Novel Isoniazid-Carborane Hybrids Active In Vitro against Mycobacterium tuberculosis

 , , and
, , and
Abstract
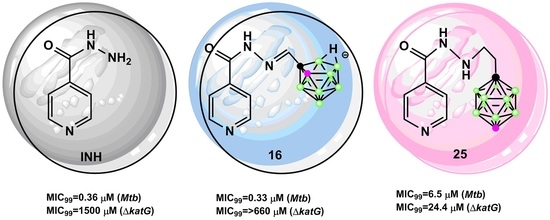
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
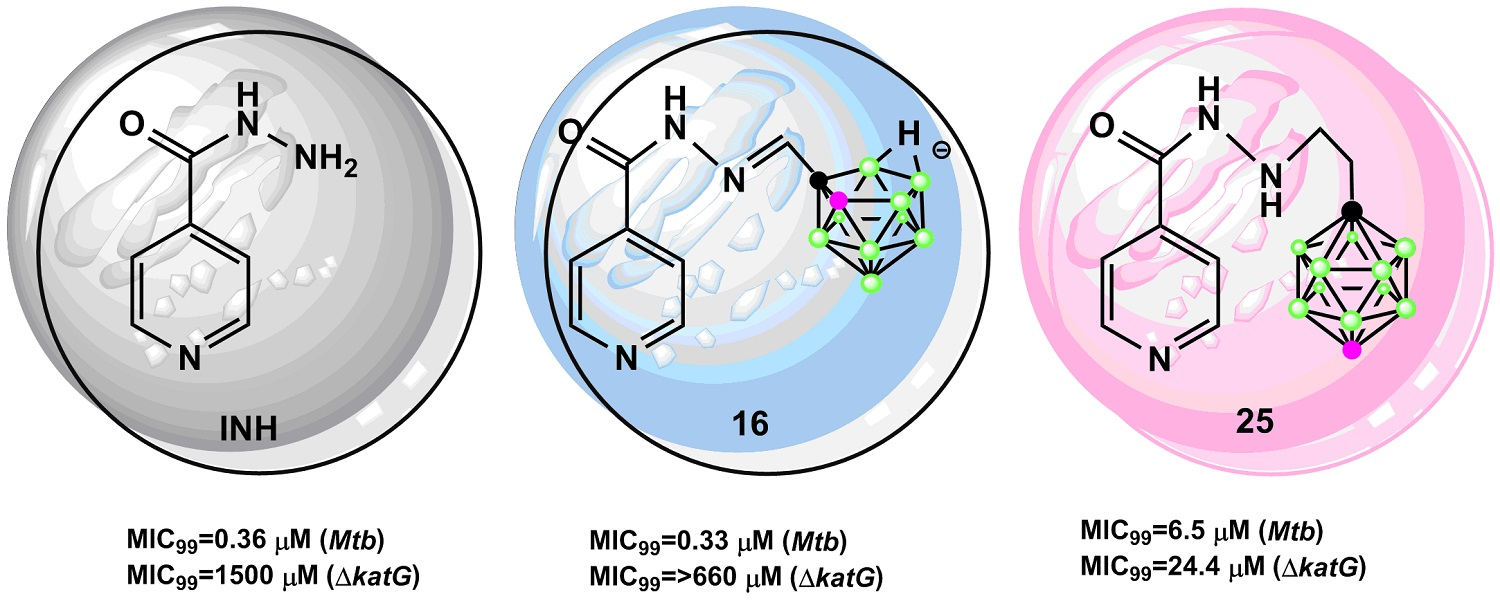
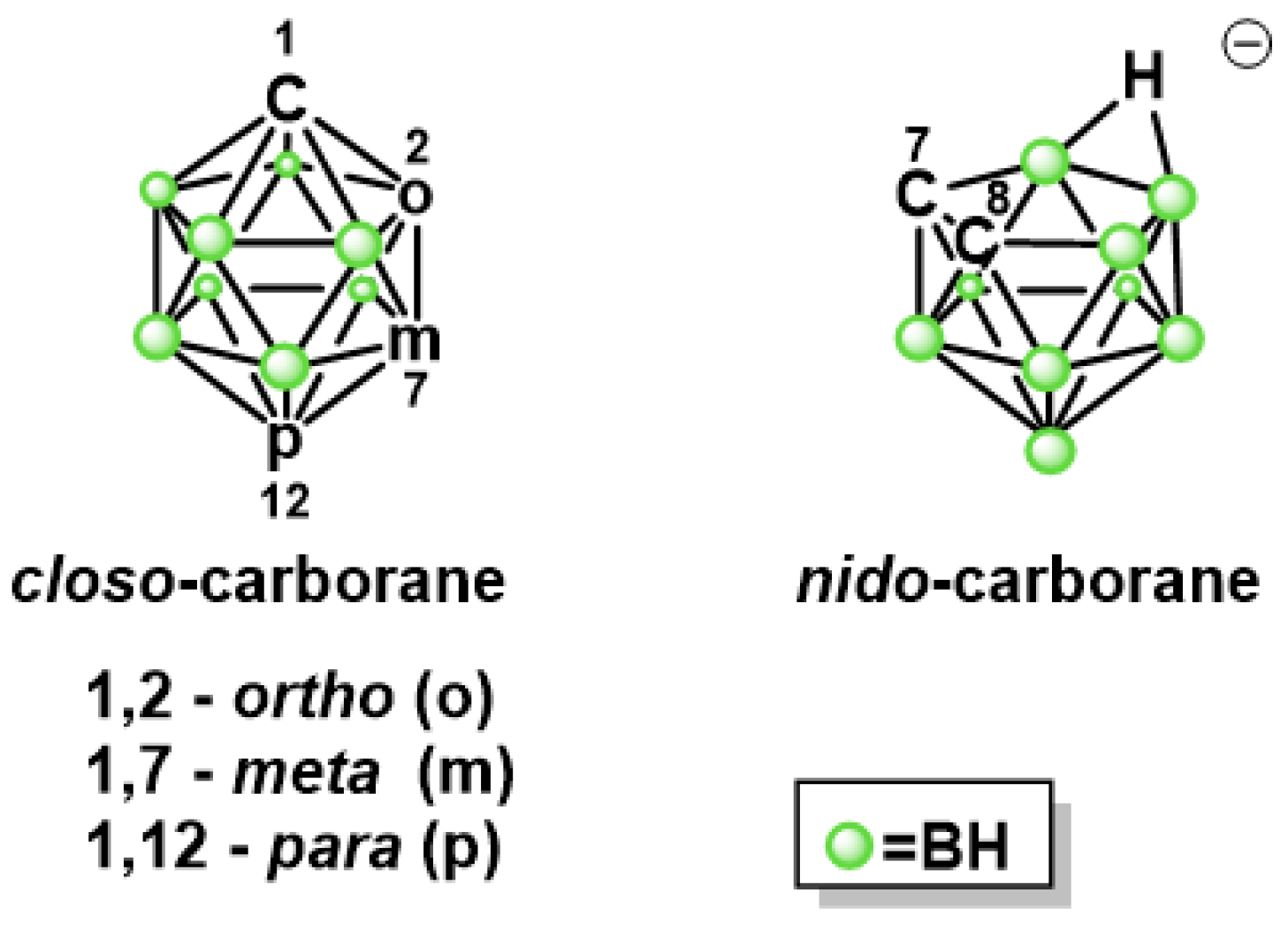
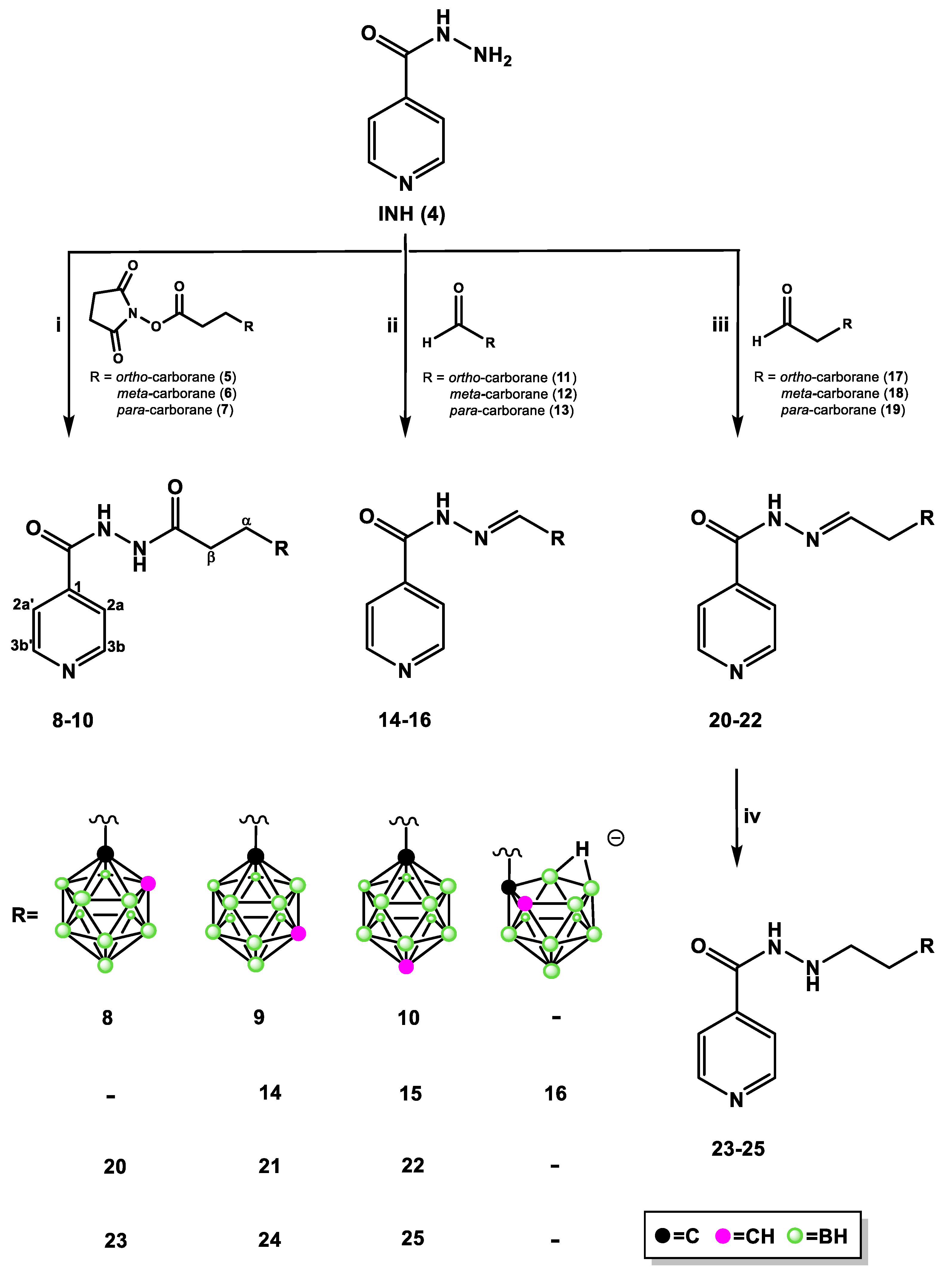
2.1.1. Synthesis of Isoniazid-Carborane Cluster Conjugates
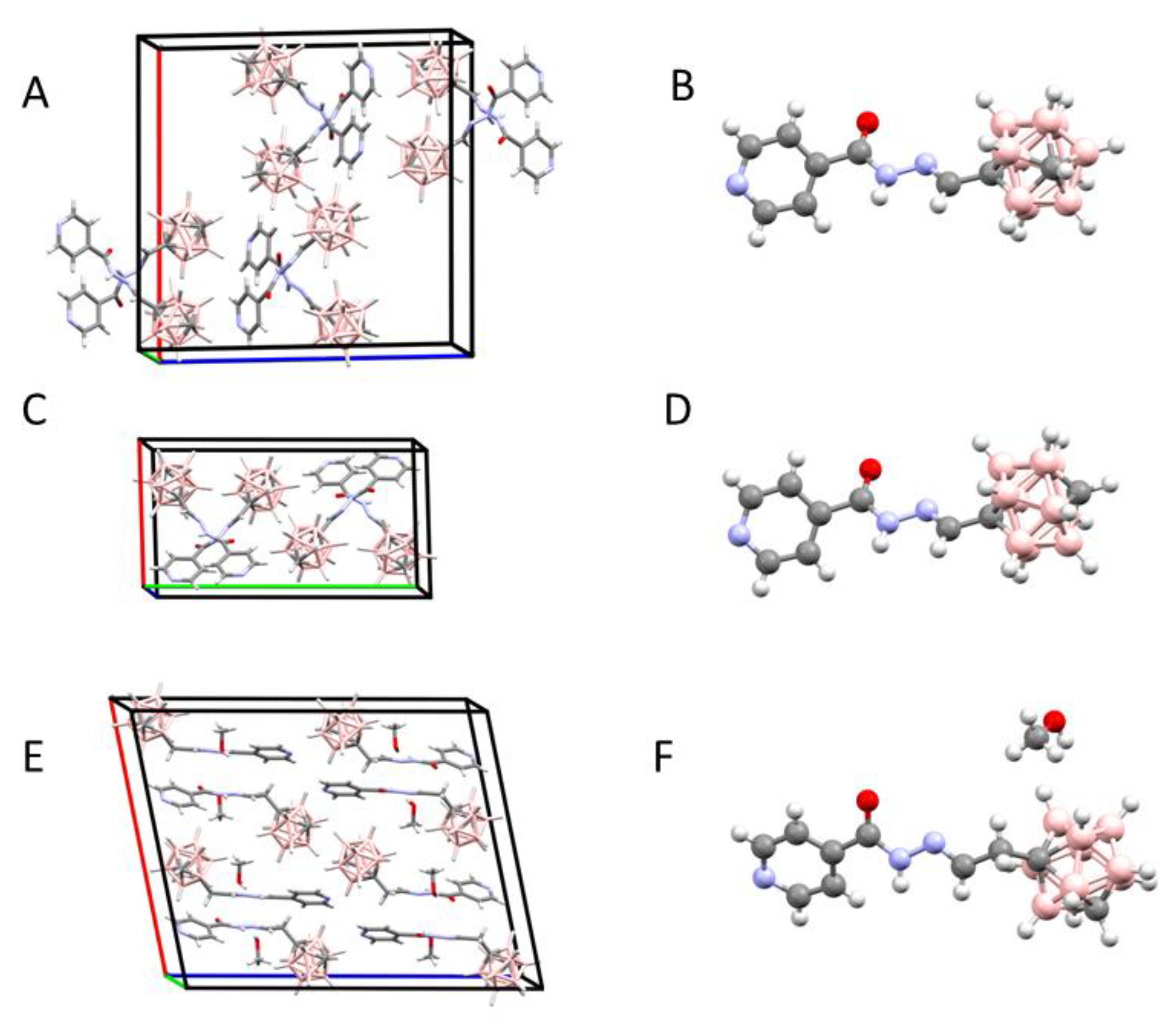
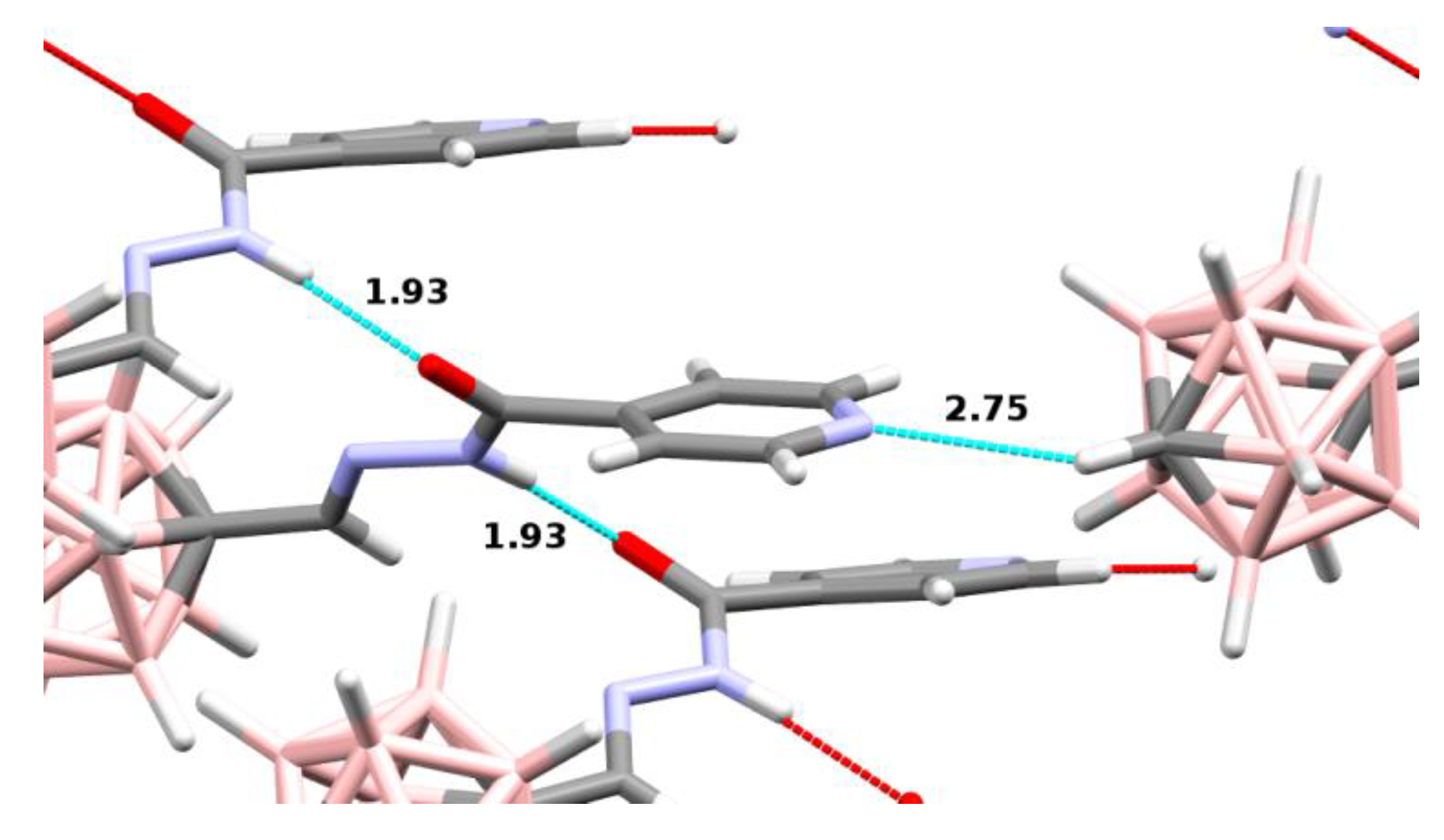
2.1.2. X-Ray Structure Analysis
2.2. Biological Investigation
2.2.1. Antimycobacterial Activity of Isoniazid-Carborane Cluster Conjugates
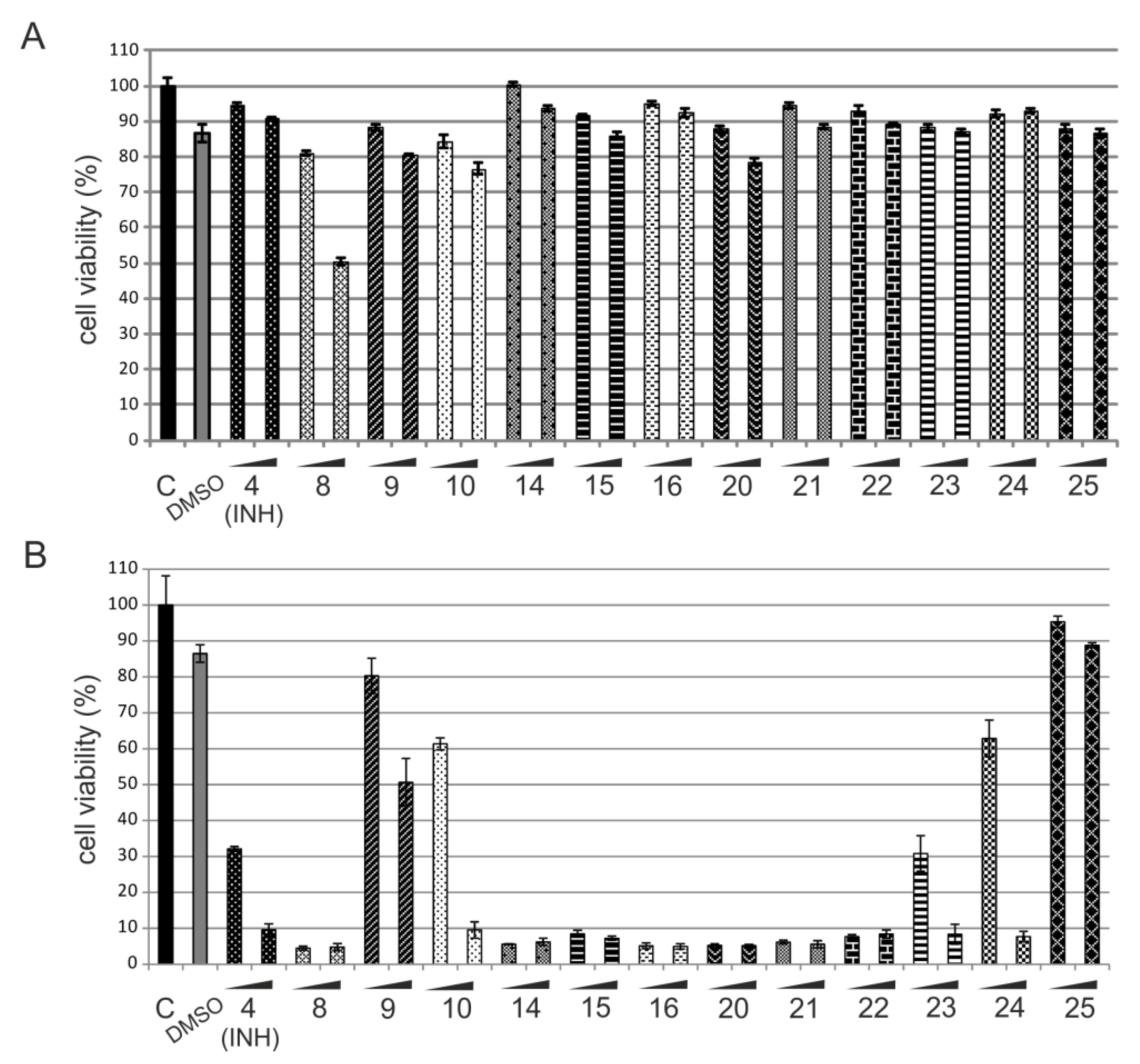
2.2.2. In Vitro Cytotoxicity Assay
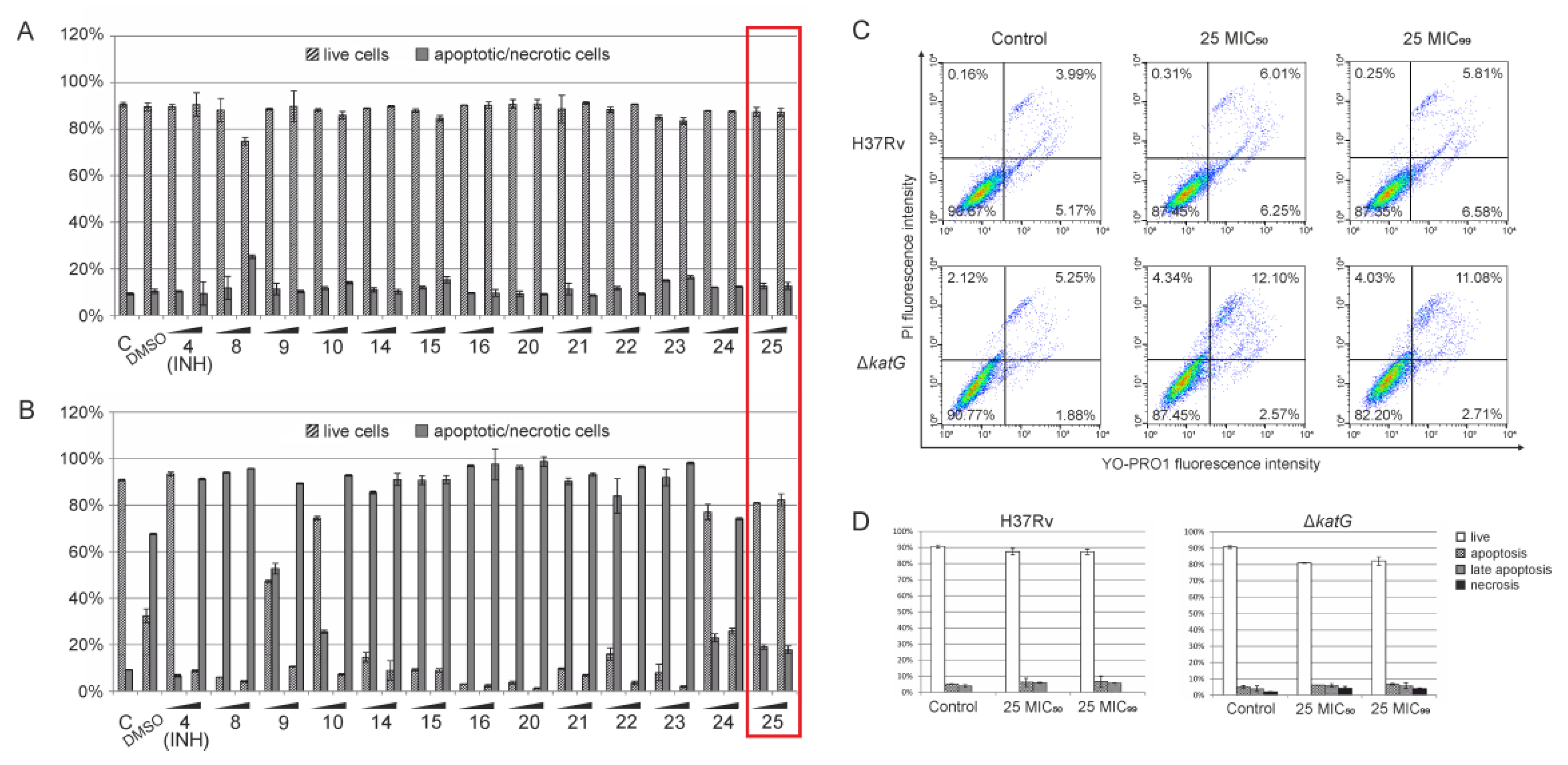
2.2.3. Apoptosis/Necrosis Assay by Flow Cytometry
2.3. Physicochemical Investigation
2.3.1. Lipophilicity Measurement
2.3.2. Parallel Artificial Membrane Permeability Measurement
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 1,12-dicarba-closo-dodecaborane-1-carboxylic acid hydrazide (3)
3.1.2. General Procedure for the Synthesis of Isonicotinyl Hydrazide 8–10
3.1.3. General Procedure for the Synthesis of Isonicotinyl Hydrazide 14–16
3.1.4. General Procedure for the Synthesis of Isonicotinyl Hydrazide 20–22
3.1.5. General Procedure for the Synthesis of Isonicotinyl Hydrazides 23–25
3.2. Biology
3.2.1. Bacterial Strain and Growth Conditions
3.2.2. M. Tuberculosis Susceptibility Tests
3.2.3. Construction of Gene Replacement Vector and Disruption of the Mtb katG Gene at its Native Chromosomal Loci
