Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers

 , ,
, ,
Abstract
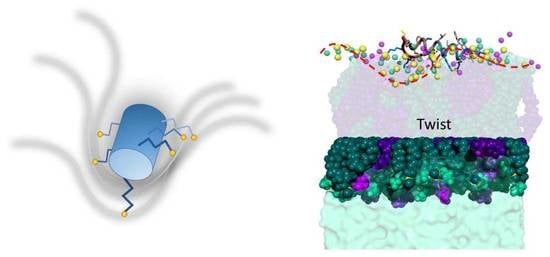
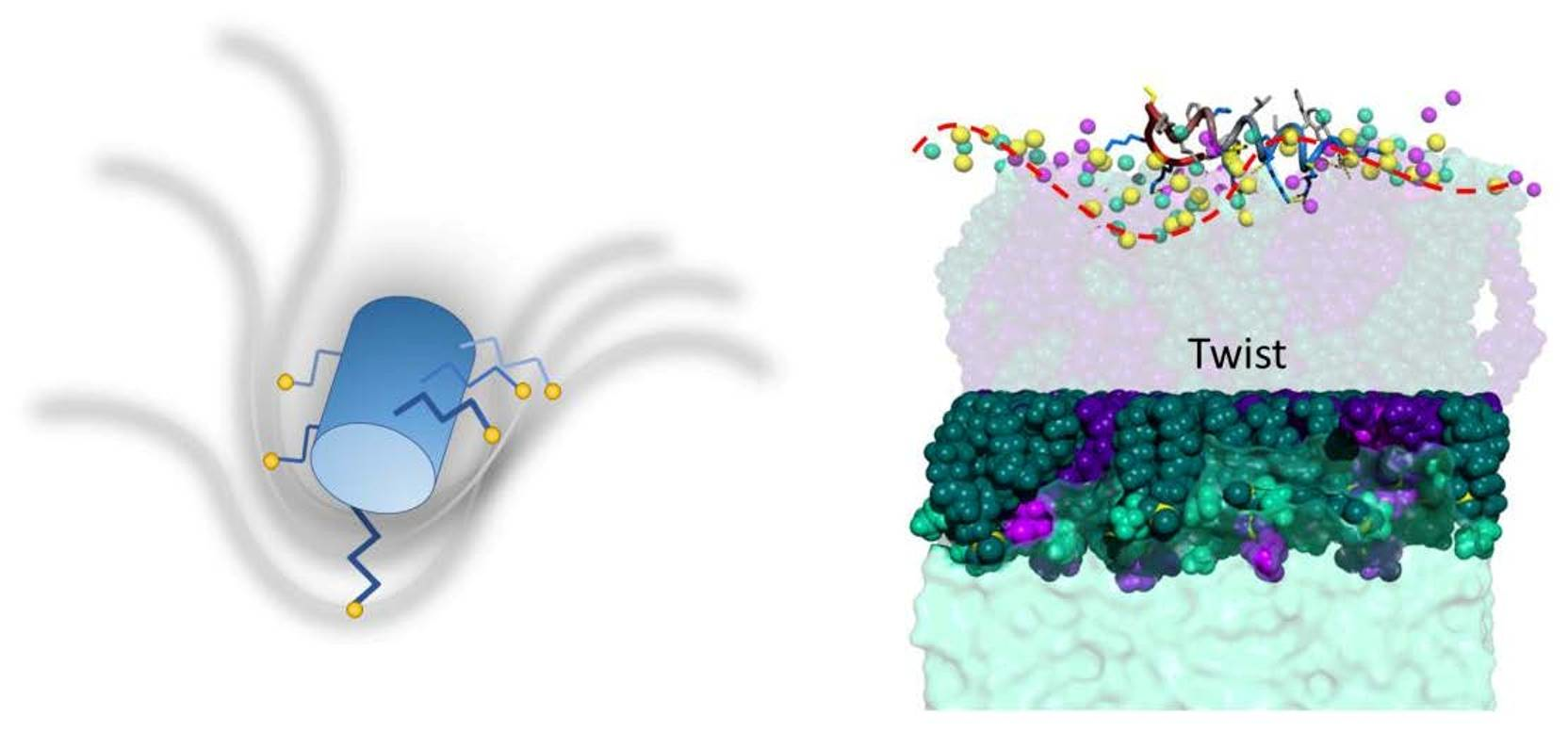
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Property-Sequence Alignment of K11 Highlight Antibacterial Motifs and Predicts Further Activities
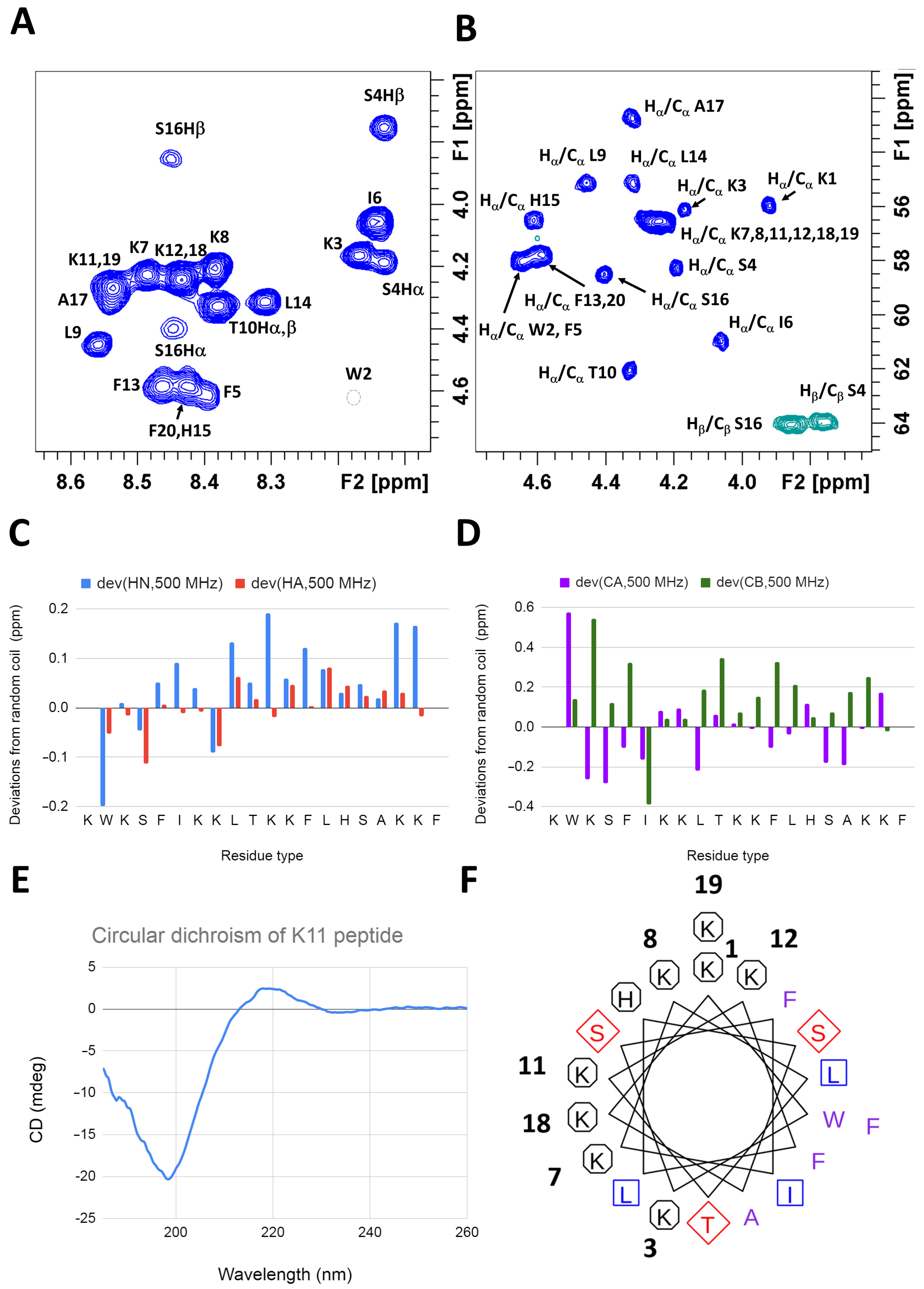
2.2. K11 Peptide Is Unstructured in Aqueous Solution
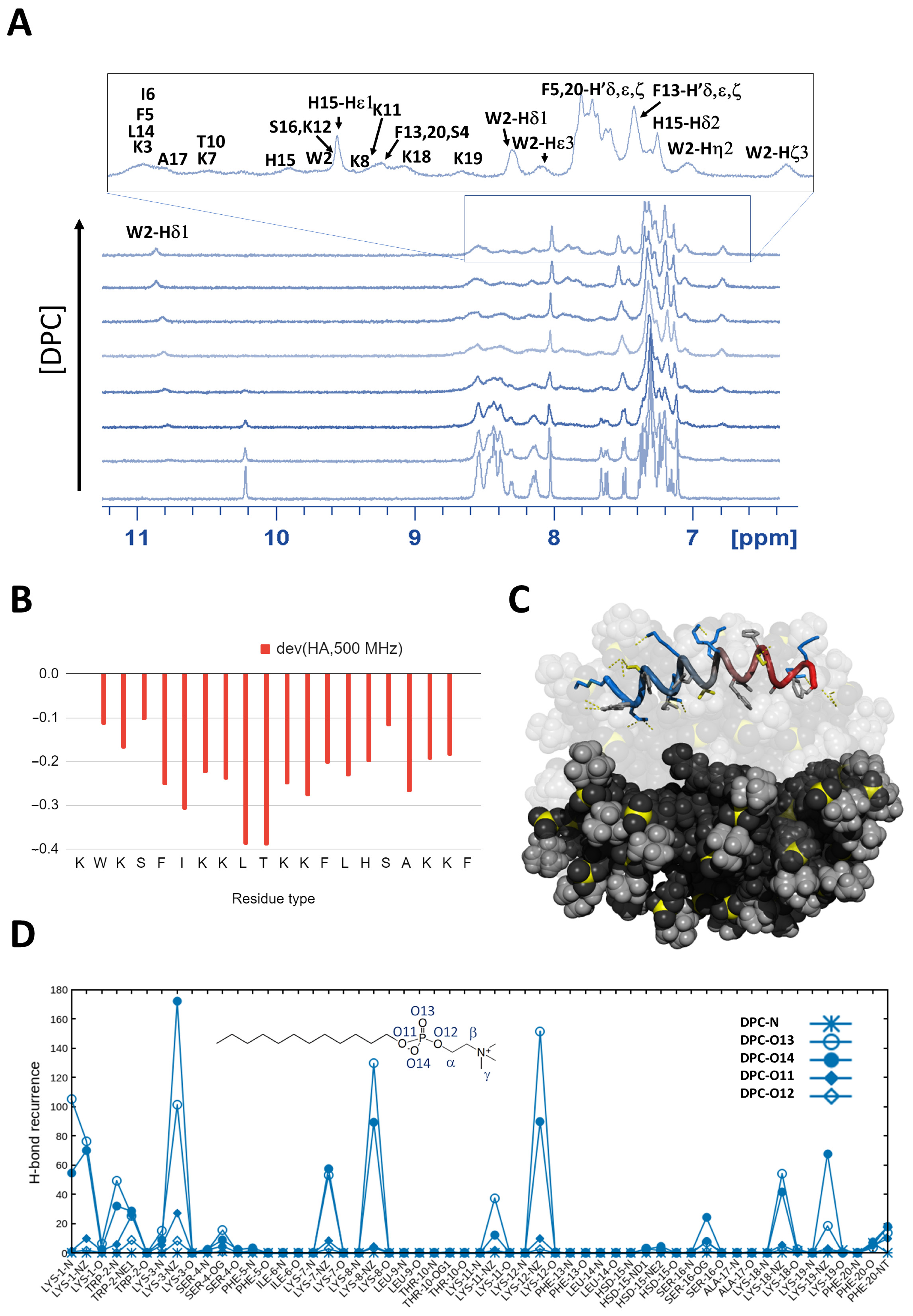
2.2.1. K11 Peptide Assumes Alpha Helical Conformation in a Lipidic Environment
2.2.2. In the Presence of Biomimetic Bicelles K11 Peptide Possibly Assumes a Conformation Similar to That Found with Micelles
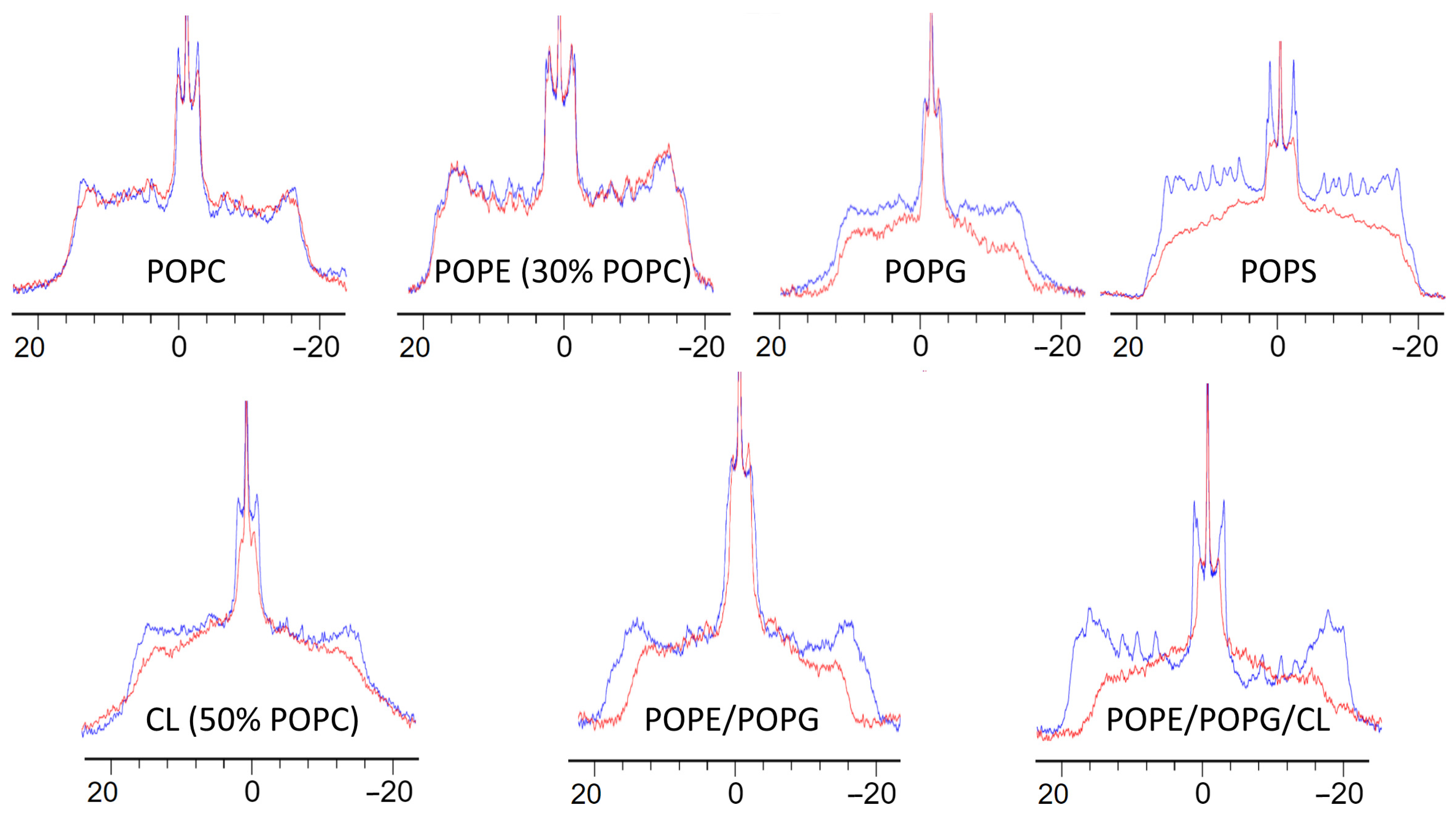
2.2.3. K11 Selectivity Perturbs the Core of Liposomes with Bacterial Phospholipid Compositions
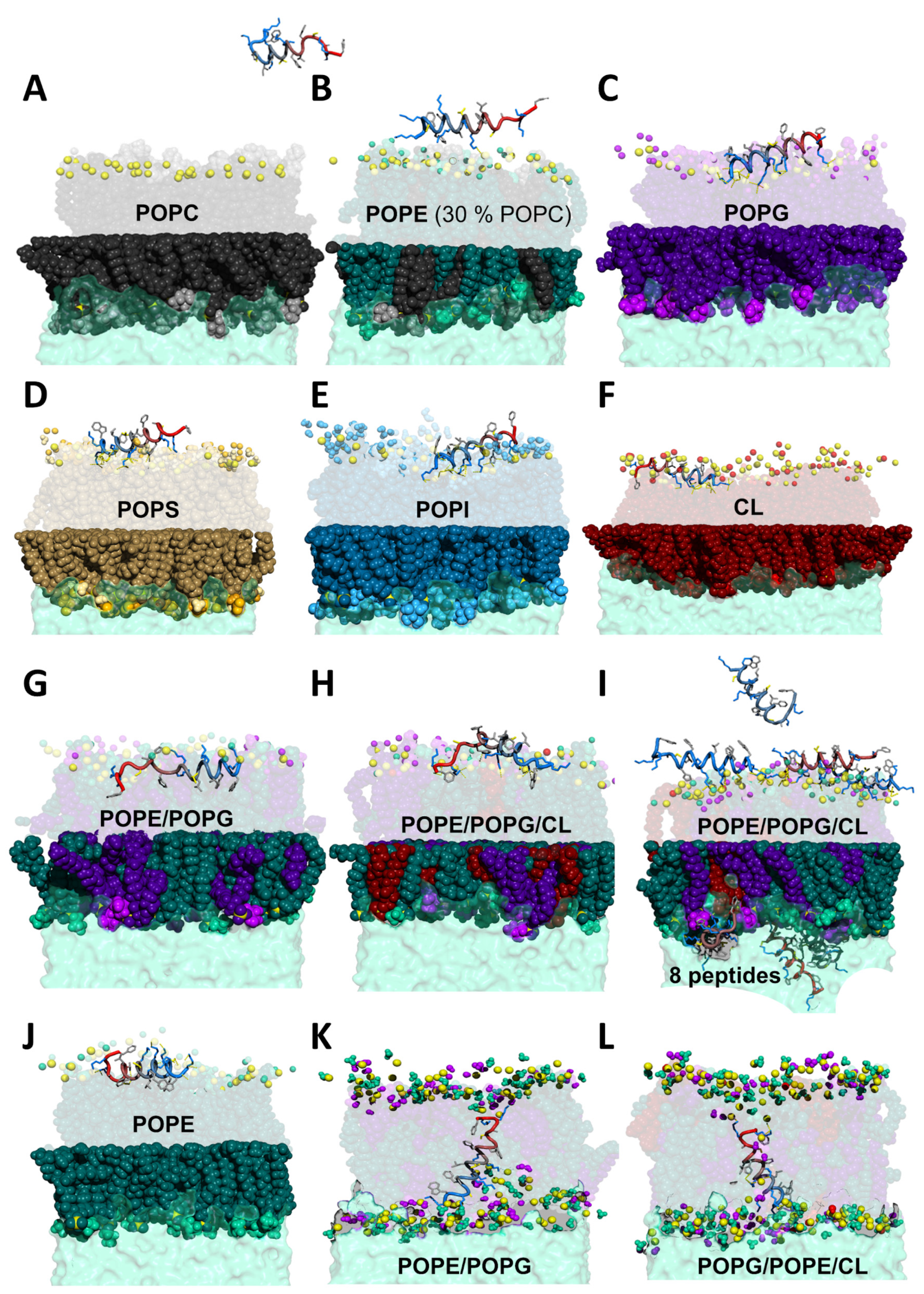
2.3. MD Simulations Provide a Molecular Picture of the Interaction
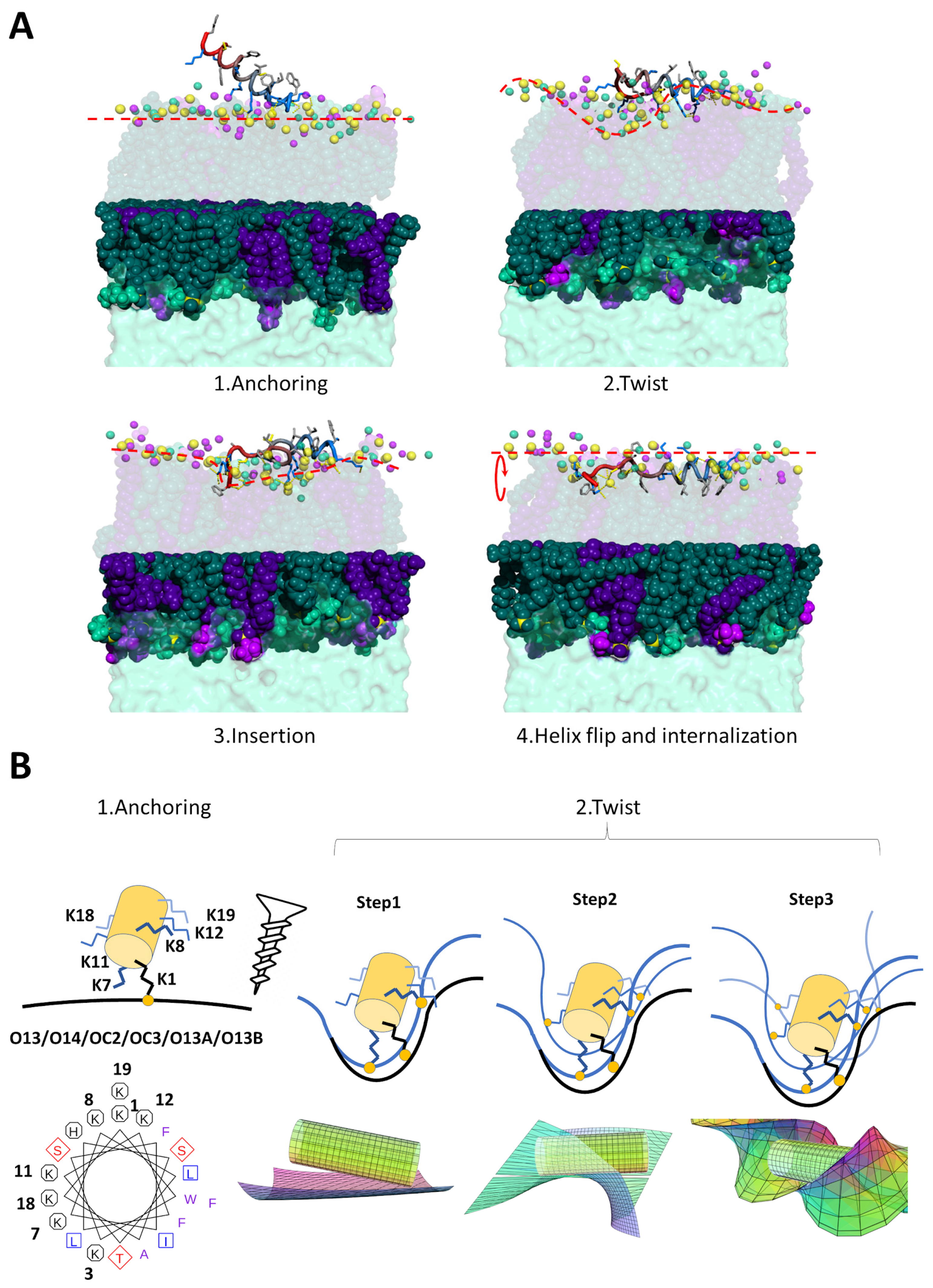
2.3.1. K11 Exerts a Twisting Effect of Its Target Membranes
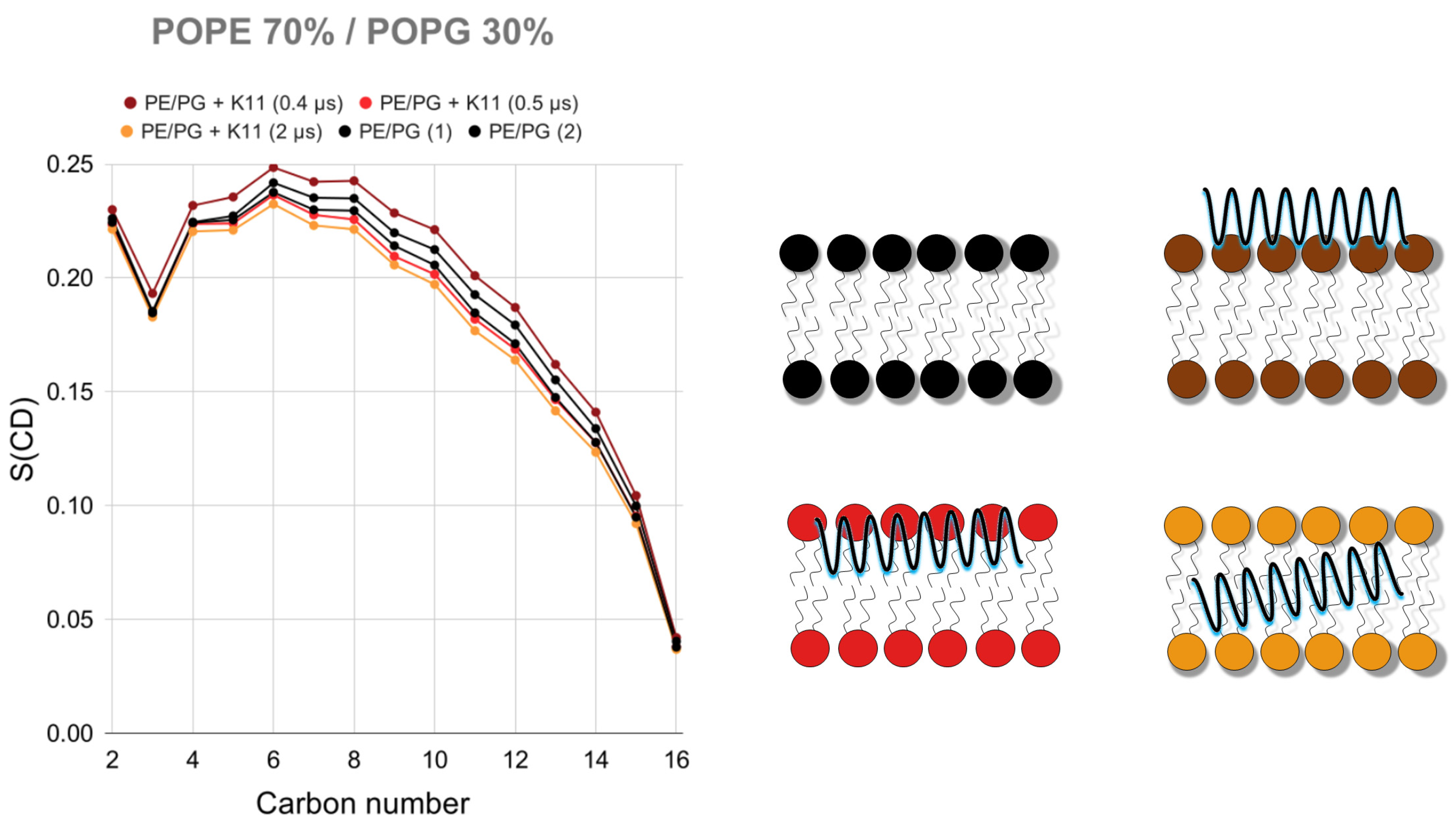
2.3.2. K11 First Rigidifies the Membrane and Subsequently Makes It More Fluid
2.3.3. K11 Approaches Phospholipids Head Groups from Opposite Leaflets Possibly Leading to Membrane Disassembly after Entering the Bilayer
2.3.4. PS Targeting Opens the Way to Possible New Biological Activities
3. Materials and Methods
3.1. Synthesis of K11 Peptide
3.2. Sequence Alignment by ADAPTABLE Web Server
3.3. Sample Preparation, NMR Experiments and Analysis
3.4. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allegranzi, B.; Bagheri Nejad, S.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care-associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef]
- Ibrahim, M.E.; Bilal, N.E.; Hamid, M.E. Increased multi-drug resistant Escherichia coli from hospitals in Khartoum state, Sudan. Afr. Health Sci. 2012, 12, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti. Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Wang, G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; CABI: Wallingford, UK, 2017; ISBN 9781786390394. [Google Scholar]
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial strategies of resistance to antimicrobial peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting drug resistance evolution: Insights from antimicrobial peptides and antibiotics. Proc. Biol. Sci. 2018, 285, 20172687. [Google Scholar] [CrossRef] [Green Version]
