
MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer


1
Department of Laboratory Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan
2
Graduate Institute of Clinical Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
3
Research Center for Environmental Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2021, 14(2), 130; https://doi.org/10.3390/ph14020130
Submission received: 15 December 2020
/
Revised: 25 January 2021
/
Accepted: 2 February 2021
/
Published: 6 February 2021
(This article belongs to the Special Issue Treatment Options and Therapeutics for Non-small Cell Lung Cancer)
Abstract
:Epidermal growth factor tyrosine kinase inhibitors (EGFR-TKIs) are currently the most effective treatment for non-small cell lung cancer (NSCLC) patients, who carry primary EGFR mutations. However, the patients eventually develop drug resistance to EGFR-TKIs after approximately one year. In addition to the acquisition of the EGFR T790M mutation, the activation of alternative receptor-mediated signaling pathways is a common mechanism for conferring the insensitivity of EGFR-TKI in NSCLC. Upregulation of the Mer receptor tyrosine kinase (MERTK), which is a member of the Tyro3-Axl-MERTK (TAM) family, is associated with a poor prognosis of many cancers. The binding of specific ligands, such as Gas6 and PROS1, to MERTK activates phosphoinositide 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) cascades, which are the signaling pathways shared by EGFR. Therefore, the inhibition of MERTK can be considered a new therapeutic strategy for overcoming the resistance of NSCLC to EGFR-targeted agents. Although several small molecules and monoclonal antibodies targeting the TAM family are being developed and have been described to enhance the chemosensitivity and converse the resistance of EGFR-TKI, few have specifically been developed as MERTK inhibitors. The further development and investigation of biomarkers which can accurately predict MERTK activity and the response to MERTK inhibitors and MERTK-specific drugs are vitally important for obtaining appropriate patient stratification and increased benefits in clinical applications.

1. Introduction
Lung cancer remains the leading cause of cancer-related death worldwide, and non-small cell lung cancer (NSCLC) represents the major histological subtype of the disease [1,2]. A subgroup of NSCLC patients, characterized by a high prevalence in never-smoker Asian females, respond well to epidermal growth factor tyrosine kinase inhibitors (EGFR-TKIs), due to the presence of sensitizing EGFR mutations, including the L858R point mutation in exon 21 and exon 19 deletions [3,4]. However, the long-term effectiveness of EGFR-TKIs is universally limited by the development of resistance. About 50% of EGFR-TKI-resistant patients can be explained by the acquisition of the EGFR T790M mutation, and the progression-free survival of these patients is a benefit of using osimertinib (AZD9291), which is a third-generation EGFR-TKI, as their second-line treatment [5]. The pathological mechanism for the other 50% of EGFR-TKI-resistant patients remains controversial. The overactivation of other pro-survival receptor tyrosine kinases (RTKs), such as HER2 and MNNG HOS transforming gene (MET) that share similar downstream signaling pathways with EGFR, is frequently observed in EGFR-TKI-resistant patients without the EGFR T790M mutation [6,7]. Many experimental settings demonstrate that targeting the “bypass” RTK-mediated signaling pathways is a potentially therapeutic strategy for overcoming the EGFR-TKI resistance (reviewed in ref. [8]).
However, the expression pattern and frequency of these receptors varies from patient to patient [9]. Thus, it is necessary to evaluate the therapeutic effect of targeting individual RTK in EGFR-TKI resistance.
The Mer receptor tyrosine kinase (MERTK), which is a member of the Tyro3-Axl-MERTK (TAM) RTK family, was initially cloned from a human B-lymphoblastoid expression library and predominantly expressed on monocytes, which are cells of epithelial and reproductive tissue [10]. MERTK is physiologically involved in natural killer-cell differentiation, the immune response, platelet aggregation, and the development of the central nervous system [11,12]. The dysregulation of MERTK has been linked to the pathology of many human diseases, such as atherosclerosis [13], retinitis pigmentosa [14], chronic obstructive pulmonary disease [15], thrombosis [16], acute liver failure [17], and diabetes [18]. In addition, MERTK contributes to the oncogenesis of a spectrum of human cancers, including hematological malignancies, glioblastoma, NSCLC, melanoma, breast cancer, colon cancer, gastric cancer, and prostate cancer [19]. Recently, evidence has showed that MERTK can function as a pro-angiogenic gene and promotes the metastasis of human cancers [20,21]. Several of these pathways were also observed to be involved in EGFR-downstream signaling, such as the activation of MAPK and PI3K/Akt cascades [8]. The collateral signaling events potentially causing the drug resistance in NSCLC are the presence of sensitizing EGFR mutations and ectopic MERTK expression (Figure 1).
TAM RTKs have been reported as novel anticancer targets through their promotion of tumor cell survival, proliferation, and migration [22,23]. Axl, which is the other member of the TAM family, has been implicated as a mechanism of resistance to EGFR-TKIs through bypassing inhibition of the original targeted oncogenic driver [8]. The DNA structure, biological ligands, and downstream signaling pathways are highly similar between Axl and MERTK [23]. Additionally, MERTK was revealed to be required for the surface accumulation of EGFR and downstream pathway activation [24]. Therefore, MERTK may represent a new target for therapeutic intervention in EGFR-TKI-resistant NSCLC.
In this review, we discuss the current understanding of mechanisms underlying EGFR-TKI resistance in NSCLC, the functions and regulation of MERTK, and the roles of MERTK in the resistance to EGFR-TKIs. We also present recent advances toward the introduction of small molecules targeting MERTK that have therapeutic potential.
2. Acquired EGFR-TKI Resistance in Lung Cancer
The epidermal growth factor receptor (EGFR/HER1/ERBB1) is a transmembrane protein consisting of an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular domain, and is composed of a juxtamembrane segment, a kinase domain, and a C-terminal regulatory tail (Figure 2). EGFR is commonly expressed in epithelial, mesenchymal, and neuronal tissues and contributes to cell proliferation, differentiation, and development [25]. Several mouse models of EGFR deficiency have been established, but the phenotypes of EGFR deficiency are strain-dependent. Some EGFR-knockout mouse strains are embryonically lethal and other survival strains show developmental abnormalities in bonding, cardiovascular tissues, skin, and eyes [26,27,28]. In adults, the dysregulation of EGFR is involved in many pathological mechanisms of human diseases, such as renal fibrosis [29], coronary artery disease [30], autoimmune disease [31], chronic obstructive pulmonary disease [32], and cancers [33,34,35,36].
The binding of EGFR to its ligands, which are characterized by a consensus EGF motif containing six spatially conserved cysteine residues (CX7, CX4–5, CX10–13, and CXCX8) [37], induces the dimerization of two EGFR monomers. In the dimeric state, 12 of 20 tyrosine residues in the intracellular domain of each monomer are trans-phosphorylated and bind to downstream effectors, such as Shc1 and Grb2 [38], leading to the activation of several signaling pathways, including Ras/Raf/MAP kinase, PI3K/Akt, JAK/STAT, and phospholipase Cγ [39]. EGFR-mediated signaling pathways regulate various cellular functions in cancer cells, including cell migration, proliferation, angiogenesis, and survival [39]. The overactivation of EGFR and downstream signaling pathways in cancer cells can be triggered by different mechanisms, such as the overproduction of ligands and EGFR proteins; deficiency of EGFR protein turnover; existence of EGFR mutations, which cause the constitutive activation of EGFR; and cross-talk with alternative cell-surface receptors [40].
Epidermal growth factor receptor mutations are frequently observed in glioblastoma multiforme, breast cancer, and NSCLC [41,42,43,44]. The two most common EGFR-activating mutations in NSCLC are in-frame deletions in exon 19 and the L858R point mutation in exon 21, accounting for 45% and 40–45% of lung adenocarcinoma patients, respectively [4,7]. These mutations result in an asymmetric configuration of the kinase domain dimer, which leads to the ligand-independent activity of EGFR [45]. Cancer cells with the L858R mutations and exon 19 deletion may respond to the constitutive activation of the EGFR, which provides strong growth and survival signaling. Accordingly, targeting EGFR becomes a valuable strategy for the management of NSCLC patients who carry these active EGFR mutations.
Gefitinib, erlotinib, and afatinib are the EGFR-TKIs that reversibly (for gefitinib and erlotinib) or irreversibly (for afatinib) bind to the ATP-binding pocket in the kinase domain and block the kinase activity of EGFR. All of these EGFR-TKIs are recommended by international guidelines as a first-line treatment for NSCLC patients with sensitizing EGFR mutations [46]. A recent meta-analysis showed that these EGFR-TKIs had comparable effects on multiple pathological parameters of NSCLC patients receiving EGFR-TKIs as their first-line treatment [47]. Despite most of the patients responding well to the EGFR target therapy, tumor recurrence eventually occurs within 9–14 months in these patients, due to the development of drug resistance [48]. Several mechanistic themes underpinning the resistance to EGFR-targeted therapy have been studied [49].
The main mechanisms involved in the acquired resistance to EGFR-TKIs include secondary mutations in EGFR, phenotypic transformation, and the activation of alternative pathways [7]. The occurrence of a second EGFR mutation, called T790M, in exon 20 represents the most frequent mechanism and accounts for 60% of these cases [50]. Threonine 790 in the ATP-binding pocket of EGFR has been considered to be a “gatekeeper” residue. The T790M mutation increases the ATP affinity to the pocket domain of wild-type EGFR and the L858R mutant, thus reducing the potency of ATP-competitive EGFR-TKIs. To fight the acquired EGFR-TKI resistance, several small molecules targeting the EGFR T790M mutant have been discovered and some of these compounds have successfully entered different phases of clinical trials [51]. Osimertinib is the first third-generation EGFR-TKI that has been approved by The Food and Drug Administration (FDA) of the United States of America and The European Medicines Agency (EMA) of The European Union [52]. As a second-line treatment, osimertinb significantly prolonged the progression-free survival (PFS) of metastatic EGFR-mutant NSCLC patients who acquired the EGFR T790M mutation to 8.5 months compared to those receiving platinum therapy plus pemetrexed (PFS = 4.2 months) [53]. Interestingly, osimertinib also exhibited a higher therapeutic efficacy compared to that of standard EGFR-TKIs (gefitinib or erlotinib) in the first-line treatment of EGFR mutation-positive (with exon 19 deletion or the L858R mutation, but no T790M mutation) advanced NSCLC patients [54]. Unfortunately, the development of drug resistance to osimertinib, which may be due to acquired mutations other than T790M, remains a fundamental challenge for the disease control of NSCLC [55].
From the perspective of EGFR-TKI resistance without acquired EGFR mutations, even though the EGFR-TKIs efficiently block EGFR-mediated signaling pathways, the activation of other RTKs that drive compensatory downstream signaling pathways to EGFR may contribute to the growth and survival of cancer cells [56]. This hypothesis has been demonstrated by several pharmacological strategies using the combination of EGFR-TKIs with specific inhibitors targeting other RTKs [57]. Amplification of the genes encoding MET and epidermal growth factor receptor 2 (HER2/ERBB2) is frequently observed in recurrent tumors of NSCLC patients who received EGFR-TKIs as a first-line treatment [58]. There is an expanding spectrum of identified alternative signaling pathways which are activated by autocrine growth factor and cytokine signaling via cognate receptors, such as MET, BRAF, HER2, HER3, and TAM receptors [59].
3. Biological Function and Regulation of MERTK
MERTK is a member of the TAM RTK family, which also includes Axl and Tyro3. This small family of RTKs are characterized by a combination of two immunoglobin-like domains and two fibronectin type III repeats in the extracellular region, a transmembrane portion, and a cytoplasmic kinase domain (reviewed in ref. [60]) (Figure 3a). In the early 1990s, each TAM receptor gene was first identified and cloned from a variety of species by independent groups. Thereafter, the TAMs were grouped into a unique RTK family in 1991 through PCR cloning their homology intracellular kinase domains [61]. The regulatory mechanism of TAM RTKs has been described in several progresses, including cell survival, proliferation, migration, adhesion, blood clot stabilization, the mediation of efferocytosis, and the modulation of inflammation (reviewed in ref. [60,62,63,64]). In normal adult tissues, MERTK, Axl, and Tyro3 display a widespread distribution pattern, with overlapping but unique expression profiles (Figure 3b). MERTK is expressed in hematopoietic cells, including monocytes, dendritic cells, natural killer cells, and platelets. High levels of MERTK expression are also found in the retina, kidney, lung, testis, and ovary. The heart, brain, and skeletal muscle also express lower levels of MERTK. Almost all adult tissue displays Axl expression. Axl is most abundantly expressed in the hippocampus and cerebellum, as well as monocytes, platelets, endothelial cells, the heart, skeletal muscle, the liver, the kidney, and the testis. Tyro3 is mainly expressed in the nervous system, and is also detected in hematopoietic cells, as well as the lung, kidney, and breast (reviewed in ref. [60]).
Unlike most of the RTKs that play essential roles in embryonic development, loss-of-function mutations in RTK genes often result in an embryonic-lethal phenotype. Even though the simultaneous genetic deletion of all three TAM RTKs is not embryonically lethal, it resulted in diverse phenotypes in a wide range of defects, such as a hyper-inflammatory state, multiple organ defects, and lymphoproliferation (reviewed in ref. [65,66]). The physiological regulatory roles of the TAM family are prominent in the innate immune control, reproductive, hematopoietic, hemostasis, vascular, and nervous systems. Disruption of the TAM receptor can lead to numerous diseases, including coagulopathy, autoimmune diseases, retinitis pigmentosa, chronic hepatitis, and neuron dysfunction (reviewed in ref. [64,65,66,67,68]).