3.2.4. Cell Culture
3.2.5. MTT Analysis of Cell Viability
3.2.6. Determination of the CC50 Value
3.2.7. Apoptosis/Necrosis Analysis by Flow Cytometry
3.3. Physicochemical Investigation
3.3.1. Partition (P) and Distribution (D) Coefficient Measurements
3.3.2. Parallel Artificial Membrane Permeability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACP | acyl carrier protein |
| CC- | cytotoxic concentration |
| COX | cyclooxygenase |
| DPBS | Dulbecco′s phosphate-buffered saline |
| EMB | ethambutol |
| FAS | fatty acid synthase |
| FTIR | Fourier Transform Infrared Spectroscopy |
| HaCaT | human keratinocytes |
| HIV | human immunodeficiency virus |
| HPLC | high-performance liquid chromatography |
| INH | isoniazid |
| KatG | catalase-peroxidase |
| MDR | multidrug-resistant TB |
| MIC | minimum inhibitory concentration |
| MS | Mass Spectrometry |
| Mtb | Mycobacterium tuberculosis |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAD | nicotinamide adenine dinucleotide |
| NOESY | Nuclear Overhauser Effect Spectroscopy |
| OD | optical density |
| PAMPA | parallel artificial membrane permeability |
| PBS | phosphate-buffered saline |
| PZA | pyrazinamide |
| RIF | rifampicin |
| RP-HPLC | reversed-phase high-performance liquid chromatography |
| SI | selectivity index |
| TEMPO | 2,2,6,6-tetramethylpiperidine-1-oxyl |
| TB | tuberculosis |
| TLC | thin layer chromatography |
| TDR | totally drug-resistant TB |
| WHO | World Health Organization |