- Kintses, B.; Méhi, O.; Ari, E.; Számel, M.; Györkei, Á.; Jangir, P.K.; Nagy, I.; Pál, F.; Fekete, G.; Tengölics, R.; et al. Phylogenetic barriers to horizontal transfer of antimicrobial peptide resistance genes in the human gut microbiota. Nat. Microbiol. 2019, 4, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the Antimicrobial Peptide Polymyxin B1 with Both Membranes of E. coli: A Molecular Dynamics Study. PLoS Comput. Biol. 2015, 11, e1004180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sani, M.-A.; Separovic, F. Interaction of cationic antimicrobial peptides from Australian frogs with lipid membranes. Pept. Sci. 2018, 110, e24061. [Google Scholar] [CrossRef]
- Giuliani, A.; Pirri, G.; Bozzi, A.; Di Giulio, A.; Aschi, M.; Rinaldi, A.C. Antimicrobial peptides: Natural templates for synthetic membrane-active compounds. Cell. Mol. Life Sci. 2008, 65, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Jin-Jiang, H.; Jin-Chun, L.; Min, L.; Qing-Shan, H.; Guo-Dong, L. The Design and Construction of K11: A Novel α-Helical Antimicrobial Peptide. Int. J. Microbiol. 2012, 2012, 764834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishi, P.; Vashist, T.; Sharma, A.; Kaur, A.; Kaur, A.; Kaur, N.; Kaur, I.P.; Tewari, R. Efficacy of designer K11 antimicrobial peptide (a hybrid of melittin, cecropin A1 and magainin 2) against Acinetobacter baumannii-infected wounds. Pathog. Dis. 2018, 76, fty072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobby, C.R.; Herndon, J.L.; Morrow, C.A.; Peters, R.E.; Symes, S.J.K.; Giles, D.K. Exogenous fatty acids alter phospholipid composition, membrane permeability, capacity for biofilm formation, and antimicrobial peptide susceptibility in Klebsiella pneumoniae. MicrobiologyOpen 2019, 8, e00635. [Google Scholar] [CrossRef]
- Benamara, H.; Rihouey, C.; Jouenne, T.; Alexandre, S. Impact of the biofilm mode of growth on the inner membrane phospholipid composition and lipid domains in Pseudomonas aeruginosa. Biochim. Biophys. Acta BBA Biomembr. 2011, 1808, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef]
- Lopalco, P.; Stahl, J.; Annese, C.; Averhoff, B.; Corcelli, A. Identification of unique cardiolipin and monolysocardiolipin species in Acinetobacter baumannii. Sci. Rep. 2017, 7, 2972. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Lee, S.H.; Yang, S.T.; Park, E.J.; Lee, D.G.; Lee, M.K.; Eom, S.H.; Song, W.K.; Kim, Y.; Hahm, K.S.; et al. Antibacterial, antitumor and hemolytic activities of alpha-helical antibiotic peptide, P18 and its analogs. J. Pept. Res. 2001, 58, 504–514. [Google Scholar] [CrossRef]
- Scott, M.G.; Yan, H.; Hancock, R.E.W. Biological Properties of Structurally Related α-Helical Cationic Antimicrobial Peptides. Infect. Immun. 1999, 67, 2005–2009. [Google Scholar] [CrossRef]
- Randle, C.L.; Albro, P.W.; Dittmer, J.C. The phosphoglyceride composition of gram-negative bacteria and the changes in composition during growth. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1969, 187, 214–220. [Google Scholar] [CrossRef]
