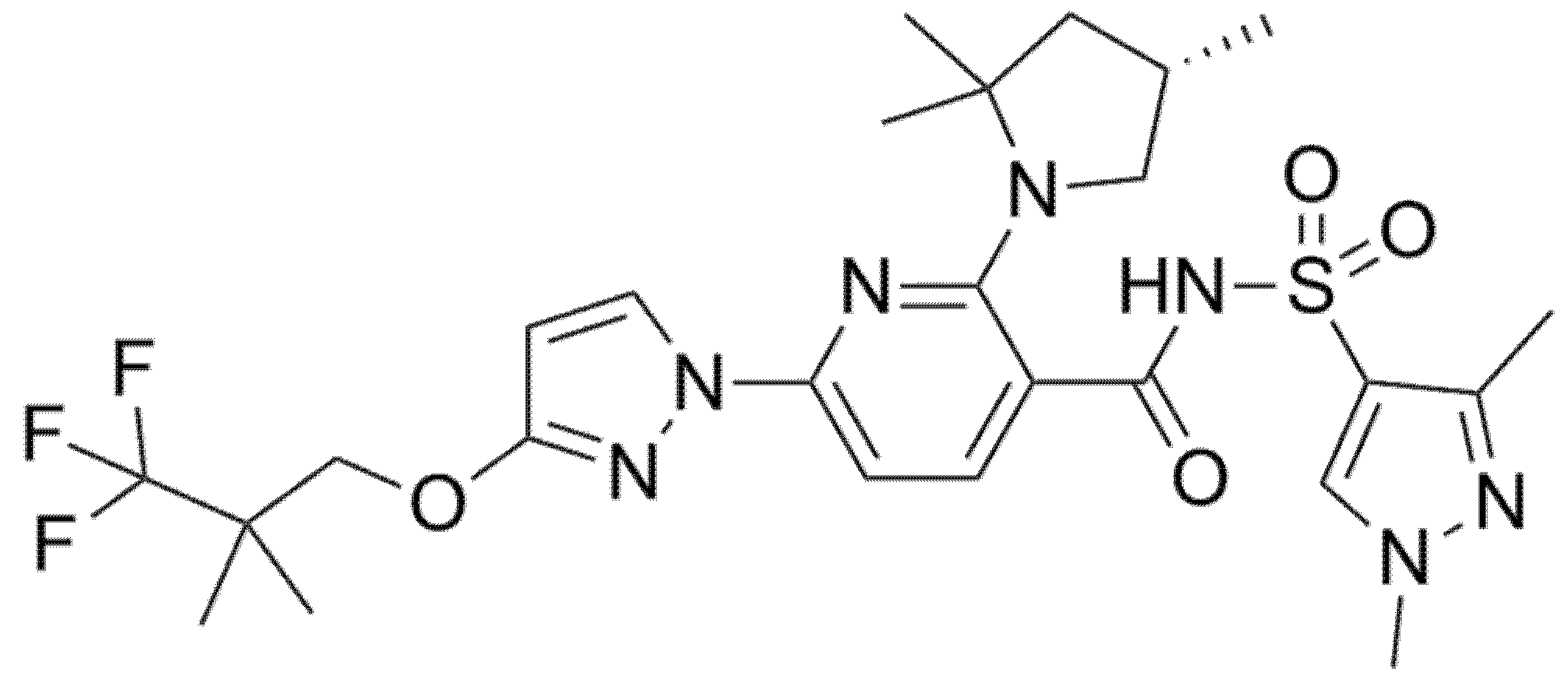
5.2. Bamocaftor (Formerly VX-659)
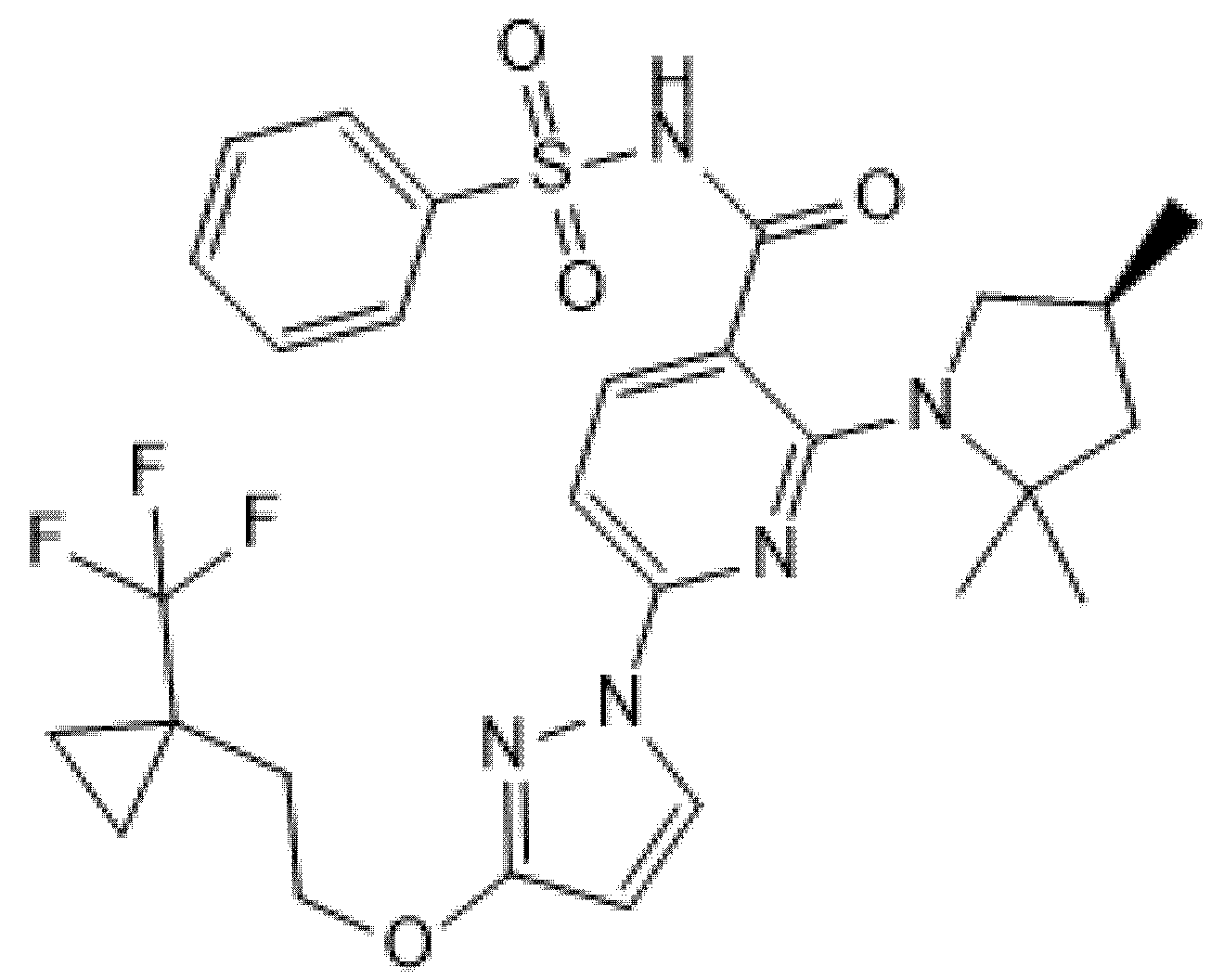
5.3. Proteostasis Pipeline
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Connett, G.J. Lumacaftor-ivacaftor in the treatment of cystic fibrosis: Design, development and place in therapy. Drug Des. Dev. Ther. 2019, 13, 2405–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Keeling, K.M.; Rowe, S.M. Pharmacological approaches for targeting cystic fibrosis nonsense mutations. Eur. J. Med. Chem. 2020, 200, 112436. [Google Scholar] [CrossRef]
- Rafeeq, M.M.; Murad, H.A.S. Cystic fibrosis: Current therapeutic targets and future approaches. J. Transl. Med. 2017, 15, 84. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef]
- McKone, E.F.; Emerson, S.S.; Edwards, K.L.; Aitken, M.L. Effect of genotype on phenotype and mortality in cystic fibrosis: A retrospective cohort study. Lancet 2003, 361, 1671–1676. [Google Scholar] [CrossRef]
- Cystic Fibrosis Trust. UK Cystic Fibrosis Registry Annual Data Report 2019. Available online: https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/2019%20Registry%20Annual%20Data%20report_Sep%202020.pdf (accessed on 14 April 2021).