| XDR | extensively drug-resistant TB |
References
- Dela Cruz, C.S.; Lyons, P.G.; Pasnick, S.; Weinstock, T.; Nahid, P.; Wilson, K.C.; Thomson, C.C. Treatment of drug-susceptible tuberculosis. Ann. Am. Thorac. Soc. 2016, 13, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.d.S.; Chin, C.M.; Dos Santos, J.L. Advances in drug discovery of new antitubercular multidrug-resistant compounds. Pharmaceuticals 2017, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; Munoz-Torrico, M.; Durate, R.; Dalcomo, M.; D′Ambrosio, L.; Migliori, G.-B. New drugs and perspectives for new anti-tuberculosis regiments. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Bahuguna, A.; Rawat, D.S. An overview of new antitubercular drugs, drug candidates, and their targets. Med. Res. Rev. 2020, 40, 263–292. [Google Scholar] [CrossRef]
- Vilcheze, C.; Jacobs, W.R., Jr. The isoniazid paradigm of killing, resistance, and persistence in Mycobacterium tuberculosis. J. Mol. Biol. 2019, 431, 3450–3461. [Google Scholar] [CrossRef]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Machado, D.; Perdigao, J.; Ramos, J.; Couto, I.; Portugal, I.; Ritter, C.; Boettger, E.C.; Viveiros, M. High level resistance to isoniazid and ethionamide in multidrug resistant Mycobacterium tuberculosis of the Lisboa family is associated with InhA double mutations. J. Antimicrob. Chemother. 2013, 68, 1728–1732. [Google Scholar] [CrossRef] [Green Version]