The best-studied TAM RTK function is the role of MERTK in the macrophage-derived clearance of apoptotic cells, known as efferocytosis (reviewed in ref. [66]). The inability of macrophages from MERTK knockdown mice to efficiently clear apoptotic cells has been associated with the production of autoantibodies and development of autoimmune diseases, such as systemic lupus erythematosus [69,70]. The typical activation of RTKs requires ligand binding to the extracellular domain and induces receptor dimerization, subsequently leading to intermolecular autophosphorylation. In the progress of efferocytosis, the two best-characterized TAM ligands, which are growth arrest-specific protein 6 (Gas6) and vitamin K-dependent protein S (PROS1), may physically link TAM rece-ptors, generally MERTK or Axl, expressed on the surface of the phagocyte [71]. In addition, these ligands have two special structural characters that are key to their bioactivity. The first is a carboxy-terminally positioned sex hormone-binding globulin (SHBG) domain. (Figure 3a) This SHBG domain can bind to the Ig domains of the receptors and induce their dimerization and subsequent kinase activity. The other is the Gla domain, which is rich in glutamic acid residues. γ-carboxylation of the Gla domain allows it to bind to the lipid phosphatidylserine (PtdSer) with its amino terminus. PtdSer is abundant in the human body, but only available to activate TAM receptors when displayed on the extracellular membrane of apoptotic cells, exosomes, and aggregated platelets. With respect to hemostasis, the MERTK receptors are located on platelets and mediate thrombogenesis, platelet aggregation, and stabilization, and reduced thrombus formation was observed in Gas or MERTK knockout mice (reviewed in ref. [62,64,66]). Furthermore, three new MERTK ligands were identified more recently, including tubby, tubby-like protein 1 (Tulp1), and galectin-3, which were also shown to be involved in the MERTK-mediated phagocytosis and innate immune response [72,73].
Many studies have demonstrated that both MERTK and Axl are linked to survival, growth, and migration, although the role of Tyro3 is less well-defined (Figure 3c). Studies using chimeric MERTK receptors have elucidated that signaling pathways downstream of the MERTK kinases include growth factor-mediated proteins such as PI3K, Ras and MAPKs. These pro-survival signaling pathways may be activated in both normal and cancer cells. The aberrant regulation of these signaling pathways plays an important role in oncogenic transformation. Therefore, the overexpression of MERTK has been detected in numerous cancers, including myeloid and lymphoblastic leukemias, breast cancer, non-small cell lung cancer, brain cancers, melanoma, prostate cancer, liver cancer, colorectal cancer, gastric cancer, ovarian cancer, and rhabdomyosarcomas (reviewed in ref. [19,66]).
Proto-oncogenes can be activated by several mechanisms, such as gene mutations, amplification, and altered protein expression. To date, the overexpression of TAM family members, rather than mutation, appears to be the predominant effect with the development of cancer (reviewed in ref. [60]). MERTKs activate prosurvival signaling pathways, which was first shown in leukemia/lymphoma cells. In a transgenic model, the ectopic expression of MERTK in lymphocytes promotes the development of lymphoblastic leukemia/lymphoma. Stimulation of these cells with Gas6 induced MERTK autophosphorylation and the subsequent activation of extracellular signal regulated kinase (ERK1/2) and Akt [74]. In human T-cell lymphoblastic leukemia cell lines, the stimulation of MERTK by the ligand Gas6 led to activation of the prosurvival proteins ERK1/2 and MERTK-dependent activation of the STAT pathway and contributed to prosurvival phenotypes of tumor cells [75]. MERTK has been implicated in metastasis and linked to migration and invasion phenotypes through the regulation of FAK1, RhoA, Rac1, and CDC42 [61]. Furthermore, MERTK inhibition decreased colony formation, invasion, and xenograft growth in a human melanoma murine xenograft model [76]. Aside from its direct roles in tumor metastasis, MERTK may have additional functions in promoting metastasis through its role in negative regulation of the immune system (reviewed in ref. [77]). TAM RTK signaling in the suppression of natural killer cell activity has been demonstrated in promoted metastasis [78], while in MERTK knockout mice, it showed a pro-inflammatory state in the tumor microenvironment and resulted in the inhibition of tumor metastasis [79]. In addition, MERTK acts as an inhibitor of the innate inflammatory response to pathogens and balances the immune system. On the other hand, it may suppress antitumor immunity. Macrophages have long been recognized as an ancient cell type involved in host defense against pathogens. More recently, macrophages are being rediscovered as key regulators in the immunosuppressive tumor microenvironment [80,81]. Many studies suggest that tumor-associated macrophages correlate with a poor prognosis in patients with cancer [82,83,84]. Tumor-associated macrophages can produce immune-modulating and wound-healing cytokines, which may decrease the antitumor immunity and increase the growth of tumor cells [85]. A previous study showed that the loss of MERTK in the tumor microenvironment of MERTK knockout mice slowed the establishment, growth, and metastasis of mammary tumors. The results in this study indicated that MERTK in the tumor microenvironment aids malignant tumor progression by suppressing antitumor immunity [79].
4. The Role of MERTK in EGFR-TKI Resistance
TAM-mediated signaling pathways are responsible for cell survival, proliferation, cell mobility, and tumor–host interactions, suggesting that the members of the TAM family may play important roles in the drug resistance of cancers. Multiple resistance mechanisms exist, including the intrinsic survival signaling in tumor cells, crosstalk of TAM RTKs as a bypass mechanism for other oncogene-targeted RTK inhibitors, and repression of the immune response in the tumor microenvironment (reviewed in ref. [22,66,86]). Much evidence strongly suggests that these receptors play major roles in resistance to conventional chemotherapy and targeted therapies (Figure 4).
The upregulation of MERTK can be observed in recurrent tumors of cancer patients who received target therapy or chemotherapy as their first-line treatment. In colorectal cancer, the upregulation of MERTK has been considered a predictive marker for resistance towards MEK1/2 inhibitors in a large clinical cohort [87]. The overexpression of MERTK was observed in melanoma cells with the BRAF V600E mutation, which are resistant to BRAF and MEK inhibitors [88]. In NSCLC, the excessive activation of Axl-mediated signaling pathways is associated with drug resistance to EGFR-targeted therapy [89,90,91]. However, the clinical relevance of MERTK and EGFR-TKI resistance is still unknown. A study using an in vitro cell culture showed that the overexpression of MERTK induced resistance to erlotinib in the PC9 cell line, which is an EGFR-TKI-sensitive cell line [92]. Furthermore, a specific MERTK inhibitor, UNC2025, induced apoptosis of NSCLC cell lines carrying the EGFR T790M mutation, leading to EGFR-TKI resistance [93]. In addition to its role in EGFR-TKI resistance, MERTK may also contribute to chemoresistance in NSCLC. The knockdown of MERTK by short hairpin RNAs (shRNAs) improved the sensitivity to chemotherapeutic agents by promoting apoptosis [94]. A monoclonal antibody targeting MERTK increased the chemosensitivity of NSCLC cells to carboplatin [95].
The contribution of the ligand-dependent canonical RTK signaling pathway of MERTK and Axl in drug resistance has been extensively explored [96]. One study demonstrated that the post-translational processing of membrane-bound Axl by γ-secretase-mediated proteolysis contributed to the nuclear localization of the cleaved intracellular domain of Axl [97]. Although the biological function of the Axl intracellular domain in the nucleus is unknown, Axl with specific mutations between amino acid 452 and 472 resisted γ-secretase and contributed to the chemotherapy of NSCLC cell lines.
Importantly, the signaling cascades activated downstream of Axl/MER are dependent on the context in which the ligands and microenvironment are presented to the TAM extracellular domains. A previous study observed that TAMs have distinct and dynamic patterns of activation by Gas6 and PROS1, representing the two best characterized TAM ligands. Gas6 binds to both MERTK and Axl, while PROS1 only binds to MERTK [96,98]. Furthermore, in the non-inflammatory microenvironment, MERTK shows weak activation toward both ligands and the affinity of Gas6 for Axl is significantly stronger than its affinity for MERTK. In contrast, in the presence of PtdSer lipid vesicle and apoptotic cells, Gas6 changes the strong binding affinity from Axl to MERTK [66,96].
The upregulation of MERTK has been observed in many cancers. Because MERTK and EGFR share PI3K/Akt and MAPK as their downstream signaling pathways, the targeting of MERTK by small molecules or monoclonal antibodies becomes a potential strategy for overcoming EGFR-TKI resistance in NSCLC. A recent study showed that the ectopic expression of MERTK induced erlotinib resistance in PC9 cells, which carry an exon19 deletion of EGFR, inducing drug resistance to erlotinib treatment [92]. It has been shown that MERTK is involved in the phenotypic and genotypic alterations of cancer cells and microenvironmental regulation. Here, the roles of MERTK in EMT, CSC maintenance, and the immunosuppressive microenvironment are discussed.
4.1. The Role of MERTK in EMT-Associated EGFR-TKI Resistance
Epithelial-to-mesenchymal transition is a biological process where epithelial cells morphologically and physiologically convert into a mesenchymal state, which is characterized by a loss of cellular polarity, reduced expression of junctional proteins (e.g., E-cadherin, Desmoglein-3, and Cytokeratin 18), greater motility and invasiveness, and the gain of mesenchymal markers (e.g., Vimentin and Matrix metalloproteinases). In addition to its roles in embryonic development and tissue repairing, EMT has been associated with metastasis, drug resistance, and stemness in multiple cancers. The upregulation of EMT inducers and downregulation of EMT inhibitors are frequently found in patients with an acquired drug resistance to chemotherapeutic agents and targeted therapies [99,100]. In the past decades, a number of intrinsic EMT inducers have been identified, including transcription factors (e.g., Snail, Slug, TWIST, and ZEB1/2), microRNAs (e.g., miR-200c and miR-34), polycomb group proteins (e.g., Bmi1 and EZH2), and long non-coding RNAs (e.g., lncRNA Val) [101]. Furthermore, many extracellular stimuli, including inflammation, hypoxia, DNA damage agents, and the extracellular matrix, are associated with EMT and tumor progression. Although the molecular mechanism of EMT-related EGFR-TKI resistance is largely unknown, reversed EMT by an ectopic expression of E-cadherin or a long-term withdrawal of EGFR-TKI sensitizes the non-T790M EGFR-TKI-resistant NSCLC cell lines to the EGFR-TKI treatment [102,103].
4.1.1. TGF-β-Induced EMT and EGFR-TKI Resistance
EMT can be triggered by a number of growth factor- and mitogen-mediated signaling pathways, which are mostly activated by RTKs. These EMT-related signaling pathways not only contribute to migration and invasion, but also promote proliferation, survival, and differentiation. Transforming growth factor β (TGF-β) is a potent EMT inducer. The binding of TGF-β to its receptors activates the canonic Smad2/3/4 signaling pathway that represses E-cadherin expression and activates Snail, Slug, and ZEB1/2 expression. Although the TGF-β receptor is a serine-threonine kinase, the phosphorylation of tyrosine residues in its cytoplasmic domain induces the activation of non-canonic PI3K/Akt/mTOR and Ras/Raf/MAP kinase pathways, which promote cell survival and proliferation. Transient exposure to TGF-β induces EMT and acquired EGFR-TKI resistance in NSCLC cell lines. TGF-β/Smad-induced EMT contributes to Axl-mediated gefitinib resistance in PC9 cells [104]. The activation of Axl or MERTK by Gas6 inhibits TGF-β-induced EMT [105].
4.1.2. Hippo Pathway-Mediated EMT and EGFR-TKI Resistance
The Hippo pathway is an evolutionarily conserved pathway that controls organ development, tissue regeneration, cell growth, and differentiation. In cancer, the Hippo pathway is involved in tumor growth, survival, migration, and stemness. In mammalian cells, the Yes-associated protein (YAP) and its paralog transcriptional coactivator with a PDZ-binding motif (TAZ) are the key transcription coactivators that translocate to the nucleus and interact with TEA domain family member (TEAD) transcription factors, leading to the activation of downstream genes that are involved in proliferation, anti-apoptosis, migration, and stemness maintenance (Figure 5). The activation of YAP/TAZ/TEAD is negatively regulated by the Hippo signal cascade. The auto-phosphorylation of MST kinases, including the MST1 and MST2 heterodimer, phosphorylates large tumor suppressor 1 and 2 kinases (LATS1 and LATS2) and MOB kinase activator 1A and 1B (MOB1A and MOB1B) and forms an MOB/LATS complex. The interaction of phosphorylated MOB1A/MOB1B with LATS1/LATS2 also leads to the auto-phosphorylation of LATS1 and LATS2 at Ser909 and Ser872, respectively. YAP and TAZ are the substrates of activated LATS1/LATS2. The phosphorylation of YAP/TAZ by LATS1/LATS2 results in their cytoplasmic sequestration through binding to 14-3-3 proteins or protein degradation by ubiquitination.
Although YAP has no DNA binding activity, it interacts with EMT-associated transcription factors that suppress the expression of epithelial markers and promote tumor progression [106]. The induction of YAP-mediated EMT contributes to proliferation, migration, invasion, and drug resistance in lung cancer [107,108]. It has been shown that YAP is upregulated in EGFR-TKI-resistant lung adenocarcinoma [109], which may be mediated by an Akt and MAPK-independent PI3K/PDK1 pathway [110]. An experimental setting showed that targeting YAP reversed EGFR-TKI resistance in NSCLC cells [111]. Combined treatment of EGFR-TKI and a YAP inhibitor prolonged survival among lung cancer patients [109]. Although the mechanism of YAP-mediated EGFR-TKI resistance is not clear, the activation of YAP is capable of promoting EMT, leading to the induction of EGFR-TKI resistance in lung cancer [112]. A recent study showed that the EMT-associated EGFR-TKI resistance was mediated by a YAP/FOXM1 axis [108].
In addition to its upstream regulators in Hippo pathways, YAP/TAZ has been shown to crosstalk with a variety of receptor-mediated signaling pathways, such as G-protein-coupled receptor, Notch, TGF-β, Wnt, Integrin, EGF, and VEGF signaling pathways. The activation of EGFR inactivates the upstream negative regulators of YAP through PI3K and PDK1-independent Akt phosphorylation. EGFR is also known to activate YAP through the Ras/Raf/MAPK signaling cascade. Upon the development of EGFR-TKI resistance, both Axl/MERTK and YAP are upregulated. It has been shown that MERTK directly interacts with and phosphorylates YAP, which is independent of upstream Hippo signaling, and contributes to tumorigenesis [113]. Gas6-mediated activation of Axl induces EMT and YAP expression, leading to EGFR-TKI resistance [114]. The results from these studies indicate that the Axl/MERTK signaling pathway is one of the positive upstream regulators of YAP that contributes to EMT-associated EGFR-TKI resistance. Interestingly, Axl may also be the downstream target of YAP that sustains YAP-driven EGFR-TKI resistance in EGFR-addicted lung cancer cell lines [115]. Both Axl/MERTK and the Hippo signaling pathway contribute to the acquired EGFR-TKI resistance, and positive feedback between Axl/MERTK expression and YAP activity may strongly sustain the EGFR-TKI resistance.