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [Green Version]
- Harman, K.; Dobra, R.; Davies, J.C. Disease-modifying drug therapy in cystic fibrosis. Paediatr. Respir. Rev. 2018, 26, 7–9. [Google Scholar] [CrossRef]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012, 287, 36639–36649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harutyunyan, M.; Huang, Y.; Mun, K.S.; Yang, F.; Arora, K.; Naren, A.P. Personalized medicine in CF: From modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L529–L543. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Kalydeco. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kalydeco (accessed on 1 September 2020).
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M.; VX11-770-110 (KONDUCT) Study Group. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: A double-blind, randomised controlled trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Robertson, S.; Green, Y.; Rosenfeld, M. An open-label study of the safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2 to 5 years with CF and CFTR gating mutation: The KIWI study. In Proceedings of the 28th Annual North American Conference of the Cystic Fibrosis Foundation, Atlanta, GA, USA, 9–11 October 2014. [Google Scholar]
- Rosenfeld, M.; Cunningham, S.; Harris, W.T.; Lapey, A.; Regelmann, W.E.; Sawicki, G.S.; Southern, K.W.; Chilvers, M.; Higgins, M.; Tian, S.; et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5 years (KLIMB). J. Cyst. Fibros. 2019, 18, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Tian, S.; Schneider, J.; Cunningham, S.; Davies, J.C.; et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): A phase 3 single-arm study. Lancet Respir. Med. 2018, 6, 545–553, Erratum in: Lancet Respir. Med. 2018, 6, e35. Erratum in: Lancet Respir. Med. 2019, 7, e15.. [Google Scholar] [CrossRef]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef]
- Van Goor, F.; Yu, H.; Burton, B.; Hoffman, B.J. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 2014, 13, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguépéroux, I.; De Braekeleer, M. The CFTR 3849+10kbC->T and 2789+5G->A alleles are associated with a mild CF phenotype. Eur. Respir. J. 2005, 25, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171, 106073. [Google Scholar] [CrossRef]
- Kerem, E.; Cohen-Cymberknoh, M.; Tsabari, R.; Wilschanski, M.; Reiter, J.; Shoseyov, D.; Gileles-Hillel, A.; Pugatsch, T.; Davies, J.C.; Short, C.; et al. Ivacaftor in People with Cystic Fibrosis and a 3849+10kb C→T or D1152H Residual Function Mutation. Ann. Am. Thorac. Soc. 2021, 18, 433–441. [Google Scholar] [CrossRef]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D.E.; VX 08-770-104 Study Group. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Uttamsingh, V.; Pilja, L.; Brummel, C.L.; Grotbeck, B.; Cassella, J.V.; Braman, G. Ctp-656 Multiple Dose Pharmacokinetic Profile Continues to Support a Once-Daily Potentiator for Cystic Fibrosis Patients with Gating Mutations. In Proceedings of the 30th Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation, Orlando, FL, USA, 27–29 October 2016. [Google Scholar]
- Vertex Pharmaceuticals Incorporated. A Phase 2 Study to Evaluate Efficacy and Safety of VX-561 in Subjects Aged 18 Years and Older with Cystic Fibrosis. NLM Identifier: NCT03911713. Available online: https://clinicaltrials.gov/ct2/show/NCT03911713 (accessed on 23 September 2020).
- Kazani, S.; Rowlands, D.J.; Bottoli, I.; Milojevic, J.; Alcantara, J.; Jones, I.; Kulmatycki, K.; Machineni, S.; Mostovy, L.; Nicholls, I.; et al. Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251). J. Cyst. Fibros. 2021, 20, 250–256. [Google Scholar] [CrossRef]
- AbbVie. A Phase 2 Study of ABBV-3067 alone and in Combination with ABBV-2222. NLM Identifier: NCT03969888. Available online: https://clinicaltrials.gov/ct2/show/NCT03969888 (accessed on 23 September 2020).