- Vilcheze, C.; Jacobs, W.R., Jr. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitao, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correira, H.F.; Aires-de-Sousa, J.; et al. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-Q.; Zhang, S.; Zhao, F.; Gao, Z.; Feng, L.-S.; Lv, Z.S.; Xu, Z.; Wu, X. Isoniazid derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 133, 255–267. [Google Scholar] [CrossRef]
- De Oliveira, P.F.M.; Guidetti, B.; Chamayou, A.; Barres, C.A.; Madecki, J.; Kordulakova, J.; Mori, G.; Orena, B.S.; Chiarelli, L.R.; Pasca, M.R.; et al. Mechanochemical synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Molecules 2017, 22, 1457. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.O.; Cantos, J.B.; D’Oca, C.R.M.; Soares, K.L.; Coelho, T.S.; Piovesan, L.A.; Russowsky, D.; da Silva, P.A.; D’Oca, M.G.M. Synthesis and antimycobacterial activity of isoniazid derivatives from renewable fatty acids. Bioorganic Med. Chem. 2013, 21, 6910–6914. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.N. Carboranes, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Leśnikowski, Z.J. Challenges and opportunities for the application of boron clusters in drug design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Leśnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef]
- Goszczyński, T.M.; Fink, K.; Boratyński, J. Icosahedral boron clusters as modifying entities for biomolecules. Expert Opin. Drug. Discov. 2018, 18, 205–213. [Google Scholar] [CrossRef]