4.2. The Role of MERTK in the Cancer Stem Cell Phenotype
Most solid tumors contain a rare population of tumor cells that harbor stem cell properties, such as self-renewal, differentiation, and drug resistance. Similar to their corresponding tissue stem cells, CSCs have greater DNA repair, anti-oxidative stress, anti-apoptosis abilities compared to differentiated cancer cells. In addition, CSCs express higher ABC transporters that can eliminate anti-cancer agents from the cells. In solid tumors, most of the CSCs are in a quiescent state, which protect them from the attack of DNA damage drugs and allows timely DNA repair. The origin of CSCs in tumors is still debated. The clonal evolution hypothesis depicts that the accumulation of oncogenic mutations contributes to the transformation of normal stem cells to CSCs. On the other hand, differentiated cancer cells may acquire CSC phenotypes by exposure to microenvironmental stress, such as inflammation, oxidative stress, and DNA damage. Recently, the CSC phenotype has been linked to acquired EGFR-TKI resistance [116]. Stemness-associated genes are upregulated in the tumors of EGFR-TKI-resistant NSCLC patients compared to their expression levels in the tumors of EGFR-TKI-sensitive patients [117]. Both stemness- and senescence-associated proteins are upregulated in gefitinib-tolerated lung adenocarcinoma cell lines [118]. The treatment of EGFR-TKIs induces YAP-mediated anti-apoptosis and dormancy of cancer cells, representing a phenotype of CSCs [119]. The activation of stemness-associated signaling pathways contributes to the acquired EGFR-TKI resistance [120]. Pharmacological and genetic approaches targeting CSC phenotypes effectively increase the drug sensitivity of EGFR-TKI-resistant NSCLC to the treatment of EGFR-TKIs [121,122].
Although it is still unknown how the CSC phenotype is provoked during the acquisition of EGFR-TKI resistance, several possible mechanisms have been proposed. It has been shown that the downregulation of E-cadherin generated the CSC phenotype and drug resistance to osimertinib in NSCLC cell lines [123]. The inhibition of NF-κB activity by Withaferin A significantly depleted CD133-positive CSCs in gefitinib-resistant lung adenocarcinoma cell lines [118]. Several histone-modifying enzymes and pro-survival signaling pathways, e.g., STAT3 and Akt/mTOR, are also involved in the acquired CSC phenotype and EGFR-TKI resistance [124,125]. So far, research on the role of MERTK in lung CSCs is lacking. It has been shown that MERTK was upregulated in glioblastoma multiforme (GBM) [126]. The upregulation of MERTK is associated with an increase in invasion, the CSC phenotype, and resistance to DNA damage-induced apoptosis in glioma cells. This MERTK-mediated CSC maintenance may be controlled by a STAT3-kRas/Src-signaling cascade [127]. In prostate cancer, the activation of TAM receptors in disseminated prostate cancer cells by the osteoblast-derived Gas6 in the bone marrow induces G1 arrest and an increase in the CSC population in the microenvironment of bone marrow [128]. However, the same research group reported a further study demonstrating that the shRNA-mediated MERTK knockdown, but not Axl and Tyro3, resulted in the upregulation of stemness-associated genes, anti-apoptosis, G1-G0 arrest, and the characteristics of dormancy in prostate cancer cells [129]. These findings suggest that the function and activity of MERTK in CSC maintenance can be regulated by microenvironmental factors. In addition, although MERTK, Axl, and Tyro3 share the same ligands, MERTK may activate specific signaling pathways that elicit unique cellular behaviors independent of Axl and Tyro3. In lung cancer, the CSC phenotype can be induced by the signaling pathways that also crosstalk with MERTK-mediated signaling pathways, e.g., STAT3, PI3K/Akt, and Ras/Raf/MAPK. Further study is required to investigate whether the activation of MERTK may induce the CSC phenotype and lead to EGFR-TKI resistance as a consequence.
4.3. The Role of MERTK in Microenvironment Remodeling
The biological and chemical interactions between tumor cells and the microenvironment critically contribute to all hallmarks of cancer, including angiogenesis, inflammation, metabolic alterations, immune suppression, metastasis, and therapeutic resistance. The stromal cells, such as tumor-associated macrophages, neutrophils, tumor-infiltrating lymphocytes, tumor-associated fibroblasts, dendritic cells, and endothelial cells, influence tumor progression and the therapeutic efficacy by directly interacting with tumor cells, secreting growth factors and cytokines, constructing the extracellular matrix, and metabolizing drugs and stress factors. MERTK is not only upregulated in tumor cells, but also excessively expressed in the surrounding monocytes, macrophages, dendritic cells, and T lymphocytes. Activation of the MERTK signaling pathway induces an immune-suppressive microenvironment via regulating inflammatory cytokine-mediated signaling pathways, the PD-1 and PD-L1 axis, and efferocytosis of macrophages (Figure 6).
4.3.1. The Role of MERTK in Immune Checkpoint Inhibitor Therapy
Programmed cell death protein-1 (PD-1), which is an immune checkpoint receptor, is expressed in lymphocytes and myeloid cells. After binding to its ligands, programmed death-ligand 1 (PD-L1), which is broadly expressed in healthy tissues and tumor cells, negatively regulates the effector functions of T cells, and promotes T-cell tolerance and escape from host immunity by downregulating CD8+ T-cell survival. Accordingly, immune checkpoint inhibitors targeting the PD-1/PD-L1 axis have been applied as an anti-cancer therapy in many cancer types, including NSCLC. Currently, four monoclonal antibodies targeting PD-1 (pembrolizumab and nivolumab) and PD-L1 (atezolizumab and durvalumab) have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced NSCLC. However, patients with EGFR-mutant advanced NSCLC were limited in or excluded from the clinical trials of the four immune checkpoint inhibitors [130,131,132,133]. So far, a clinical trial investigating the therapeutic effect of immune checkpoint inhibitors in EGFR-TKI-resistant NSCLC patients is lacking. More studies are required to examine the regulation of the PD-1/PD-L1 interaction and immune microenvironment in EGFR-resistant tumors.
The Regulation of PD-L1 Expression by MERTK
PD-L1 is frequently upregulated in many cancer cells, including NSCLC cells. The high levels of PD-L1 expression can be explained by the activation of oncogenic signaling pathways, such as PI3K/Akt/mTOR and Ras/Raf/MAPKs, which are also the downstream signaling pathways of EGFR and MERTK [134,135]. The correlation between the EGFR status, EGFR-TKI resistance, and PD-1/PD-L1 expression has recently been revealed in NSCLC. In NSCLC patients who received gefitinib as their first-line treatment, the PD-L1 expression in tumor cells was markedly increased after gefitinib treatment [136]. In addition, the induction of PD-L1 expression by acquired EGFR-TKI resistance contributes to the immune escape of NSCLC cells [137]. These findings suggest that the mechanisms contributing to EGFR-TKI resistance may also trigger PD-L1 expression and promote the immune escape of tumor cells. It has been shown that the activation of MERTK-mediated signaling pathways through binding to Gas6 or ectopic expression of a constitutively activated MERTK significantly induces PD-L1 mRNA and protein expression [138]. In contrast, the reduction of MERTK and its downstream signaling pathway by shRNA-mediated knockdown, treatment of the MERTK inhibitor, BMS-777607, or the PI3K inhibitor decreases PD-L1 expression. In addition to the regulation of PD-L1 expression in cancer cells, the expression of PD-L1 in monocytes and macrophages was downregulated in MERTK-deficient mice or in MERTK-wild-type mice which were treated with an MERTK inhibitor called MRX-2843 [139]. Although MERTK is not dominantly expressed in T cells, the inhibition of MERTK indirectly upregulates PD-1 expression in CD4+ and CD8+ T cells, leading to T cell activation [139]. These findings suggest that targeting MERTK may enhance the therapeutic effect of immune checkpoint inhibitors. So far, there has been no study or clinical trial evaluating the therapeutic effect of the combination of MERTK inhibitors and anti-PD-1/PD-L1 agents in NSCLC, especially in patients with TKI-sensitive EGFR mutations. However, in a recent animal study of triple-negative breast cancer, the combination of the MERTK inhibitor BMS-777607 and an anti-PD-1 monoclonal antibody synergistically decreased tumor growth and the incidence of lung metastasis [140]. Since the excessive activation of MERTK-mediated signaling pathways is involved in EGFR-TKI resistance in NSCLC, the combination of MERTK inhibitors and anti-PD-1/PD-L1 agents may shed light on overcoming EGFR-TKI resistance in NSCLC.
The Regulation of MERTK by PD-L1-Mediated EGFR-TKI Resistance
In addition to its immune-suppressive role in cancer progression, the direct function of PD-L1 in tumor cells has been neglected thus far. A cohort study showed that high PD-L1 expression (>50% of immunohistochemistry reactivity) in treatment-naïve tumors predicts a poor EGFR-TKI response and prognosis of NSCLC patients with TKI-sensitive mutations [141], suggesting that PD-L1 may contribute to EGFR-TKI resistance in NSCLC. Although the PD-L1 protein lacks enzyme activity, the intracellular domain of PD-L1 contains three evolutionarily conserved motifs, known as the RMLDVEKC, DTSSK, and QFEET motifs, which can interact with some signaling molecules that are involved in the activation of pro-survival signaling pathways or the inhibition of some pro-apoptotic signaling pathways [142]. One study showed that the overexpression of PD-L1 led to YAP-mediated EGFR-TKI resistance in NSCLC cells, although the detailed molecular mechanism is not clear [143]. In addition, the ectopic expression of PD-L1 promotes TGF-β-mediated EMT and EGFR-TKI resistance in NSCLC cells through upregulating Smad3 phosphorylation [144]. Interestingly, PD-L1 may translocate into the nucleus and interact with Sp1, which promotes Gas6 expression and secretion, leading to activation of the MERTK signaling pathway and the proliferation of NSCLC cells [145]. Although direct evidence of the PD-L1-regulated MERTK signaling pathway is lacking, a study of the PD-L1 signalosome showed that the intracellular domain of PD-L1 may interact with molecules that are involved in Akt/mTOR, the DNA damage response, and the anti-apoptosis pathway [142,146]. Since the activation of MERTK is one of the EGFR-TKI-resistant mechanisms, especially to the non-T790M EGFR mutation, the PD-L1-induced MERTK signaling pathway may explain the PD-L1-mediated EGFR-TKI resistance.
4.3.2. The Role of MERTK in Efferocytosis
Phagocytosis is a cellular process for removing infectious microbes, foreign substances, and apoptotic cells by macrophages. In cancer, monocytes and macrophages are recruited to the tumor microenvironment, through a process guided by the tumor or stromal cell-derived chemokines (Figure 7). The infiltrated monocytes differentiate into macrophages and acquire a specific tumor-associated macrophage phenotype. Most of the tumor-associated macrophages resemble M2-like macrophages. Unlike M1-like macrophages with pro-inflammatory and anti-cancer activities, the M2-like macrophages function as immunosuppressive cells contributing to tumor initiation, angiogenesis, drug resistance, and metastasis [147,148]. The M2 macrophages help the immunosuppressive microenvironment by releasing anti-inflammatory cytokines, such as IL-20, TGF-β, and HGF [149], or by preventing the release of intracellular antigens from apoptotic cancer cells via efferocytosis [150].
MERTK is involved in the innate immune response by regulating the efferocytosis of macrophages. Although MERTK is not expressed in circulating monocytes, it is upregulated during the transformation of monocytes into tumor-associated macrophages, especially M2 macrophages [151]. The PtdSer, which is exposed on the apoptotic cancer cells, binds to PROS and Gas6 and transmits an “eat me” signal to the macrophages, which express abundant MERTK [13]. The upregulation of MERTK in M2 macrophages enhances their efferocytosis [152]. As such, the activation of MERTK increased the production of anti-inflammatory cytokines [151]. It is worth noting that Gas6 is produced and released from cancer-associated fibroblasts and M2 macrophages, suggesting a positive feedback loop that drastically reinforces the immunosuppressive environment in tumors [153].
Since M2 macrophages contribute to maintenance of the tumor immunosuppressive microenvironment that promotes drug resistance and immune escape through activation of the MERTK signaling pathway, targeting MERTK has become a potential therapeutic strategy for fighting NSCLC. It has been shown that the phagocytosis of Mertk knockout (Mertk−/−) macrophages was reduced and the cells failed to engulf apoptotic cells [70]. The MERTK-knockout-mediated inhibition of phagocytosis may be due to the disruption of cytoskeleton organization [154].
5. Development of Biological Agents and Small Molecules Targeting MERTK
TAM-mediated signaling pathways are responsible for the regulation of multiple biological processes, including survival, proliferation, cell mobility, and immunosuppression. The three members of the TAM family play important roles in the onset and progression of many diseases. Much evidence strongly suggests that these receptors play major roles in drug resistance to conventional chemotherapy and targeted therapies in NSCLC. Because MERTK and EGFR share similar downstream signaling pathways, targeting MERTK is a potential strategy for overcoming EGFR-TKI resistance in NSCLC.