- Shokri, A.; Larsson, G. Characterisation of the Escherichia coli membrane structure and function during fedbatch cultivation. Microb. Cell Fact. 2004, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modak, M.J.; Nair, S.; Venkataraman, A. Studies on the Fatty Acid Composition of some Salmonellas. J. Gen. Microbiol. 1970, 60, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanth, C.; Abbassi, F.; Lequin, O.; Ayala-Sanmartin, J.; Ladram, A.; Nicolas, P.; Amiche, M. Mechanism of antibacterial action of dermaseptin B2: Interplay between helix-hinge-helix structure and membrane curvature strain. Biochemistry 2009, 48, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Shivaji, S.; Chaturvedi, P.; Suresh, K.; Reddy, G.S.N.; Dutt, C.B.S.; Wainwright, M.; Narlikar, J.V.; Bhargava, P.M. Bacillus aerius sp. nov., Bacillus aerophilus sp. nov., Bacillus stratosphericus sp. nov. and Bacillus altitudinis sp. nov., isolated from cryogenic tubes used for collecting air samples from high altitudes. Int. J. Syst. Evol. Microbiol. 2006, 56, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, J.A.F.; Houtsmuller, U.M.T.; Van Deenen, L.L.M. On the phospholipids of Bacillus megaterium. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1965, 106, 438–441. [Google Scholar] [CrossRef]
- Bishop, D.G.; Op den Kamp, J.A.; van Deenen, L.L. The distribution of lipids in the protoplast membranes of Bacillus subtilis. A study with phospholipase C and trinitrobenzenesulphonic acid. Eur. J. Biochem. 1977, 80, 381–391. [Google Scholar] [CrossRef]
- Komaratat, P.; Kates, M. The lipid composition of a halotolerant species of Staphylococcus epidermidis. Biochim. Biophys. Acta 1975, 398, 464–484. [Google Scholar] [CrossRef]
- Kanemasa, Y.; Yoshioka, T.; Hayashi, H. Alteration of the phospholipid composition of Staphylococcus aureus cultured in medium containing NaCl. Biochim. Biophys. Acta 1972, 280, 444–450. [Google Scholar]
- De Bony, J.; Lopez, A.; Gilleron, M.; Welby, M.; Lanéelle, G.; Rousseau, B.; Beaucourt, J.P.; Tocanne, J.F. Transverse and lateral distribution of phospholipids and glycolipids in the membrane of the bacterium Micrococcus luteus. Biochemistry 1989, 28, 3728–3737. [Google Scholar] [CrossRef]
- Tari, A.; Huang, L. Structure and function relationship of phosphatidylglycerol in the stabilization of phosphatidylethanolamine bilayer. Biochemistry 1989, 28, 7708–7712. [Google Scholar] [CrossRef] [PubMed]