- Hey-Hawkins, E.; Vinas Teixidor, C. Boron-Based Compounds: Potential and Emerging Applications in Medicine, 1st ed.; Wiley: Oxford, UK, 2018. [Google Scholar]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sarosi, M.B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska, A.; Matuszewski, M.; Zwoliński, K.; Ignaczak, A.; Olejniczak, A.B. Insight into lipophilicity of deoxyribonucleoside-boron cluster conjugates. Eur. J. Pharm. Sci. 2018, 111, 226–237. [Google Scholar] [CrossRef]
- Ji, B.; Peacock, G.; Lu, D.R. Synthesis of cholesterol-carborane conjugate for targeted drug delivery. Bioorganic Med. Chem. Lett. 2002, 12, 2455–2458. [Google Scholar] [CrossRef]
- Różycka, D.; Leśnikowski, Z.J.; Olejniczak, A.B. Synthesis of boron cluster analogs of penicillin and their antibacterial activity. J. Organomet. Chem. 2019, 881, 19–24. [Google Scholar] [CrossRef]
- Marshall, J.; Hooton, J.; Han, Y.; Creamer, A.; Ashraf, R.S.; Porte, Y.; Anthopoulos, T.D.; Stavrinou, P.N.; Mclachlan, M.A.; Bronstein, H.; et al. Polythiophenes with vinylene linked ortho, meta, and para-carborane sidechains. Polym. Chem. 2014, 5, 6190–6199. [Google Scholar] [CrossRef] [Green Version]
- Wiesboeck, R.A.; Hawthorne, M.F. Dicarbaundecaborane(13) and derivatives. J. Am. Chem. Soc. 1964, 86, 1642–1643. [Google Scholar] [CrossRef]
- Yoo, J.; Hwang, J.-W.; Do, Y. Facile and mild deboronation of O-carboranes using cesium fluoride. Inorg. Chem. 2001, 40, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Svantesson, E.; Pettersson, J.; Olin, Å.; Markides, K.; Sjöberg, S. A kinetic study of the self-degradation of O-carboranylalanine to nido-carboranylalanine in solution. Acta Chem. Scand. 