- Conrath, K.; Gesson, C.; Allamassey, L.; Van de Steen, O.; Kanters, D.; De Kock, H.; De Boek, C.; Davies, J.C. Glpg1837 in subjects with cystic fibrosis and the S1251n or G551d mutation: Results from phase 2a studies (Saphira 1 and 2). In Proceedings of the 31st Annual North American Cystic Fibrosis Conference Cystic Fibrosis Foundation, Indianapolis, IN, USA, 2–4 November 2017. [Google Scholar]
- Guerra, L.; Favia, M.; Di Gioia, S.; Laselva, O.; Bisogno, A.; Casavola, V.; Colombo, C.; Conese, M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discov. 2020, 15, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phuan, P.W.; Yang, B.; Knapp, J.M.; Wood, A.B.; Lukacs, G.L.; Kurth, M.J.; Verkman, A.S. Cyanoquinolines with independent corrector and potentiator activities restore ΔPhe508-cystic fibrosis transmembrane conductance regulator chloride channel function in cystic fibrosis. Mol. Pharmacol. 2011, 80, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T.; VX11-661-101 Study Group. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Shiferaw, D.; Faruqi, S. Profile of tezacaftor/ivacaftor combination and its potential in the treatment of cystic fibrosis. Ther. Clin. Risk Manag. 2019, 15, 1029–1040, Erratum in: Ther. Clin. Risk Manag. 2019, 15, 1207. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A.M.; Vortherms, T.; et al. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid (ABBV/GLPG-2222), a Potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for the Treatment of Cystic Fibrosis. J. Med. Chem. 2018, 61, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Galapagos, N.V. First-in-Human Single and Multiple Dose of GLPG2222. NLM Identifier: NCT02662452. Available online: https://clinicaltrials.gov/ct2/show/NCT02662452 (accessed on 29 September 2020).
- Bell, S.C.; Barry, P.J.; De Boeck, K.; Drevinek, P.; Elborn, J.S.; Plant, B.J.; Minić, P.; Van Braeckel, E.; Verhulst, S.; Muller, K.; et al. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J. Cyst. Fibros. 2019, 18, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Flatley Discovery Lab LLC. A Study to Evaluate Safety, PK and PD of FDL169 in Cystic Fibrosis Subjects. NLM Identifier: NCT03093714. Available online: https://clinicaltrials.gov/ct2/show/NCT03093714 (accessed on 30 September 2020).
- Flatley Discovery Lab LLC. Bioavailability and Pharmacokinetics Study of FDL169 in Healthy Subjects and Subjects with Cystic Fibrosis. NLM Identifier: NCT02767297. Available online: https://clinicaltrials.gov/ct2/show/NCT02767297 (accessed on 30 September 2020).
- European Medicines Agency. Orkambi. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/orkambi (accessed on 1 September 2020).
- Boyle, M.P.; Bell, S.C.; Konstan, M.W.; McColley, S.A.; Rowe, S.M.; Rietschel, E.; Huang, X.; Waltz, D.; Patel, N.R.; Rodman, D.; et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial. Lancet Respir. Med. 2014, 2, 527–538. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.; Niknian, M.; Gilmartin, G.; Pilewski, J.M.; VX11-770-901 Investigators. Effect of ivacaftor in patients with advanced cystic fibrosis and a G551D-CFTR mutation: Safety and efficacy in an expanded access program in the United States. J. Cyst. Fibros. 2016, 15, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- Milla, C.E.; Ratjen, F.; Marigowda, G.; Liu, F.; Waltz, D.; Rosenfeld, M.; VX13-809-011 Part B Investigator Group. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am. J. Respir. Crit. Care Med. 2017, 195, 912–920. [Google Scholar] [CrossRef] [Green Version]
- McNamara, J.J.; McColley, S.A.; Marigowda, G.; Liu, F.; Tian, S.; Owen, C.A.; Stiles, D.; Li, C.; Waltz, D.; Wang, L.T.; et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: An open-label phase 3 study. Lancet Respir. Med. 2019, 7, 325–335. [Google Scholar] [CrossRef]
- Hubert, D.; Chiron, R.; Camara, B.; Grenet, D.; Prévotat, A.; Bassinet, L.; Dominique, S.; Rault, G.; Macey, J.; Honoré, I.; et al. Real-life initiation of lumacaftor/ivacaftor combination in adults with cystic fibrosis homozygous for the Phe508del CFTR mutation and severe lung disease. J. Cyst. Fibros. 2017, 16, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther CRJr Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef] [Green Version]
- Schneider, E.K. Cytochrome P450 3A4 Induction: Lumacaftor versus Ivacaftor Potentially Resulting in Significantly Reduced Plasma Concentration of Ivacaftor. Drug Metab. Lett. 2018, 12, 71–74. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Symkevi. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/symkevi (accessed on 1 September 2020).