- Murzyn, K.; Róg, T.; Pasenkiewicz-Gierula, M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys. J. 2005, 88, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Burra, S.S.; Murthy, P.S. Correlation between calmodulin-like protein, phospholipids, and growth in glucose-grown Mycobacterium phlei. Can. J. Microbiol. 1992, 38, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Kenis, H.; Reutelingsperger, C. Targeting phosphatidylserine in anti-cancer therapy. Curr. Pharm. Des. 2009, 15, 2719–2723. [Google Scholar] [CrossRef]
- Zwaal, R.F.A.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. CMLS Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martín, F.; Annaval, T.; Buchoux, S.; Sarazin, C.; D’Amelio, N. ADAPTABLE: A comprehensive web platform of antimicrobial peptides tailored to the user’s research. Life Sci. Alliance 2019, 2, e201900512. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Lee, M.K.; Kim, K.L.; Hahm, K.S. Structure-antitumor and hemolytic activity relationships of synthetic peptides derived from cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J. Pept. Res. 1997, 50, 279–285. [Google Scholar] [CrossRef]
- Park, Y.; Lee, D.G.; Hahm, K.-S. Antibiotic activity of Leu-Lys rich model peptides. Biotechnol. Lett. 2003, 25, 1305–1310. [Google Scholar] [CrossRef]
- Gray, M.; Gong, J.; Nguyen, V.; Osada, T.; Hartman, Z.; Hutchins, J.; Freimark, B.; Lyerly, K. Targeting of phosphatidylserine by monoclonal antibodies augments the activity of paclitaxel and anti-PD1/PD-L1 therapy in the murine breast model E0771. J. ImmunoTher. Cancer 2015, 3, P357. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target-phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar] [PubMed]
- Hasim, S.; Vaughn, E.N.; Donohoe, D.; Gordon, D.M.; Pfiffner, S.; Reynolds, T.B. Influence of phosphatidylserine and phosphatidylethanolamine on farnesol tolerance in Candida albicans. Yeast 2018, 35, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, N.K.; Sarkar, P.; Gaur, N.A.; Chattopadhyay, A.; Prasad, R. Phosphatidylserine decarboxylase governs plasma membrane fluidity and impacts drug susceptibilities of Candida albicans cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Cassilly, C.; Reynolds, T. PS, It’s Complicated: The Roles of Phosphatidylserine and Phosphatidylethanolamine in the Pathogenesis of Candida albicans and Other Microbial Pathogens. J. Fungi 2018, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef] [Green Version]
- Lösel, D.M. Lipids in the Structure and Function of Fungal Membranes. In Biochemistry of Cell Walls and Membranes in Fungi; Kuhn, P.J., Ed.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 119–133. [Google Scholar]