The biological activities of MERTK can be mediated by its extracellular domain, which is required for the immune regulatory function and its intracellular domain, harboring kinase activity. Accordingly, anti-MERTK agents can be categorized as biological agents against the extracellular domain and small molecules targeting its intracellular kinase activity (Table 1). It has been shown that the treatment of a polyclonal anti-MERTK antibody (ab70693; Abcam, Cambridge, MA, USA) or homemade soluble MERTK ectodomain reduced the infection of classical swine fever virus and promoted the innate immune response [155]. A metastatic mouse model of lung adenocarcinoma showed that the combination of the anti-MERTK monoclonal antibody (4E9.E6; Bristol Myers Squibb, New York, NY) and anti-PD-1 monoclonal antibody (4H2; Bristol Myers Squibb) sensitized the tumors to radiotherapy by increasing the retention of CD8+CD103+ tissue-resident memory T cells in the tumors [156]. In addition, a preclinical study showed that Mer590, which is a monoclonal antibody, inhibits the MERTK-mediated signaling pathway, reduces colony formation, and sensitizes NSCLC cells to chemotherapy [95]. A homemade MERTK monoclonal antibody, which was derived by the extracellular domain of MERTK, significantly inhibited glioblastoma multiform migration [157]. In addition to the oncogenic roles of MERTK in cancer cells, MERTK expressed in stromal cells contributes to the immunosuppressive microenvironment. The inhibition of MERTK by the monoclonal antibody AF591 (R&D Systems, Inc., Minneapolis, MN, USA) blocks the apoptotic cell-induced inhibition of dendritic cells [158]. Interestingly, treatment of the MERTK monoclonal antibody has a limited effect on the bacteria-induced inflammatory response in macrophages, suggesting that MERTK plays a unique fundamental role in the tumor microenvironment, in addition to its role in systematic inflammation [159]. It is worth noting that although the three members of TAM receptors share the same downstream signaling pathways, the antibodies targeting MERTK, Axl, and Tyro3 may have different therapeutic efficacies or biological effects [160,161,162].
Interestingly, antibody-mediated MERTK activation has also recently been reported [160]. In contrast to Gas6 that can activate all three TAM receptors, the MERTK-activating antibody (AF591) and the Axl-activating antibody (AF854; R&D Systems) have absolute receptor specificity. An intravenous injection of the Axl-activating antibody strongly activates Axl receptors, leading to cleavage of the Axl extracellular domain from the cell membrane and the loss of steady-state Axl as a consequence in the spleen and lung, while the MERTK-activating antibody intensively activates the MERTK receptor, but is not associated with cleavage or loss of the MERTK protein, in the liver and lung of mice. However, the antibody alone is not able to induce apoptotic cell phagocytosis. In contrast, the antibody inhibits Gas6-mediated phagocytosis by competing with Gas6 for receptor binding [160]. In a mouse model of autoimmune encephalomyelitis, the Axl-activating antibody alone, but not the MERTK-activating antibody alone, reduced the number of inflammatory macrophages/microglia and the extent of demyelination, while the production of proinflammatory cytokines was not changed after the antibody treatment [161].
The upregulation of MERTK can be observed in recurrent tumors of cancer patients who received target therapy or chemotherapy as their first-line treatment. In colorectal cancer, the upregulation of MERTK has been considered as a predictive marker for resistance towards MEK1/2 inhibitors in a large clinical cohort [84]. The overexpression of MERTK was observed in melanoma cells with the BRAF V600E mutation, which are resistant to BRAF and MEK inhibitors [85]. In NSCLC, the excessive activation of Axl-mediated signaling pathways is associated with drug resistance to EGFR-targeted therapy [86,87,88]. A study using an in vitro cell culture showed that the overexpression of MERTK induced resistance to erlotinib in the PC9 cell line, which is an EGFR-TKI-sensitive cell line [89]. So far, clinical studies that form the basis of the different rationales for these combinations have been launched (Table 2).
Due to the conserved sequences of the kinase domain, many compounds not only inhibit their primary RTK targets, but also exhibit biochemically inhibitory activity against TAM receptors. Several inhibitors were designed according to the molecular structure of MERTK, such as UNC569, UNC1062, UNC2025, and MRX-2843. Among these, UNC2025 induced the apoptosis of NSCLC cell lines carrying the EGFR T790M mutation, leading to EGFR-TKI resistance [93]. The application of the MERTK-specific inhibitor UNC569 sensitized EGFR-TKI-insensitive H1975 cells, which carry the EGFR T790M mutation, to the erlotinib treatment [93]. In addition, UNC1666 and UNC2250 are pyrrolopyrimidine- and pyridinepyrimidine-based MERTK inhibitors that inhibit the colony formation of NSCLC cells. Some other structurally-distinct MERTK inhibitors have been developed, such as CT413, RXDX-106, SGI-7079, UNC2541, and UNC3133. However, the therapeutic effects of these synthetic compounds have not been evaluated in a preclinical study of NSCLC.
The prominent expression and functions of TAM receptors across the immune system also suggest their potential role in mediating resistance to immune checkpoint inhibitors in cancer therapy. The activation of MERTK in macrophages promotes the phagocytosis of apoptotic tumor cells, which contributes to a macrophage-mediated immunosuppressive tumor environment [180]. The genetic depletion of MERTK or inhibition of MERTK by UNC2025 reduced the number of macrophages in tumors and sensitized the response of tumor cells to radiotherapy [156]. UNC4241and MRX-2843 alter the immune checkpoint pathway and increase T cell activation in a mouse model [181]. Furthermore, JNJ-28312141 and UNC2025 modulate inflammation and the transformation of macrophages [182]. Although the developed MERTK inhibitors display convincing therapeutic activities, there is no MERTK-targeting compound that has moved to a phase III clinical trial. In addition, the tumors of NSCLC exhibit heterogeneous expression patterns of MERTK [94]. Accordingly, the side effects of MERTK inhibitors in clinical application must be considered, due to the complicated crosstalk between MERTK and other RTK-mediated signaling pathways.
6. Conclusions
In the past decade, the contribution of the ligand-dependent canonical RTK-signaling pathway of MERTK in drug resistance has been extensively explored. Importantly, the activation of MERTK signaling pathways depends on the context in which the ligands and microenvironment are presented to the TAM extracellular domains. The complexity of ligand–receptor interactions reflects the difficulty of drug development for overcoming EGFR-TKI resistance in NSCLC. Recently, the combination of MERTK inhibitors and other therapeutic agents, such as immune checkpoint inhibitors, TKIs, and chemotherapy agents, has become the most promising strategy for fighting NSCLC. Although clinical studies have shown that anti-MERTK therapies potentially prolong the survival of NSCLC patients, the effectiveness of these compounds is not satisfactory. Future study may focus on the therapeutic efficacy among NSCLC patients with different genetic alterations of MERTK and with different EGFR-TKI-resistant mechanisms.
Author Contributions
Conceptualization, C.-J.C. and Y.-P.L.; writing—original draft preparation, C.-J.C. and Y.-P.L.; writing—review and editing, Y.-P.L.; supervision, Y.-P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by the Research Center for Environmental Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan and by Kaohsiung Medical University Research Center Grant (KMU-TC108A01) and Kaohsiung Medical University Hospital (KMUH108-8R76).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: http://seer.cancer.gov/csr/1975_2012/ (accessed on 31 January 2021).
- Ghafoor, Q.; Baijal, S.; Taniere, P.; O’Sullivan, B.; Evans, M.; Middleton, G. Epidermal Growth Factor Receptor (EGFR) Kinase Inhibitors and Non-Small Cell Lung Cancer (NSCLC)—Advances in Molecular Diagnostic Techniques to Facilitate Targeted Therapy. Pathol. Oncol. Res. 2017, 24, 723–731. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Maione, P.; Sacco, P.C.; Sgambato, A.; Casaluce, F.; Rossi, A.; Gridelli, C. Overcoming resistance to targeted therapies in NSCLC: Current approaches and clinical application. Ther. Adv. Med. Oncol. 2015, 7, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Arcila, M.E.; Chmielecki, J.; Ladanyi, M.; Miller, V.A.; Pao, W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011, 17, 5530–5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632. [Google Scholar] [CrossRef]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, S.; Liu, W.; Yang, Y.; Zhang, L.; Zhang, S.; Jiang, Z.; Yang, Z.; Zhu, X. Targeted Next-Generation Sequencing Reveals a Novel Frameshift Mutation in the MERTK Gene in a Chinese Family with Retinitis Pigmentosa. Genet. Test. Mol. Biomark. 2018, 22, 165–169. [Google Scholar] [CrossRef]
- Kim, W.J.; Park, H.J.; Choi, Y.J.; Kwon, E.Y.; Kim, B.M.; Lee, J.H.; Chang, J.H.; Lee Kang, J.; Choi, J.H. Association between Genetic Variations of MERTK and Chronic Obstructive Pulmonary Disease in Koreans. J. Korean Med. Sci. 2018, 33, e56. [Google Scholar] [CrossRef]
- Branchford, B.R.; Stalker, T.J.; Law, L.; Acevedo, G.; Sather, S.; Brzezinski, C.; Wilson, K.M.; Minson, K.; Lee-Sherick, A.B.; Davizon-Castillo, P.; et al. The small-molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J. Thromb. Haemost. 2018, 16, 352–363. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; De Michieli, F.; Paschetta, E.; Pinach, S.; Saba, F.; Bongiovanni, D.; Framarin, L.; Berrutti, M.; Leone, N.; et al. MERTK rs4374383 variant predicts incident nonalcoholic fatty liver disease and diabetes: Role of mononuclear cell activation and adipokine response to dietary fat. Hum. Mol. Genet. 2017, 26, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, C.T.; Deryckere, D.; Earp, H.S.; Graham, D.K. Molecular pathways: MERTK signaling in cancer. Clin. Cancer Res. 2013, 19, 5275–5280. [Google Scholar] [CrossRef] [Green Version]
- Png, K.J.; Halberg, N.; Yoshida, M.; Tavazoie, S.F. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 2011, 481, 190–194. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Drake, J.M.; Clark, P.M.; Smith, B.A.; Zong, Y.; Volpe, C.; Mathis, C.; Morrissey, C.; Castor, B.; Huang, J.; et al. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E172–E181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vouri, M.; Hafizi, S. TAM Receptor Tyrosine Kinases in Cancer Drug Resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Warner, S.L.; Vankayalapati, H.; Bearss, D.J.; Sharma, S. Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 2011, 10, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komurov, K.; Padron, D.; Cheng, T.; Roth, M.; Rosenblatt, K.P.; White, M.A. Comprehensive mapping of the human kinome to epidermal growth factor receptor signaling. J. Biol. Chem. 2010, 285, 21134–21142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Kondo, K.; Yamaguchi, M.; Richmond, G.; Hutchison, M.; Wakeling, A.; Averbuch, S.; Wadsworth, P. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. 2003, 23, 3639–3650. [Google Scholar] [PubMed]
- Wang, K.; Yamamoto, H.; Chin, J.R.; Werb, Z.; Vu, T.H. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J. Biol. Chem. 2004, 279, 53848–53856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.C.; Threadgill, D.W. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 2009, 47, 85–92. [Google Scholar] [CrossRef]
- Schreier, B.; Rabe, S.; Schneider, B.; Bretschneider, M.; Rupp, S.; Ruhs, S.; Neumann, J.; Rueckschloss, U.; Sibilia, M.; Gotthardt, M.; et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 2013, 61, 333–340. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef]
- Ghatge, M.; Nair, J.; Sharma, A.; Vangala, R.K. Integrative gene ontology and network analysis of coronary artery disease associated genes suggests potential role of ErbB pathway gene EGFR. Mol. Med. Rep. 2018, 17, 4253–4264. [Google Scholar] [CrossRef]
- Spindler, V.; Waschke, J. Pemphigus—A Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front. Immunol. 2018, 9, 136. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, T.; Wang, T.; Hao, N.; Shen, Y.; Wu, Y.; Yuan, Z.; Chen, L.; Wen, F. Phloretin attenuates mucus hypersecretion and airway inflammation induced by cigarette smoke. Int. Immunopharmacol. 2018, 55, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef] [Green Version]
- Mitsudomi, T.; Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. 2017, 28, 687–694. [Google Scholar]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. EGF Recept. Fam. 2003, 284, 2–13. [Google Scholar]
- Begley, M.J.; Yun, C.H.; Gewinner, C.A.; Asara, J.M.; Johnson, J.L.; Coyle, A.J.; Eck, M.J.; Apostolou, I.; Cantley, L.C. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nat. Struct. Mol. Biol. 2015, 22, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Rappoport, J.Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int. J. Biochem. Cell Biol. 2014, 51, 23–28. [Google Scholar] [CrossRef]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell. Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef]
- Komotar, R.J.; Starke, R.M.; Connolly, E.S.; Sisti, M.B. Alteration in NFKBIA and EGFR in glioblastoma multiforme. Neurosurgery 2011, 68, N14–N15. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Yang, X.X.; Liu, M.F.; Yao, G.Y.; Dong, J.Y.; Ye, C.S.; Li, M. Mutational analysis of key EGFR pathway genes in Chinese breast cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 5599–5603. [Google Scholar] [CrossRef] [Green Version]
- Lv, N.; Xie, X.; Ge, Q.; Lin, S.; Wang, X.; Kong, Y.; Shi, H.; Xie, X.; Wei, W. Epidermal growth factor receptor in breast carcinoma: Association between gene copy number and mutations. Diagn. Pathol. 2011, 6, 118. [Google Scholar] [CrossRef] [Green Version]