- Food and Drug Administration. Symdeko. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210491lbl.pdf (accessed on 1 September 2020).
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Rowe, S.M.; McColley, S.A.; Rietschel, E.; Li, X.; Bell, S.C.; Konstan, M.W.; Marigowda, G.; Waltz, D.; Boyle, M.P.; VX09-809-102 Study Group. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann. Am. Thorac. Soc. 2017, 14, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.; Flume, P.; McNamara, J.; Solomon, M.; Chilvers, M.; Chmiel, J.; Harris, R.S.; Haseltine, E.; Stiles, D.; Li, C.; et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 708–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southern, K.W.; Murphy, J.; Sinha, I.P.; Nevitt, S.J. Corrector therapies (with or without potentiators) for people with cystic fibrosis with class II CFTR gene variants (most commonly F508del). Cochrane Database Syst. Rev. 2020, 12, CD010966. [Google Scholar] [CrossRef]
- Munck, A.; Kerem, E.; Ellemunter, H.; Campbell, D.; Wang, L.T.; Ahluwalia, N.; Owen, C.A.; Wainwright, C. Tezacaftor/ivacaftor in people with cystic fibrosis heterozygous for minimal function CFTR mutations. J. Cyst. Fibros. 2020, 19, 962–968. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Mall, M.A.; Ramsey, B.W.; McKone, E.F.; Tullis, E.; Marigowda, G.; McKee, C.M.; Waltz, D.; Moskowitz, S.M.; Savage, J.; et al. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508delalleles. ERJ Open Res. 2019, 5, 00082–02019. [Google Scholar] [CrossRef] [Green Version]
- Vertex Pharmaceuticals Incorporated. A Study Evaluating the Safety of VX-152 Combination Therapy in Adults with Cystic Fibrosis. NLM Identifier: NCT02951195. Available online: https://clinicaltrials.gov/ct2/show/NCT02951195 (accessed on 30 September 2020).
- Vertex Pharmaceuticals Incorporated. A Study Evaluating the Safety and Efficacy of VX-440 Combination Therapy in Subjects with Cystic Fibrosis. NLM Identifier: NCT02951182. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02951182 (accessed on 30 September 2020).
- European Medicines Agency. Kaftrio. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio (accessed on 28 September 2020).
- Vertex Pharmaceuticals Inc. Trikafta™ (Elexacaftor, Tezacaftor and Ivacaftor Tablets; Ivacaftor Tablets): US Prescribing Information. 2019. Available online: https://pi.vrtx.com/files/uspi_elexacaftor_tezacaftor_ivacaftor.pdf (accessed on 15 November 2020).
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948, Erratum in: Lancet 2020, 395, 1694. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-tezacaftor-ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of ELX/TEZ/IVA in Children 6 through 11 Years of Age with CF and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021, 203. [Google Scholar] [CrossRef]
- Van Goor, F. Precision Medicine in Cystic Fibrosis: Ivacaftor in Rare Mutations. In Proceedings of the 30th North American Cystic Fibrosis Conference. Cystic Fibrosis Foundation, Orlando, FL, USA, 27–29 October 2016. [Google Scholar]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Vertex Pharmaceuticals Incorporated. A Study of VX-659 Combination Therapy in CF Subjects Homozygous for F508del (F/F). NLM Identifier: NCT03460990. Available online: https://clinicaltrials.gov/ct2/show/NCT03460990 (accessed on 1 October 2020).
- Vertex Pharmaceuticals Incorporated. A Phase 3 Study of VX-659 Combination Therapy in Subjects with Cystic Fibrosis Heterozygous for the F508del Mutation and a Minimal Function Mutation (F/MF). NLM Identifier: NCT03447249. Available online: https://clinicaltrials.gov/ct2/show/NCT03447249 (accessed on 1 October 2020).