1999, 53, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Kononov, L.O.; Orlova, A.V.; Zinin, A.I.; Kimer, B.G.; Sivaev, I.B.; Bregadze, V.I. Conjugates of polyhedral boron compounds with carbohydrates. 2. Unexpected easy closo- to nido-transformation of a carborane-carbohydrate in neutral aqueous solution. J. Organomet. Chem. 2005, 690, 2769–2774. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, S.; Huang, P.; Chen, G. Synthesis of 1,2-bis(aminomethyl)1,2-dicarba-closo-dodecaborane hydrochloride its easy degradation research. Inorg. Chem. Commun. 2011, 14, 924–936. [Google Scholar] [CrossRef]
- Wyrzykiewicz, E.; Błaszczyk, A. New isomeric N-substituted hydrazones of 2-, 3- and 4-pyridinecarboxaldexydes and methyl-3-pyridylketone. J. Heterocycl. Chem. 2000, 37, 975–981. [Google Scholar] [CrossRef]
- Lane, C.F. Sodium cyanoborohydride—a highly selective reducing agent for organic functional groups. Synthesis 1975, 3, 135–146. [Google Scholar] [CrossRef]
- Hardie, M.J.; Raston, C.L. Crystalline hydrogen bonded complexes of O-carborane. CrystEngComm 2001, 3, 162–164. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Lepšik, M.; Horinek, D.; Havlas, Z.; Hobza, P. Interaction of carboranes with biomolecules: Formation of dihydrogen bonds. ChemPhysChem 2006, 7, 1100–1105. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Hnyk, D.; Lepšík, M.; Hobza, P. Interaction of heteroboranes with biomolecules: Part 2. The effect of various metal vertices and exo-substitutions. Phys. Chem. Chem. Phys. 2007, 9, 2085–2093. [Google Scholar] [CrossRef]