- Metro, G.; Crino, L. Advances on EGFR mutation for lung cancer. Transl. Lung Cancer Res. 2012, 1, 5–13. [Google Scholar]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Tosoian, J.J.; Mamawala, M.; Epstein, J.I.; Landis, P.; Wolf, S.; Trock, B.J.; Carter, H.B. Intermediate and longer-term outcomes from a prospective active-surveillance program for favorable-risk prostate cancer. J. Clin. Oncol. 2015, 33, 3379–3385. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Janne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, C.; Gasper, H.; Sahin, K.B.; Tang, M.; Kulasinghe, A.; Adams, M.N.; Richard, D.J.; O’Byrne, K.J. Epidermal Growth Factor Receptor (EGFR)-Mutated Non-Small-Cell Lung Cancer (NSCLC). Pharmaceuticals 2020, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.; Franchina, T.; Ricciardi, G.R.R.; Smiroldo, V.; Picciotto, M.; Zanghi, M.; Rolfo, C.; Adamo, V. Third generation EGFR TKIs in EGFR-mutated NSCLC: Where are we now and where are we going. Crit. Rev. Oncol. Hematol. 2017, 117, 38–47. [Google Scholar] [CrossRef]
- Planchard, D. Adjuvant Osimertinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1780–1782. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Mok, T.S.; Han, J.Y.; Ahn, M.J.; Delmonte, A.; Ramalingam, S.S.; Kim, S.W.; Shepherd, F.A.; Laskin, J.; He, Y.; et al. Osimertinib versus platinum-pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann. Oncol. 2020, 31, 1536–1544. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2020, 40, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kwok, H.F. Current Strategies for Treating NSCLC: From Biological Mechanisms to Clinical Treatment. Cancers 2020, 12, 1587. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Chen, X.; Zhou, F. EGFR-mutant NSCLC: Emerging novel drugs. Curr. Opin. Oncol. 2021, 33, 87–94. [Google Scholar] [CrossRef]
- Chen, J.A.; Riess, J.W. Advances in targeting acquired resistance mechanisms to epidermal growth factor receptor tyrosine kinase inhibitors. J. Thorac. Dis. 2020, 12, 2859–2876. [Google Scholar] [CrossRef]
- Shah, R.; Lester, J.F. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non-Small-Cell Lung Cancer: A Clash of the Generations. Clin. Lung Cancer 2020, 21, e216–e228. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Lai, C.; Lemke, G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron 1991, 6, 691–704. [Google Scholar] [CrossRef]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Nguyen, K.Q.; Tsou, W.I.; Kotenko, S.; Birge, R.B. TAM receptors in apoptotic cell clearance, autoimmunity, and cancer. Autoimmunity 2013, 46, 294–297. [Google Scholar] [CrossRef]
- Van der Meer, J.H.; van der Poll, T.; van’t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Leighton, J.A.; Ghosh, S. Tyro3, Axl, and Mertk receptor signaling in inflammatory bowel disease and colitis-associated cancer. Inflamm. Bowel Dis. 2014, 20, 1472–1480. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.Q.; Tsou, W.I.; Kotenko, S.; Birge, R.B. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar]
- Ji, R.; Meng, L.; Li, Q.; Lu, Q. TAM receptor deficiency affects adult hippocampal neurogenesis. Metab. Brain Dis. 2015, 30, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Hanayama, R.; Kawane, K. Autoimmunity and the clearance of dead cells. Cell 2010, 140, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A.K.; Salzberg, D.B.; Sather, S.; Liang, X.; Nickoloff, S.; Anwar, A.; Deryckere, D.; Hill, K.; Joung, D.; Sawczyn, K.K.; et al. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene 2006, 25, 6092–6100. [Google Scholar] [CrossRef] [Green Version]
- Brandao, L.N.; Winges, A.; Christoph, S.; Sather, S.; Migdall-Wilson, J.; Schlegel, J.; McGranahan, A.; Gao, D.; Liang, X.; Deryckere, D.; et al. Inhibition of MerTK increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J. 2013, 3, e101. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, J.; Sambade, M.J.; Sather, S.; Moschos, S.J.; Tan, A.C.; Winges, A.; DeRyckere, D.; Carson, C.C.; Trembath, D.G.; Tentler, J.J.; et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Invest. 2013, 123, 2257–2267. [Google Scholar] [CrossRef] [Green Version]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnevi, E.; Andersson, R.; Rosendahl, A.H. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 2014, 92, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Burbridge, M. Key Roles of Axl and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [Green Version]
- Frejno, M.; Zenezini Chiozzi, R.; Wilhelm, M.; Koch, H.; Zheng, R.; Klaeger, S.; Ruprecht, B.; Meng, C.; Kramer, K.; Jarzab, A.; et al. Pharmacoproteomic characterisation of human colon and rectal cancer. Mol. Syst. Biol. 2017, 13, 951. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Kohler, R.; Tang, F.; Hynx, D.; Wang, Y.; Orso, F.; Pretre, V.; Ritschard, R.; Hirschmann, P.; Cron, P.; et al. mTORC1/autophagy-regulated MerTK in mutant BRAFV600 melanoma with acquired resistance to BRAF inhibition. Oncotarget 2017, 8, 69204–69218. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the Axl kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Strategies to Overcome Bypass Mechanisms Mediating Clinical Resistance to EGFR Tyrosine Kinase Inhibition in Lung Cancer. Mol. Cancer Ther. 2017, 16, 265–272. [Google Scholar]
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219. [Google Scholar] [CrossRef] [Green Version]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, C.T.; Linger, R.M.; Cohen, R.A.; Sather, S.; Kirkpatrick, G.D.; Davies, K.D.; DeRyckere, D.; Earp, H.S.; Graham, D.K. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget 2014, 5, 10434–10445. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, Tyro3, Axl, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the Axl receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2017, 31, 1382–1397. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14, 1073–1090. [Google Scholar] [CrossRef]
- Fraunhoffer, N.A.; Abuelafia, A.M.; Bigonnet, M.; Gayet, O.; Roques, J.; Telle, E.; Santofimia-Castano, P.; Borrello, M.T.; Chuluyan, E.; Dusetti, N.; et al. Evidencing a Pancreatic Ductal Adenocarcinoma Subpopulation Sensitive to the Proteasome Inhibitor Carfilzomib. Clin. Cancer Res. 2020, 26, 5506–5519. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, X.; Ren, S.; Su, C.; Li, C.; Li, W.; Yu, J.; Cheng, N.; Zhou, C. HOTAIR induces EGFR-TKIs resistance in non-small cell lung cancer through epithelial-mesenchymal transition. Lung Cancer 2020, 147, 99–105. [Google Scholar] [CrossRef]
- Tian, H.; Lian, R.; Li, Y.; Liu, C.; Liang, S.; Li, W.; Tao, T.; Wu, X.; Ye, Y.; Yang, X.; et al. Akt-induced lncRNA VAL promotes EMT-independent metastasis through diminishing Trim16-dependent Vimentin degradation. Nat. Commun. 2020, 11, 5127. [Google Scholar] [CrossRef]
- Lee, A.F.; Chen, M.C.; Chen, C.J.; Yang, C.J.; Huang, M.S.; Liu, Y.P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e0180383. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, Q.; Zhang, X.; Zheng, J.; Xie, B.; Xu, Z.; Zhang, W. Sensitivity to chemotherapeutics of NSCLC cells with acquired resistance to EGFR-TKIs is mediated by T790M mutation or epithelial-mesenchymal transition. Oncol. Rep. 2018, 39, 1783–1792. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Sun, L.; Liu, T.; Zhu, J.; Zeng, Y.; Zhang, Y.; Wang, X.; Liu, Z.; Huang, J.A. The miR6253p/Axl axis induces nonT790M acquired resistance to EGFRTKI via activation of the TGF-β/Smad pathway and EMT in EGFRmutant nonsmall cell lung cancer. Oncol. Rep. 2020, 44, 185–195. [Google Scholar]
- Jung, J.; Lee, Y.J.; Choi, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Gas6 Prevents Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells via Production of PGE2, PGD2 and Their Receptors. Cells 2019, 8, 643. [Google Scholar] [CrossRef] [Green Version]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bonisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef]
- Keller, M.; Dubois, F.; Teulier, S.; Martin, A.P.J.; Levallet, J.; Maille, E.; Brosseau, S.; Elie, N.; Hergovich, A.; Bergot, E.; et al. NDR2 kinase contributes to cell invasion and cytokinesis defects induced by the inactivation of RASSF1A tumor-suppressor gene in lung cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L.; et al. A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci. Transl. Med. 2020, 12, eaaz4589. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.F.; Tseng, Y.C.; Nguyen, P.A.; Li, Y.C.; Ho, C.C.; Wu, C.W. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef]
- Xia, H.; Dai, X.; Yu, H.; Zhou, S.; Fan, Z.; Wei, G.; Tang, Q.; Gong, Q.; Bi, F. EGFR- PI3K -PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: The mechanism and its implications in targeted therapy. Cell Death Dis. 2018, 9, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhu, G.Y.; Cao, D.; Pan, H.; Li, Y.W. Gossypol overcomes EGFR-TKIs resistance in non-small cell lung cancer cells by targeting YAP/TAZ and EGFR(L858R/T790M). Biomed. Pharmacother. 2019, 115, 108860. [Google Scholar] [CrossRef]
- Kaowinn, S.; Yawut, N.; Koh, S.S.; Chung, Y.H. Cancer upregulated gene (CUG)2 elevates YAP1 expression, leading to enhancement of epithelial-mesenchymal transition in human lung cancer cells. Biochem. Biophys. Res. Commun. 2019, 511, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Nouri, K.; Janse van Rensburg, H.J.; Maritan, S.M.; Wu, L.; Hao, Y.; Montminy, T.; Yu, J.; Khanal, P.; Mulligan, L.M.; et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020, 39, 334–355. [Google Scholar] [CrossRef] [PubMed]
- Saab, S.; Chang, O.S.; Nagaoka, K.; Hung, M.C.; Yamaguchi, H. The potential role of YAP in Axl-mediated resistance to EGFR tyrosine kinase inhibitors. Am. J. Cancer Res. 2019, 9, 2719–2729. [Google Scholar] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent Axl Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Lv, H.; Zhong, D.S.; Zhou, Q.H. EGFR-TKI resistance and MAP17 are associated with cancer stem cell like properties. Oncol. Lett. 2018, 15, 6655–6665. [Google Scholar] [CrossRef]
- Hu, F.; Li, C.; Zheng, X.; Zhang, H.; Shen, Y.; Zhou, L.; Yang, X.; Han, B.; Zhang, X. Lung adenocarcinoma resistance to therapy with EGFRtyrosine kinase inhibitors is related to increased expression of cancer stem cell markers SOX2, OCT4 and NANOG. Oncol. Rep. 2020, 43, 727–735. [Google Scholar]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef] [Green Version]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Togashi, Y.; Hayashi, H.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Nakagawa, K.; Nishio, K. Inhibition of beta-Catenin enhances the anticancer effect of irreversible EGFR-TKI in EGFR-mutated non-small-cell lung cancer with a T790M mutation. J. Thorac. Oncol. 2015, 10, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, G.; Liu, Y.; Ma, M.; Song, K.; Li, H.; Zhu, D.; Tang, X.; Kong, J.; Yuan, X. Targeting histone deacetylase SIRT1 selectively eradicates EGFR TKI-resistant cancer stem cells via regulation of mitochondrial oxidative phosphorylation in lung adenocarcinoma. Neoplasia 2020, 22, 33–46. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, Y.; Yan, A.; Wang, M.; Han, Q.; Wang, K.; Wang, J.; Qiao, C.; Pan, Z.; Chen, C.; et al. 1,25-dihydroxyvitamin D3 signaling-induced decreases in IRX4 inhibits NANOG-mediated cancer stem-like properties and gefitinib resistance in NSCLC cells. Cell Death Dis. 2020, 11, 670. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef]
- Chang, Y.F.; Lim, K.H.; Chiang, Y.W.; Sie, Z.L.; Chang, J.; Ho, A.S.; Cheng, C.C. STAT3 induces G9a to exacerbate HER3 expression for the survival of epidermal growth factor receptor-tyrosine kinase inhibitors in lung cancers. BMC Cancer 2019, 19, 959. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Ma, Y.; Bi, J.W.; Xiong, Y.; Lv, C.; Li, S.; Wu, N.; Yang, Y. Shisa3 brakes resistance to EGFR-TKIs in lung adenocarcinoma by suppressing cancer stem cell properties. J. Exp. Clin. Cancer Res. 2019, 38, 481. [Google Scholar] [CrossRef]
- Wang, Y.; Moncayo, G.; Morin, P., Jr.; Xue, G.; Grzmil, M.; Lino, M.M.; Clement-Schatlo, V.; Frank, S.; Merlo, A.; Hemmings, B.A. Mer receptor tyrosine kinase promotes invasion and survival in glioblastoma multiforme. Oncogene 2013, 32, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, H.; Eom, H.; Kaushik, N.; Yoo, K.C.; Shim, J.K.; Kwon, M.; Choi, M.Y.; Yoon, T.; Kang, S.G.; Lee, S.J. MerTK mediates STAT3-kRas/Src-signaling axis for glioma stem cell maintenance. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 2), 87–95. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Decker, A.M.; Wang, J.; Lee, E.; Kana, L.A.; Yumoto, K.; Cackowski, F.C.; Rhee, J.; Carmeliet, P.; Buttitta, L.; et al. Endogenous Gas6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 2016, 7, 25698–25711. [Google Scholar] [CrossRef] [Green Version]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the Akt-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Minchom, A.; Thavasu, P.; Ahmad, Z.; Stewart, A.; Georgiou, A.; O’Brien, M.E.R.; Popat, S.; Bhosle, J.; Yap, T.A.; de Bono, J.; et al. A study of PD-L1 expression in kRas mutant non-small cell lung cancer cell lines exposed to relevant targeted treatments. PLoS ONE 2017, 12, e0186106. [Google Scholar] [CrossRef] [Green Version]
- Han, J.J.; Kim, D.W.; Koh, J.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Change in PD-L1 Expression After Acquiring Resistance to Gefitinib in EGFR-Mutant Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2016, 17, 263–270.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Wang, R.; Zhang, X.; Ma, Y.; Zhong, L.; Li, K.; Nishiyama, A.; Arai, S.; Yano, S.; Wang, W. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol. Cancer 2019, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates Akt-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.Y.; Liao, W.Y.; Ho, C.C.; Chen, K.Y.; Tsai, T.H.; Hsu, C.L.; Su, K.Y.; Chang, Y.L.; Wu, C.T.; Hsu, C.C.; et al. Association between programmed death-ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur. J. Cancer 2020, 124, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Tung, J.N.; Lin, P.L.; Wang, Y.C.; Wu, D.W.; Chen, C.Y.; Lee, H. PD-L1 confers resistance to EGFR mutation-independent tyrosine kinase inhibitors in non-small cell lung cancer via upregulation of YAP1 expression. Oncotarget 2018, 9, 4637–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zeng, Y.; Liu, T.; Du, W.; Zhu, J.; Liu, Z.; Huang, J.A. The canonical TGF-β/Smad signalling pathway is involved in PD-L1-induced primary resistance to EGFR-TKIs in EGFR-mutant non-small-cell lung cancer. Respir. Res. 2019, 20, 164. [Google Scholar] [CrossRef]