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discov. 2018, 23, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modulators | Commercial Name | Approval Year | Responsive Mutations | Approved Ages |
|---|---|---|---|---|
| Ivacaftor | Kalydeco® (EU/USA) | 2012 | G551D, S549N, G1244E, G178R, S1251N, G551S, G1349D, S1255P, R117H, E56K, K1060T, P67L, E193K, A1067T, R74W, L206W, G1069R, D110E, R347H, D579G, R1070Q, D1270N, D110H, R352Q, S945L, R1070W, R117C, A455E, S977F, F1074L, F1052V, D115H; 3849+10 kb C>T, 2789+5G>A, 3273-26A>G, 711+3A>G, E831X | ≥4 months |
| Lumacaftor-Ivacaftor | Orkambi® (EU/USA) | 2015 | Two copy of F508del | ≥2 years |
| Tezacaftor-Ivacaftor | Symkevi® (EU) Symdeko® (USA) | 2018 | Two copy of F508del One copy of F508del in association with E56K, K1060T, P67L, E193K, A1067T, R74W, L206W, D110E, D110H, R347H, D579G, R1070Q, D1270N, R352Q, S945L, R1070W, R117C, A455E, S977F, F1074L, F1052V, D1152H, 3849+10 kb C>T, 2789+5G>A, 327326A>G, 711+3A>G | ≥6 years |
| Elexacaftor-Tezacaftor-Ivacaftor | Kaftrio® (EU) Trikafta® (USA) | 2020 (EU) 2019 (USA) | One copy of F508del | ≥12 years |
| Study | Patients Characteristics | Outcomes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study Identifier (NCT) | Name | First Author, Year | Phase | Investigational Drug | Age (Years) | Genotype | Number of Patients | ppFEV1 (%) | Sweat Chloride (mmol/L) | CFQ-R Score (Points) |
| NCT03227471 | VX16-445-001 | Keating, 2018 | 2 | ELX/TEZ/IVA | ≥18 | F508del/F508del | 28 | +11 (*) | −39.6 (*) | +20.7 (*) |
| F508del/MF | 95 | +13.8 (*) | −39.1 (*) | +25.7 (*) | ||||||
| NCT03224351 | VX16-659-101 | Davies, 2018 | 2 | VX-659/TEZ/IVA | ≥18 | F508del/F508del | 29 | +9.7 (*) | −42.2 (*) | +19.5 (*) |
| F508del/MF | 88 | +13.3 (*) | −51.4 (*) | +24.6 (*) | ||||||
| NCT03525548 | VX17-445-103 | Heijerman, 2019 | 3 | ELX/TEZ/IVA | ≥12 | F508del/F508del | 107 | +10 (*) | −45.1 (*) | +17.4 (*) |
| NCT03525444 | VX17-445-102 | Middleton, 2019 | 3 | ELX/TEZ/IVA | ≥12 | F508del/MF | 403 | +13.8 (*) | −41.8 (**) | +20.2 (**) |
| NCT03460990 | VX17-659-103 | - | 3 | VX-659/TEZ/IVA | ≥12 | F508del/F508del | 111 | +9.9 (*) | −48.7 (*) | +13.5 (*) |
| NCT03447249 | VX17-659-102 | - | 3 | VX-659/TEZ/IVA | ≥12 | F508del/MF | 382 | +14 (*) | −44.6 (**) | +20.1 (**) |
| NCT03691779 | VX18-445-106 | Zemanick, 2021 | 3 | ELX/TEZ/IVA | 6–11 | F508del/F508del | 29 | +11.2 (**) | −70.4 (**) | +7 (**) |
| F508del/MF | 37 | +9.1 (**) | −55.1 (**) | +6.9 (**) | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meoli, A.; Fainardi, V.; Deolmi, M.; Chiopris, G.; Marinelli, F.; Caminiti, C.; Esposito, S.; Pisi, G. State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy. Pharmaceuticals 2021, 14, 928. https://doi.org/10.3390/ph14090928
Meoli A, Fainardi V, Deolmi M, Chiopris G, Marinelli F, Caminiti C, Esposito S, Pisi G. State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy. Pharmaceuticals. 2021; 14(9):928. https://doi.org/10.3390/ph14090928
Chicago/Turabian StyleMeoli, Aniello, Valentina Fainardi, Michela Deolmi, Giulia Chiopris, Francesca Marinelli, Caterina Caminiti, Susanna Esposito, and Giovanna Pisi. 2021. "State of the Art on Approved Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators and Triple-Combination Therapy" Pharmaceuticals 14, no. 9: 928. https://doi.org/10.3390/ph14090928