- Popiołek, Ł. Hydrazide-hydrazones as potential antimicrobial agents: Overview of the literature science 2010. Med. Chem. Res. 2017, 26, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmaniye, D.; Levent, S.; Karaduman, A.B.; Ilgýn, S.; Özkay, Y.; Kaplanikli, Z.A. Synthesis of new benzothiazole acylhydrazones as anticancer agents. Molecules 2018, 23, 1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, P.C.; Lima, L.M.; da Silva, K.C.M.; Leda, P.H.O.; de Miranda, A.L.P.; Fraga, C.A.M.; Barreiro, E.J. Synthesis and analgesic activity of novel N-acylarylhydrazones derivatives. Bioorganic Med. Chem. 2004, 12, 3149–3158. [Google Scholar]
- Dimmock, J.R.; Vashishtha, S.C.; Stables, J.P. Anticonvulsant properties of various acetylhydrazones, oxamoylhydrazones and semicarbazones derived from aromatic and unsaturated carbonyl compounds. Eur. J. Med. Chem. 2000, 35, 187–203. [Google Scholar] [CrossRef]
- Abdel-Aal, M.T.; El-Sayed, W.A.; El-Ashry, E.S.H. Synthesis and antiviral evaluation of some sugar arylglycinoylhyrdazones and their axadiazoline derivatives. Arch. Pharm. 2006, 339, 656–663. [Google Scholar] [CrossRef]
- Rollas, S.; Kucukguzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [Green Version]