- Du, W.; Zhu, J.; Zeng, Y.; Liu, T.; Zhang, Y.; Cai, T.; Fu, Y.; Zhang, W.; Zhang, R.; Liu, Z.; et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Liu, N.; Ma, X.; Qin, J.; Liu, Y.; Wang, X. M2 macrophages reduce the effect of gefitinib by activating Akt/mTOR in gefitinib-resistant cell lines HCC827/GR. Thorac. Cancer 2020, 11, 3289–3298. [Google Scholar] [CrossRef]
- Hwang, I.; Kim, J.W.; Ylaya, K.; Chung, E.J.; Kitano, H.; Perry, C.; Hanaoka, J.; Fukuoka, J.; Chung, J.Y.; Hewitt, S.M. Tumor-associated macrophage, angiogenesis and lymphangiogenesis markers predict prognosis of non-small cell lung cancer patients. J. Transl. Med. 2020, 18, 443. [Google Scholar] [CrossRef]
- Trus, E.; Basta, S.; Gee, K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: Competing factors in macrophage polarization. Cytokine 2020, 127, 154939. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Reinoso, V.; Baek, D.Y.; Kurutz, A.; Rubin, J.R.; Koh, A.J.; McCauley, L.K.; Roca, H. Unique Pro-Inflammatory Response of Macrophages during Apoptotic Cancer Cell Clearance. Cells 2020, 9, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroud, P.; Renaudineau, S.; Gudefin, L.; Calcei, A.; Menguy, T.; Rozan, C.; Mizrahi, J.; Caux, C.; Duong, V.; Valladeau-Guilemond, J. Expression of TAM-R in Human Immune Cells and Unique Regulatory Function of MerTK in IL-10 Production by Tolerogenic DC. Front. Immunol. 2020, 11, 564133. [Google Scholar] [CrossRef]
- Lai, Y.S.; Putra, R.; Aui, S.P.; Chang, K.T. M2C Polarization by Baicalin Enhances Efferocytosis via Upregulation of MERTK Receptor. Am. J. Chin. Med. 2018, 46, 1899–1914. [Google Scholar] [CrossRef]
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Yang, L.; et al. Interaction with CD68 and Regulation of Gas6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 3892–3905. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, S.; Liu, Q.; Xie, J.; Zhang, J.; Han, D.; Lu, Q.; Lu, Q. Mertk deficiency affects macrophage directional migration via disruption of cytoskeletal organization. PLoS ONE 2015, 10, e0117787. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Li, L.F.; Zhang, Y.; Qu, L.; Wang, W.; Li, M.; Yu, S.; Zhou, M.; Luo, Y.; Sun, Y.; et al. MERTK is a host factor that promotes classical swine fever virus entry and antagonizes innate immune response in PK-15 cells. Emerg. Microbes Infect. 2020, 9, 571–581. [Google Scholar] [CrossRef]
- Caetano, M.S.; Younes, A.I.; Barsoumian, H.B.; Quigley, M.; Menon, H.; Gao, C.; Spires, T.; Reilly, T.P.; Cadena, A.P.; Cushman, T.R.; et al. Triple Therapy with MerTK and PD1 Inhibition Plus Radiotherapy Promotes Abscopal Antitumor Immune Responses. Clin. Cancer Res. 2019, 25, 7576–7584. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.E.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2012, 31, 4171–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S., Jr.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, H.; Fang, L.; Ding, P.; Xu, K.; Yang, Q.; Liu, R. MerTK Does Not Mediate Phagocytosis of Staphylococcus aureus but Attenuates Inflammation Induced by Staphylococcal Lipoteichoic Acid Through Blocking NF-kappaB Activation. Inflammation 2017, 40, 1543–1552. [Google Scholar] [CrossRef]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuBois, J.C.; Ray, A.K.; Davies, P.; Shafit-Zagardo, B. Anti-Axl antibody treatment reduces the severity of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 324. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, A.; Wang, Y.; Chen, F.; Zhao, Z.; Davra, V.; Suzuki-Inoue, K.; Ozaki, Y.; Birge, R.B.; Lu, Q.; et al. Tyro3, Axl, and Mertk receptors differentially participate in platelet activation and thrombus formation. Cell Commun. Signal. 2018, 16, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, W.; Stashko, M.A.; Deryckere, D.; Cummings, C.T.; Hunter, D.; Yang, C.; Jayakody, C.N.; Cheng, N.; Simpson, C.; et al. UNC1062, a new and potent Mer inhibitor. Eur. J. Med. Chem. 2013, 65, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, D.; Stashko, M.A.; DeRyckere, D.; Hunter, D.; Kireev, D.; Miley, M.J.; Cummings, C.; Lee, M.; Norris-Drouin, J.; et al. Pseudo-cyclization through intramolecular hydrogen bond enables discovery of pyridine substituted pyrimidines as new Mer kinase inhibitors. J. Med. Chem. 2013, 56, 9683–9692. [Google Scholar] [CrossRef] [Green Version]
- Roohullah, A.; Cooper, A.; Lomax, A.J.; Aung, J.; Barge, A.; Chow, L.; McHale, M.; Desai, J.; Whittle, J.R.; Tran, B.; et al. A phase I trial to determine safety and pharmacokinetics of ASLAN002, an oral MET superfamily kinase inhibitor, in patients with advanced or metastatic solid cancers. Invest. New Drugs 2018, 36, 886–894. [Google Scholar] [CrossRef]
- Arkenau, H.T.; Plummer, R.; Molife, L.R.; Olmos, D.; Yap, T.A.; Squires, M.; Lewis, S.; Lock, V.; Yule, M.; Lyons, J.; et al. A phase I dose escalation study of AT9283, a small molecule inhibitor of aurora kinases, in patients with advanced solid malignancies. Ann. Oncol. 2012, 23, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Krishnamurthi, S.S.; Saleh, M.N.; Gitlitz, B.J.; Borad, M.J.; Gold, P.J.; Chiorean, E.G.; Springett, G.M.; Abbas, R.; Agarwal, S.; et al. Phase I study of bosutinib, a Src/Abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, J.A.; Hung, C.N.; DeArmond, D.T.; Chen, M.; Lin, C.L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.M.; Lucio, N.D.; Chou, C.W.; et al. Single-Cell Proteomic Profiling Identifies Combined Axl and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020, 80, 1551–1563. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Tsao, M.S.; Liu, G.; Tu, D.; Ho, C.; Shepherd, F.A.; Murray, N.; Goffin, J.R.; Nicholas, G.; Sakashita, S.; et al. A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND.196. Oncotarget 2017, 8, 69651–69662. [Google Scholar] [CrossRef] [Green Version]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mirzapoiazova, T.; Carol Tan, Y.H.; Pang, K.M.; Pozhitkov, A.; Wang, Y.; Wang, Y.; Mambetsariev, B.; Wang, E.; Nasser, M.W.; et al. Inhibiting crosstalk between MET signaling and mitochondrial dynamics and morphology: A novel therapeutic approach for lung cancer and mesothelioma. Cancer Biol. Ther. 2018, 19, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Padda, S.; Neal, J.W.; Wakelee, H.A. MET inhibitors in combination with other therapies in non-small cell lung cancer. Transl. Lung Cancer Res. 2012, 1, 238–253. [Google Scholar] [PubMed]
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S., 3rd; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancers Expressing Wild-type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, L.; Bahleda, R.; Tolaney, S.M.; Kwak, E.L.; Cleary, J.M.; Pandya, S.S.; Hollebecque, A.; Abbas, R.; Ananthakrishnan, R.; Berkenblit, A.; et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J. Clin. Oncol. 2014, 32, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Okura, N.; Nishioka, N.; Yamada, T.; Taniguchi, H.; Tanimura, K.; Katayama, Y.; Yoshimura, A.; Watanabe, S.; Kikuchi, T.; Shiotsu, S.; et al. ONO-7475, a Novel Axl Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2244–2256. [Google Scholar] [CrossRef]
- Clémenson, C.; Chargari, C.; Liu, W.; Mondini, M.; Ferté, C.; Burbridge, M.F.; Cattan, V.; Jacquet-Bescond, A.; Deutsch, E. The MET/Axl/FGFR Inhibitor S49076 Impairs Aurora B Activity and Improves the Antitumor Efficacy of Radiotherapy. Mol. Cancer Ther. 2017, 16, 2107–2119. [Google Scholar] [CrossRef] [Green Version]
- Kerkemeyer, K.L.S.; Lai, F.Y.X.; Mar, A. Lichen planus pemphigoides during therapy with tislelizumab and sitravatinib in a patient with metastatic lung cancer. Australas. J. Dermatol. 2020, 61, 180–182. [Google Scholar] [CrossRef]
- Fiedler, W.; Giaccone, G.; Lasch, P.; van der Horst, I.; Brega, N.; Courtney, R.; Abbattista, A.; Shalinsky, D.R.; Bokemeyer, C.; Boven, E. Phase I trial of SU14813 in patients with advanced solid malignancies. Ann. Oncol. 2011, 22, 195–201. [Google Scholar] [CrossRef]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Crittenden, M.R.; Baird, J.; Friedman, D.; Savage, T.; Uhde, L.; Alice, A.; Cottam, B.; Young, K.; Newell, P.; Nguyen, C.; et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 2016, 7, 78653–78666. [Google Scholar] [CrossRef] [Green Version]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef]
- Illig, C.R.; Manthey, C.L.; Wall, M.J.; Meegalla, S.K.; Chen, J.; Wilson, K.J.; Ballentine, S.K.; Desjarlais, R.L.; Schubert, C.; Crysler, C.S.; et al. Optimization of a potent class of arylamide colony-stimulating factor-1 receptor inhibitors leading to anti-inflammatory clinical candidate 4-cyano-N-[2-(1-cyclohexen-1-yl)-4-[1-[(dimethylamino)acetyl]-4-piperidinyl]pheny l]-1H-imidazole-2-carboxamide (JNJ-28312141). J. Med. Chem. 2011, 54, 7860–7883. [Google Scholar] [PubMed]
Figure 1.
Effects of the Mer receptor tyrosine kinase (MERTK)-mediated signaling pathway in epidermal growth factor tyrosine kinase inhibitors (EGFR-TKI) resistance. For EGFR-TKI resistance without the acquired T790M mutation in EGFR, the downstream signaling pathways of EGFR are still blocked by EGFR-TKIs. Alternative RTK-mediated signaling pathways (bypass) contribute to the survival of the EGFR-TKI-resistant cancer cells. Through binding to their ligands, MERTK-induced signaling pathways may crosstalk with other RTK-mediated signaling pathways that promote cell survival.
Figure 1.
Effects of the Mer receptor tyrosine kinase (MERTK)-mediated signaling pathway in epidermal growth factor tyrosine kinase inhibitors (EGFR-TKI) resistance. For EGFR-TKI resistance without the acquired T790M mutation in EGFR, the downstream signaling pathways of EGFR are still blocked by EGFR-TKIs. Alternative RTK-mediated signaling pathways (bypass) contribute to the survival of the EGFR-TKI-resistant cancer cells. Through binding to their ligands, MERTK-induced signaling pathways may crosstalk with other RTK-mediated signaling pathways that promote cell survival.

Figure 2.
The mechanisms of acquired EGFR-TKI resistance. (a) The illustration of EGFR structure. (b) The downstream signaling pathways of EGFR contribute to cancer progression. (c) The EGFR gene amplification, deficiency of protein degradation, acquired T790M mutation, and different EGFR constitution are the major causes of EGFR-TKI resistance.
Figure 2.
The mechanisms of acquired EGFR-TKI resistance. (a) The illustration of EGFR structure. (b) The downstream signaling pathways of EGFR contribute to cancer progression. (c) The EGFR gene amplification, deficiency of protein degradation, acquired T790M mutation, and different EGFR constitution are the major causes of EGFR-TKI resistance.

Figure 3.
The biological role of MERTK in cancer cells. (a) The illustration of MERTK structure. (b) A model depicts the interaction of Tyro3-Axl-MERTK (TAM) receptors with their ligands (Gas6 and PROS1). The thickness of the arrows reflects the activation strength of each ligand to the TAM receptors. (c) Binding of ligands to the TAM receptors activates the downstream signaling pathways involving in cancer progression and drug resistance.
Figure 3.
The biological role of MERTK in cancer cells. (a) The illustration of MERTK structure. (b) A model depicts the interaction of Tyro3-Axl-MERTK (TAM) receptors with their ligands (Gas6 and PROS1). The thickness of the arrows reflects the activation strength of each ligand to the TAM receptors. (c) Binding of ligands to the TAM receptors activates the downstream signaling pathways involving in cancer progression and drug resistance.

Figure 4.
The mechanisms of MERTK-mediated EGFR-TKI resistance. The activation of MERTK contributes to epithelial-to-mesenchymal transition (EMT), cancer stem cell (CSC) maintenance, and the immunosuppressive microenvironment, which are the major biological factors involved in the development of EGFR-TKI resistance.
Figure 4.
The mechanisms of MERTK-mediated EGFR-TKI resistance. The activation of MERTK contributes to epithelial-to-mesenchymal transition (EMT), cancer stem cell (CSC) maintenance, and the immunosuppressive microenvironment, which are the major biological factors involved in the development of EGFR-TKI resistance.

Figure 5.
The role of MERTK in Yes-associated protein (YAP)-mediated EGFR-TKI resistance. Activation of hippo pathway blocks the nuclear translocation of YAP or promotes YAP degradation. In the EGFR-TKI resistant cancer cells, MERTK and Axl are upregulated. EGFR-mediated PI3K-independent Akt and MAPK signaling pathways contribute to the activation of YAP. In addition, MERTK may directly interact with YAP and phosphorylate YAP at Y444 residue, which promotes its nuclear localization and transcriptional activity.
Figure 5.
The role of MERTK in Yes-associated protein (YAP)-mediated EGFR-TKI resistance. Activation of hippo pathway blocks the nuclear translocation of YAP or promotes YAP degradation. In the EGFR-TKI resistant cancer cells, MERTK and Axl are upregulated. EGFR-mediated PI3K-independent Akt and MAPK signaling pathways contribute to the activation of YAP. In addition, MERTK may directly interact with YAP and phosphorylate YAP at Y444 residue, which promotes its nuclear localization and transcriptional activity.

Figure 6.
The role of MERTK in microenvironment remodeling. MERTK expressed in cancer cells promotes cell proliferation and survival. In the tumor microenvironment, MERTK-mediated signaling pathways enhance PD-L1 expression in cancer cells leading to the immunosuppression of CD8+ T cells. In addition, apoptotic cancer cells can be eliminated by macrophages through efferocytosis via a PtdSer-Gas6-MERTK, so called “eat me”, signal. Inhibition of MERTK activity by small molecules or monoclonal antibodies may enhance the therapeutic efficacy of anti-cancer agents.