- Mahto, K.K.; Singh, A.; Khandelwal, N.K.; Bhardwaj, N.; Jha, J.; Prasad, R. An assessment of growth media enrichment on lipid metabolome and the concurrent phenotypic properties of Candida albicans. PLoS ONE 2014, 9, e113664. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13 C Chemical-Shift Index: A simple method for the identification of protein secondary structure using 13 C chemical-shift data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef]
- Wishart, D.S. Interpreting protein chemical shift data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef]
- Beswick, V.; Guerois, R.; Cordier-Ochsenbein, F.; Coïc, Y.M.; Tam, H.D.; Tostain, J.; Noël, J.P.; Sanson, A.; Neumann, J.M. Dodecylphosphocholine micelles as a membrane-like environment: New results from NMR relaxation and paramagnetic relaxation enhancement analysis. Eur. Biophys. J. 1999, 28, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Springer Publishing: New York, NY, USA, 1981; pp. 331–342. [Google Scholar]
- Porcelli, F.; Ramamoorthy, A.; Barany, G.; Veglia, G. On the role of NMR spectroscopy for characterization of antimicrobial peptides. Methods Mol. Biol. 2013, 1063, 159–180. [Google Scholar] [PubMed] [Green Version]
- van Dam, L.; Karlsson, G.; Edwards, K. Direct observation and characterization of DMPC/DHPC aggregates under conditions relevant for biological solution NMR. Biochim. Biophys. Acta 2004, 1664, 241–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, I.; Auger, M. Bicelles as model membranes for solid- and solution-state NMR studies of membrane peptides and proteins. Concepts Magn. Reson. Part A 2005, 24A, 17–37. [Google Scholar] [CrossRef]
- Davis, J.H. The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta BBA Rev. Biomembr. 1983, 737, 117–171. [Google Scholar] [CrossRef]
- Molugu, T.R.; Lee, S.; Brown, M.F. Concepts and Methods of Solid-State NMR Spectroscopy Applied to Biomembranes. Chem. Rev. 2017, 117, 12087–12132. [Google Scholar] [CrossRef]
- Salnikov, E.S.; Mason, A.J.; Bechinger, B. Membrane order perturbation in the presence of antimicrobial peptides by (2)H solid-state NMR spectroscopy. Biochimie 2009, 91, 734–743. [Google Scholar] [CrossRef]
- Romantsov, T.; Guan, Z.; Wood, J.M. Cardiolipin and the osmotic stress responses of bacteria. Biochim. Biophys. Acta 2009, 1788, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Sendecki, A.M.; Poyton, M.F.; Baxter, A.J.; Yang, T.; Cremer, P.S. Supported Lipid Bilayers with Phosphatidylethanolamine as the Major Component. Langmuir 2017, 33, 13423–13429. [Google Scholar] [CrossRef]
- Lewis, R.N.A.H.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- Szoka, F., Jr.; Papahadjopoulos, D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef] [PubMed]
- Papahadjopoulos, D.; Miller, N. Phospholipid model membranes. I. Structural characteristics of hydrated liquid crystals. Biochim. Biophys. Acta 1967, 135, 624–638. [Google Scholar] [CrossRef]