Figure 6.
The role of MERTK in microenvironment remodeling. MERTK expressed in cancer cells promotes cell proliferation and survival. In the tumor microenvironment, MERTK-mediated signaling pathways enhance PD-L1 expression in cancer cells leading to the immunosuppression of CD8+ T cells. In addition, apoptotic cancer cells can be eliminated by macrophages through efferocytosis via a PtdSer-Gas6-MERTK, so called “eat me”, signal. Inhibition of MERTK activity by small molecules or monoclonal antibodies may enhance the therapeutic efficacy of anti-cancer agents.

Figure 7.
In tumor, M2-like macrophages scavenge the apoptotic cancer cells through a process of efferocytosis. Gas6 or PROS binds to the PtdSer presented on the membrane of apoptotic cells. The MERTK expressed in M2-like macrophages interacts with the Gas6/PtdSer complex, and induces cytoskeleton reorganization. In addition, MERTK expressed in macrophage can contribute to the release of anti-inflammatory cytokines that build an immunosuppressive microenvironment.
Figure 7.
In tumor, M2-like macrophages scavenge the apoptotic cancer cells through a process of efferocytosis. Gas6 or PROS binds to the PtdSer presented on the membrane of apoptotic cells. The MERTK expressed in M2-like macrophages interacts with the Gas6/PtdSer complex, and induces cytoskeleton reorganization. In addition, MERTK expressed in macrophage can contribute to the release of anti-inflammatory cytokines that build an immunosuppressive microenvironment.


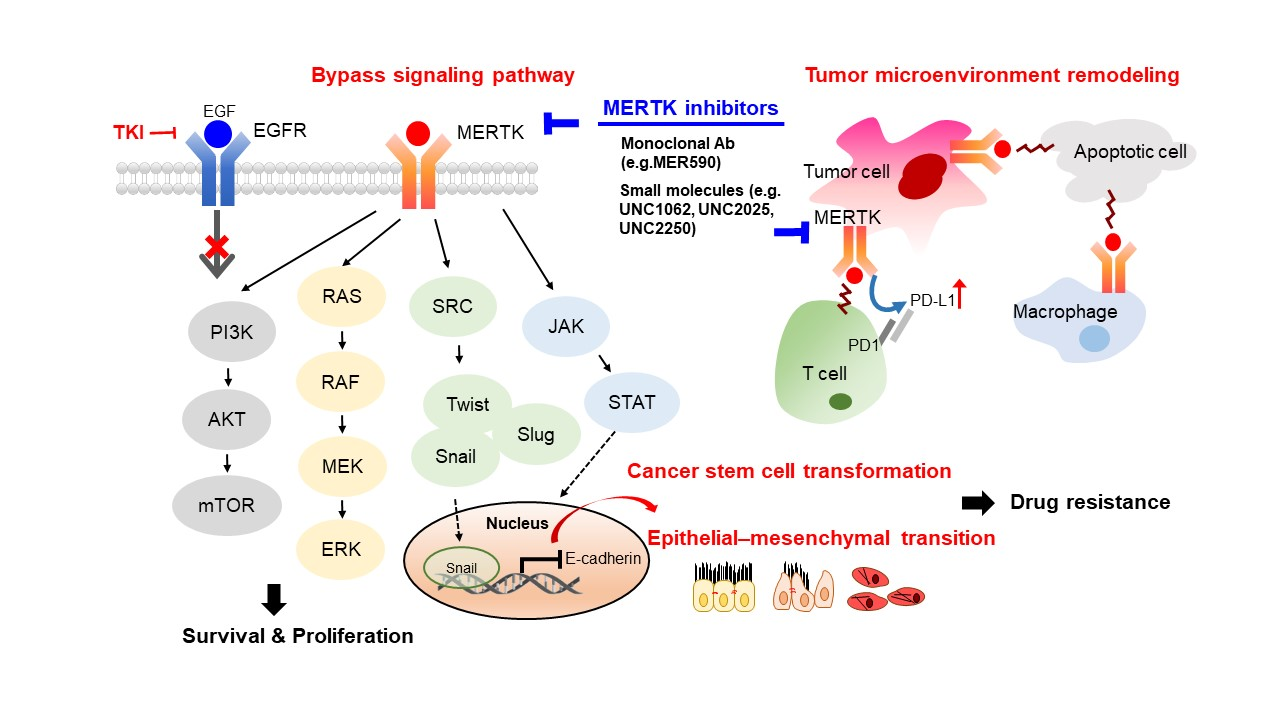{kind=link}
{kind=link}
{kind=link}
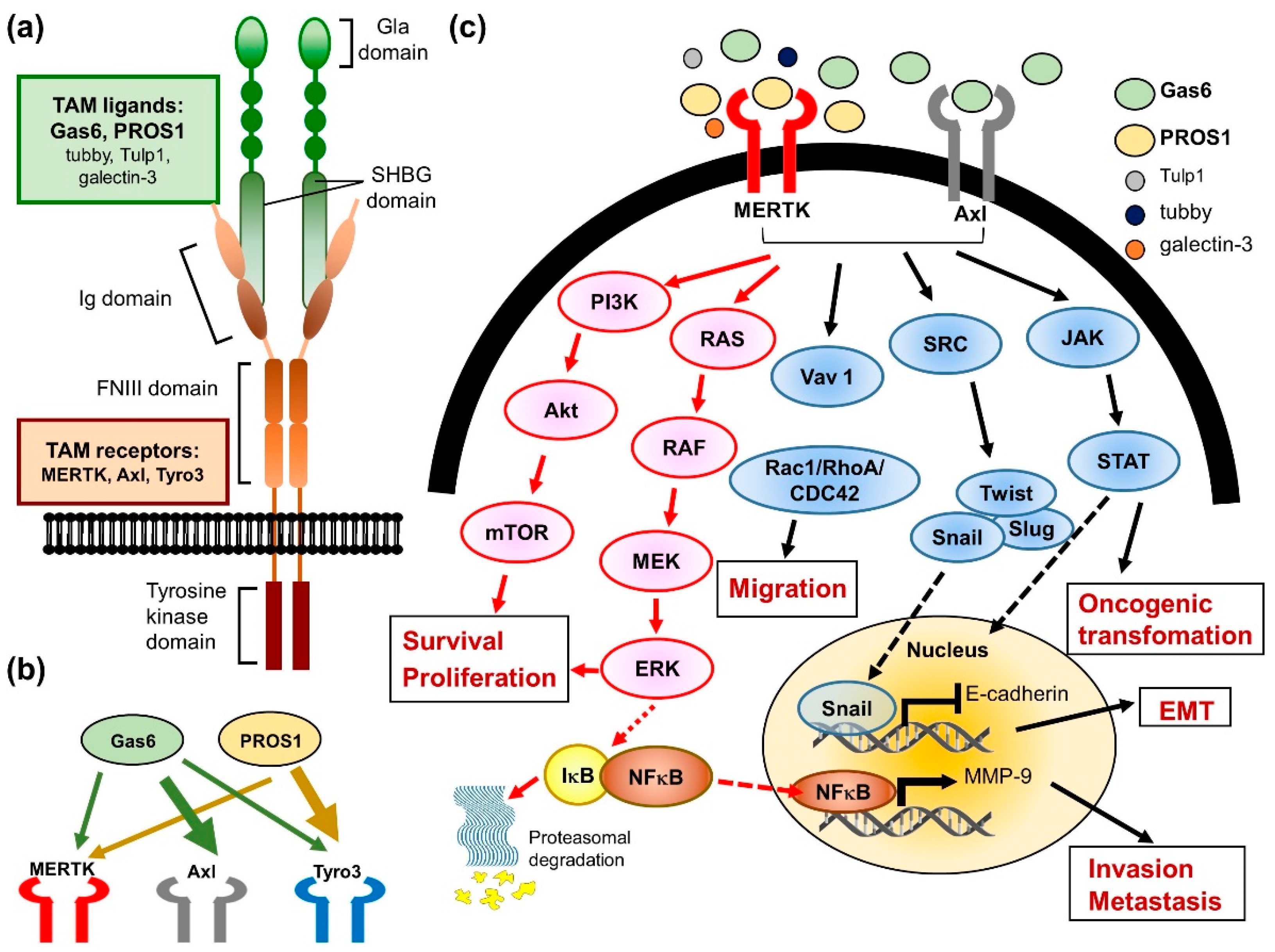{kind=link}
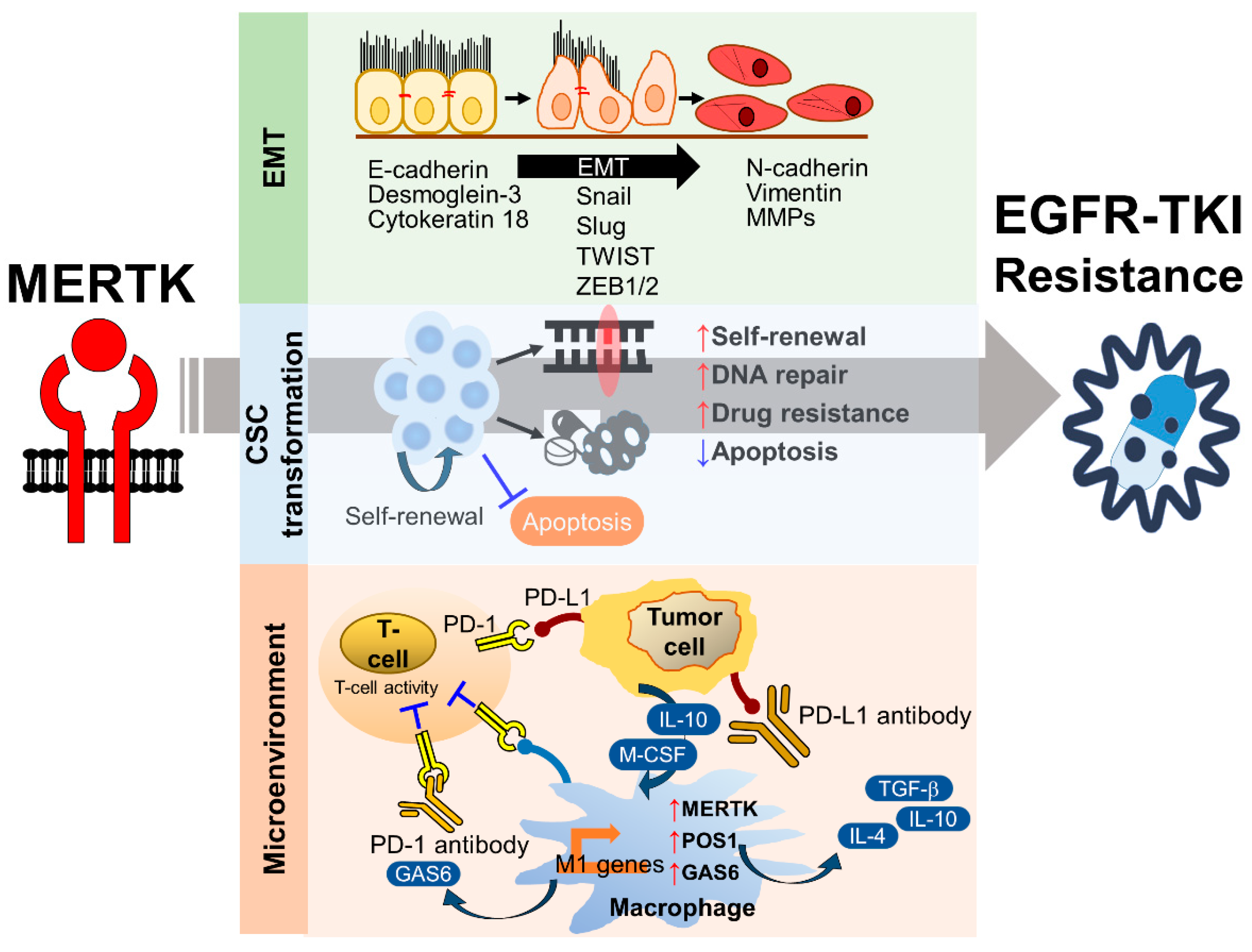{kind=link}
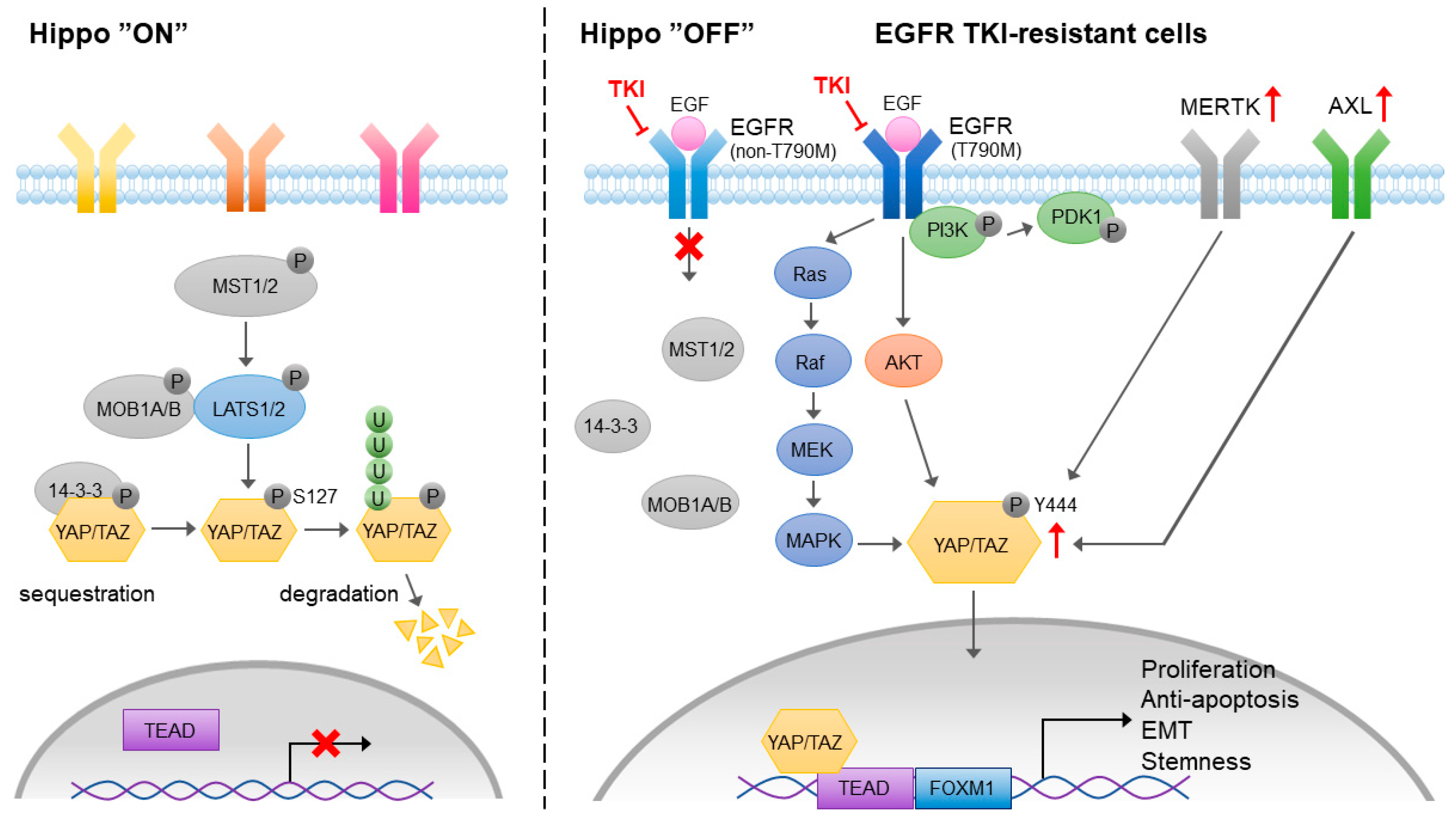{kind=link}
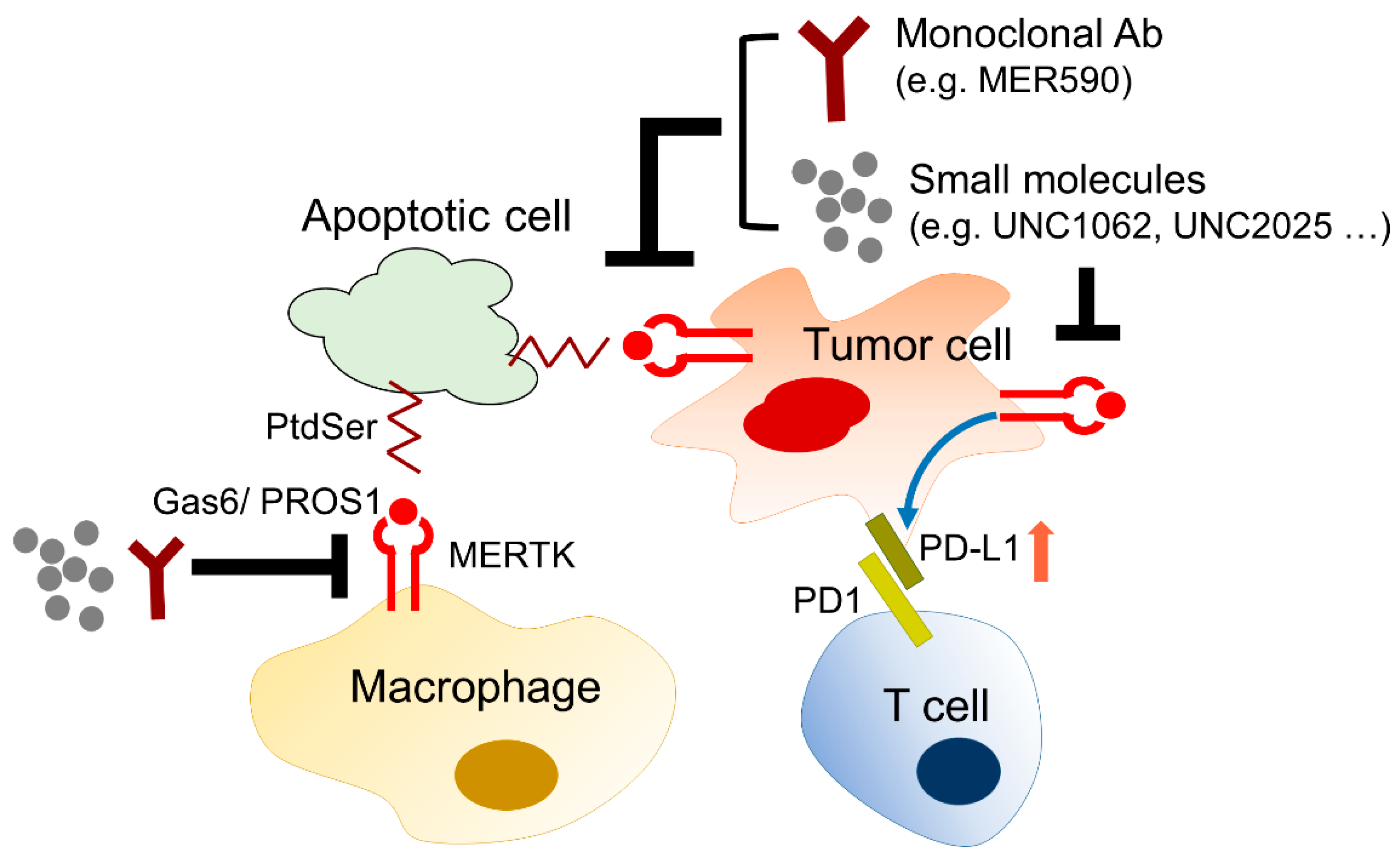{kind=link}
{kind=link}
Table 1.
MERTK inhibitors in preclinical development for NSCLC.
| Compounds | Target(s) | Experimental Models | Outcomes |
|---|---|---|---|
| Mer590 1 | MERTK/Tyro3 | Cell culture | Reduction of MERTK protein levels, decrease in colony formation and chemosensitivity [95] |
| UNC1062 | MERTK/Axl/Tyro3 | Cell culture | Inhibition of A549 colony formation [163] |
| UNC2025 | MERTK/Axl/Tyro3 | Cell culture/ subcutaneous xenograft in mice | Inhibition of colony formation and tumor growth using multiple NSCLC cell lines [93] |
| UNC2250 | MERTK/Axl/Tyro3 | Cell culture | Inhibition of Colo699 colony formation [164] |
1 Mer590 is a monoclonal antibody.
Table 2.
The compounds harboring inhibitory activity of MERTK in clinical trials for NSCLC. 1 The inhibitory effect of individual compounds to corresponding primary targets has been approved by experimental data from biochemical, cell lines and animal studies. 2 The binding activities of compounds to their targets are screened by biochemical kinase selectivity assays. The inhibitory activities and cytotoxicity of some compounds are examined in cell lines. 3 Sources of information on clinical trials include ClinicalTrials.Gov (https://clinicaltrials.gov/ (accessed on 31 January 2021)), Canadian Cancer Trials Group Investigational New Drug (IND), International Standard Randomised Controlled Trial Number Register (ISRCTN) (https://www.isrctn.com/ (accessed on 31 January 2021)), and TrialTrove (https://citeline.com/ (accessed on 31 January 2021)).
Table 2.
The compounds harboring inhibitory activity of MERTK in clinical trials for NSCLC. 1 The inhibitory effect of individual compounds to corresponding primary targets has been approved by experimental data from biochemical, cell lines and animal studies. 2 The binding activities of compounds to their targets are screened by biochemical kinase selectivity assays. The inhibitory activities and cytotoxicity of some compounds are examined in cell lines. 3 Sources of information on clinical trials include ClinicalTrials.Gov (https://clinicaltrials.gov/ (accessed on 31 January 2021)), Canadian Cancer Trials Group Investigational New Drug (IND), International Standard Randomised Controlled Trial Number Register (ISRCTN) (https://www.isrctn.com/ (accessed on 31 January 2021)), and TrialTrove (https://citeline.com/ (accessed on 31 January 2021)).
| Name | Primary Target(s) 1 | Other Target(s) 2 | Development Phase (Trials Registry Number 3) | Tumor Type 1 |
|---|---|---|---|---|
| ASLAN002/BMS-777607 | MET and RON | Axl, Flt3, MERTK and Tyro3 | Phase I (NCT01721148) | Advanced or Metastatic Solid Tumours [165] |