- Litman, B.J. Lipid model membranes. Characterization of mixed phospholipid vesicles. Biochemistry 1973, 12, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Tinker, D.O.; Pinteric, L. On the identification of lamellar and hexagonal phases in negatively stained phospholipid-water systems. Biochemistry 1971, 10, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Junger, E.; Reinauer, H. Liquid crystalline phases of hydrated phosphatidylethanolamine. Biochim. Biophys. Acta 1969, 183, 304–308. [Google Scholar] [CrossRef]
- Yesylevskyy, S.O.; Rivel, T.; Ramseyer, C. The influence of curvature on the properties of the plasma membrane. Insights from atomistic molecular dynamics simulations. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Brown, M.F. Curvature forces in membrane lipid-protein interactions. Biochemistry 2012, 51, 9782–9795. [Google Scholar] [CrossRef] [Green Version]
- Klebe, G. Protein–Ligand Interactions as the Basis for Drug Action. In Drug Design; Springer: Berlin/Heidelberg, Germany, 2013; pp. 61–88. [Google Scholar]
- Seeburger, P. Calcplot3d, an Exploration Environment for Multivariable Calculus-Taylor Polynomials of a Function of Two Variables (1st and 2nd Degree); Convergence; MAA: Washington, DC, USA, 2011. [Google Scholar]
- Teilum, K.; Kunze, M.B.A.; Erlendsson, S.; Kragelund, B.B. (S)Pinning down protein interactions by NMR. Protein Sci. 2017, 26, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Choi, H.; Weisshaar, J.C. Melittin-Induced Permeabilization, Re-sealing, and Re-permeabilization of E. coli Membranes. Biophys. J. 2018, 114, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Orsi, M.; Noro, M.G.; Essex, J.W. Dual-resolution molecular dynamics simulation of antimicrobials in biomembranes. J. R. Soc. Interface 2011, 8, 826–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isralewitz, B.; Baudry, J.; Gullingsrud, J.; Kosztin, D.; Schulten, K. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 2001, 19, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J. Umbrella sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Domański, J.; Hedger, G.; Best, R.B.; Stansfeld, P.J.; Sansom, M.S.P. Convergence and Sampling in Determining Free Energy Landscapes for Membrane Protein Association. J. Phys. Chem. B 2017, 121, 3364–3375. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200–212. [Google Scholar] [CrossRef]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta 2016, 1858, 1635–1651. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Aisenbrey, C.; Marquette, A.; Bechinger, B. The Mechanisms of Action of Cationic Antimicrobial Peptides Refined by Novel Concepts from Biophysical Investigations. Adv. Exp. Med. Biol. 2019, 1117, 33–64. [Google Scholar]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellet, M.; Bernard, G.; Voyer, N.; Auger, M. Insights on the interactions of synthetic amphipathic peptides with model membranes as revealed by 31P and 2H solid-state NMR and infrared spectroscopies. Biophys. J. 2006, 90, 4071–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Azevedo, H.S.; Orsi, M. Interaction of Antimicrobial Lipopeptides with Bacterial Lipid Bilayers. J. Membr. Biol. 2019, 252, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufourc, E.J.; Dufourcq, J.; Birkbeck, T.H.; Freer, J.H. Delta-haemolysin from Staphylococcus aureus and model membranes. A solid-state 2H-NMR and 31P-NMR study. Eur. J. Biochem. 1990, 187, 581–587. [Google Scholar] [CrossRef]
- Dufourc, E.J.; Smith, I.C.; Dufourcq, J. Molecular details of melittin-induced lysis of phospholipid membranes as revealed by deuterium and phosphorus NMR. Biochemistry 1986, 25, 6448–6455. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Martinez, G.V.; Brown, M.F.; Ramamoorthy, A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry 2004, 43, 8459–8469. [Google Scholar] [CrossRef]
- Zhuang, X.; Makover, J.R.; Im, W.; Klauda, J.B. A systematic molecular dynamics simulation study of temperature dependent bilayer structural properties. Biochim. Biophys. Acta 2014, 1838, 2520–2529. [Google Scholar] [CrossRef] [Green Version]
- Smondyrev, A.M.; Berkowitz, M.L. Structure of Dipalmitoylphosphatidylcholine/Cholesterol Bilayer at Low and High Cholesterol Concentrations: Molecular Dynamics Simulation. Biophys. J. 1999, 77, 2075–2089. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zhang, F.; Liu, Z.; Ma, J.; Yang, J. Expression and characterization of cecropinXJ, a bioactive antimicrobial peptide from (Bombycidae, Lepidoptera) in Escherichia coli. Exp. Ther. Med. 2013, 5, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Romoli, O.; Mukherjee, S.; Mohid, S.A.; Dutta, A.; Montali, A.; Franzolin, E.; Brady, D.; Zito, F.; Bergantino, E.; Rampazzo, C.; et al. Enhanced Silkworm Cecropin B Antimicrobial Activity against from Single Amino Acid Variation. ACS Infect. Dis. 2019, 5, 1200–1213. [Google Scholar] [CrossRef]
- Liu, D.; Liu, J.; Li, J.; Xia, L.; Yang, J.; Sun, S.; Ma, J.; Zhang, F. A potential food biopreservative, CecXJ-37N, non-covalently intercalates into the nucleotides of bacterial genomic DNA beyond membrane attack. Food Chem. 2017, 217, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Springer Publishing: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Nielsen, J.T.; Mulder, F.A.A. POTENCI: Prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Monnier, N.; Furlan, A.L.; Buchoux, S.; Deleu, M.; Dauchez, M.; Rippa, S.; Sarazin, C. Exploring the Dual Interaction of Natural Rhamnolipids with Plant and Fungal Biomimetic Plasma Membranes through Biophysical Studies. Int. J. Mol. Sci. 2019, 20, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlan, A.L.; Castets, A.; Nallet, F.; Pianet, I.; Grélard, A.; Dufourc, E.J.; Géan, J. Red wine tannins fluidify and precipitate lipid liposomes and bicelles. A role for lipids in wine tasting? Langmuir 2014, 30, 5518–5526. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.L.; Jobin, M.-L.; Pianet, I.; Dufourc, E.J.; Géan, J. Flavanol/lipid interaction: A novel molecular perspective in the description of wine astringency & bitterness and antioxidant action. Tetrahedron 2015, 71, 3143–3147. [Google Scholar]
- Grélard, A.; Guichard, P.; Bonnafous, P.; Marco, S.; Lambert, O.; Manin, C.; Ronzon, F.; Dufourc, E.J. Hepatitis B subvirus particles display both a fluid bilayer membrane and a strong resistance to freeze drying: A study by solid-state NMR, light scattering, and cryo-electron microscopy/tomography. FASEB J. 2013, 27, 4316–4326. [Google Scholar] [CrossRef]
- Davis, J.H.; Jeffrey, K.R.; Bloom, M.; Valic, M.I.; Higgs, T.P. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 1976, 42, 390–394. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Jo, S.; Lee, H.S.; Klauda, J.B.; Im, W. CHARMM-GUI micelle builder for pure/mixed micelle and protein/micelle complex systems. J. Chem. Inf. Model. 2013, 53, 2171–2180. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.J.; Klauda, J.B.; Sodt, A.J. Simulation Best Practices for Lipid Membranes [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5966. [Google Scholar] [CrossRef]
- Lemkul, J. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Buchoux, S. FATSLiM: A fast and robust software to analyze MD simulations of membranes. Bioinformatics 2017, 33, 133–134. [Google Scholar] [CrossRef] [Green Version]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Janert, P.K. Gnuplot in Action: Understanding Data with Graphs; Manning Publications: Shelter Island, NY, USA, 2016; ISBN 9781933988399. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals 2021, 14, 1. https://doi.org/10.3390/ph14010001
Ramos-Martín F, Herrera-León C, Antonietti V, Sonnet P, Sarazin C, D’Amelio N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals. 2021; 14(1):1. https://doi.org/10.3390/ph14010001
Chicago/Turabian StyleRamos-Martín, Francisco, Claudia Herrera-León, Viviane Antonietti, Pascal Sonnet, Catherine Sarazin, and Nicola D’Amelio. 2021. "Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers" Pharmaceuticals 14, no. 1: 1. https://doi.org/10.3390/ph14010001