| AT9283 | Aurora kinases | GSK3β, FGFR2, JAK2/3, MERTK, RET, RSK2/3, VEGFR3 (Flt4), Tyk2 and Yes | Phase I (NCT00443976) Phase I (NCT00985868) | Advanced or metastatic solid tumors [166] Relapsed and refractory solid tumors |
| Bemcentinib/BGB324/R428 | Axl | MERTK and Tyro3 | Phase II (NCT03184571) | Advanced NSCLC |
| Bosutinib/SKI-606/PF-5208763 | Src and Abl | MERTK, Axl, Tyro3, EphA2, Kit, Ste20, Tec and Trk | Phase I (NCT03023319) Phase I (NCT01001936) Phase II (NCT03297606) | NSCLC [167] Advanced malignant solid tumors Advanced solid tumors |
| Crizotinib/PF-2341066 | ALK, MET and ROS1 | MERTK, Axl, Tyro3, and RON | Multiple trials | MET+, ROS1+ or ALK+ advanced NSCLC |
| Dubermatinib/TP-0903 | Axl | MERTK, Tyro3, AURKA, Flt3, AURKB, JAK2 and Abl | Phase I (NCT02729298) | EGFR+ NSCLC [168] |
| Foretinib/GSK1363089/XL880/EXEL-2880 | MET and VEGFR2 | MERTK, Axl, Tyro3, RON, PDGFR, KDR, Kit, Flt3 and TIE2 | Phase II (NCT02034097) Phase I (NCT00742261) Phase I (NCT01068587) Phase II (IND.196) | NSCLC Solid Tumors Advanced or Metastatic NSCLC Advanced or Metastatic NSCLC [169] |
| INCB081776 | Axl and MERTK | Phase I (NCT03522142) | Advanced solid tumors | |
| Merestinib/LY2801653 | MET | MERTK, Axl, Tyro3, MET, Flt3, RON, ROS1, Tek, DDR1/2, MKNK1/2 | Phase I (NCT03027284) Phase II (NCT02920996) Phase I (NCT02745769) | Advanced or metastatic cancers [170] NSCLC Advanced cancers |
| Glesatinib/MGCD265 | MET and VEGFR2 | MERTK, Axl, Tyro3, Tek and RON | Phase II (NCT02544633) Phase I (NCT00975767) | NSCLC [171] Advanced NSCLC |
| MK2461 | MET | MERTK, RON, Flt1, Flt3 and PDGFR | Phase I/II (NCT00496353) Phase I (NCT00518739) | Advanced solid tumors [172] Advanced cancers |
| MRX-2843 | MERTK and Flt3 | Axl and Tyro3 | Phase I (NCT03510104) | Advanced and metastatic solid tumors [173] |
| Neratinib/HKI-272 | HER2 and EGFR | MERTK, Axl and Tyro3 | Phase II (NCT01827267) Phase II (NCT00266877) | HER2-activating mutations in NSCLC [174] Advanced NSCLC |
| ONO-7475 | Axl and MERTK | Phase I (NCT03730337) | Advanced or metastatic solid tumors [175] | |
| S49076 | MET | Axl, MERTK and FGFR | Phase I (ISRCTN00759419) | NSCLC [176] |
| Sitravatinib/MGCD516 | spectrum-selective RTKs | MERTK, Axl, Tyro3, VEGFR2, PDGFR, Kit and MET | Phase I (NCT02219711) Phase III (NCT03906071) Phase II (NCT02954991) Phase I (NCT03666143) Phase II (NCT02664935) | Advanced cancers [177] Metastatic non-squamous NSCLC NSCLC Advanced solid tumors. NSCLC |
| SU-14813 | multi-targeted RTKs | MERTK, Axl, Tyro3, FLT3, VEGFR, PDGFR, Kit | Phase I (NCT00982267) | Advanced solid tumors [178] |
| Vandetanib/ZD6474 | multi-targeted RTKs | MERTK, Axl, Tyro3, VEGFR2, VEGFR3, EGFR and RET | Multiple trials | NSCLC [179] |
Abbreviation: MET, MNNG HOS transforming gene; RON, recepteur d’origine nantais receptor; Flt3, Fms-like tyrosine kinase 3; GSK3β, glycogen synthase kinase 3 beta; JAK, Janus kinase; RSK, ribosomal s6 kinase; AURKA, Aurora Kinase A; AURKB, Aurora Kinase B; RET, rearranged during transfection; VEGFR, vascular endothelial growth factor receptor; Tyk2, tyrosine Kinase 2; Yes, tyrosine-protein kinase Yes; Src, v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; Abl, v-abl Abelson murine leukemia viral oncogene homolog; EphA2, erythropoietin-producing hepatoma (Eph) receptor tyrosine kinase A2; Ste20, sterile 20 protein kinase; Tec, Tec Protein Tyrosine Kinase; Trk, tropomyosin receptor kinase; ROS1, c-ros oncogene 1; ALK, anaplastic lymphoma kinase; VEGFR2, vascular endothelial growth factor receptor 2; KDR, kinase insert domain receptor; Tie2, tunica interna endothelial cell kinase 2; DDR, discoidin domain receptor; Tek, TEK receptor tyrosine kinase; MKNK1/2, MAP kinase-interacting serine/threonine-protein kinase 1/2; PDGFR, platelet-derived growth factor receptor; Kit, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; and FGFR, fibroblast growth factor receptor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.-J.; Liu, Y.-P. MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Pharmaceuticals 2021, 14, 130. https://doi.org/10.3390/ph14020130
AMA Style
Chen C-J, Liu Y-P. MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Pharmaceuticals. 2021; 14(2):130. https://doi.org/10.3390/ph14020130
Chicago/Turabian StyleChen, Chao-Ju, and Yu-Peng Liu. 2021. "MERTK Inhibition: Potential as a Treatment Strategy in EGFR Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer" Pharmaceuticals 14, no. 2: 130. https://doi.org/10.3390/ph14020130
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.