- Neuman, W.; Xu, S.; Sarosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banrjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate induces potent and selective inhibition of cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, R.C.; Campbell, S.R.; Fairchild, R.G.; Kisliuk, R.L.; Micca, P.L.; Quneer, S.F.; Riordan, J.M.; Sedwick, W.D.; Waud, W.R.; Leung, A.K.W.; et al. Novel Boron-Containing, Nonclassical Antifolates: Synthesis and Preliminary Biological and Structural Evaluation. J. Med. Chem. 2007, 50, 3283–3289. [Google Scholar] [CrossRef]
- Adamska, A.; Rumijowska-Galewicz, A.; Ruszczynska, A.; Studzińska, M.; Jabłońska, A.; Paradowska, E.; Bulska, E.; Munier-Lehmann, H.; Dziadek, J.; Leśnikowski, Z.J.; et al. Anti-mycobacterial activity of thymine derivatives bearing boron clusters. Eur. J. Med. Chem. 2016, 121, 71–81. [Google Scholar] [CrossRef]
- Zhao, X.B.; Yu, S.W.; Wang, F.; Sacchettini, J.C.; Magliozzo, R.S. Hydrogen peroxide-mediated isoniazid activation catalyzed by Mycobacterium tuberculosis catalase-peroxidase (KatG) and its S315T mutant. Biochemistry 2006, 45, 4131–4140. [Google Scholar] [CrossRef]
- Crich, D.; Chen, C.; Hwang, J.-T.; Yuan, H.; Papadatos, A.; Walter, R.I. Photoinduced free radical chemistry of the acyl tellurides: Generation, inter-and intramolecular trapping, and ESR spectroscopic identification of acyl radicals. J. Am. Chem. Soc. 1994, 116, 8937–8951. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Choi, S.Y.; Hu, B.; Smith, J.A. Evidence for a higher oxidation state of manganese in the reaction of dinuclear manganese complexes with oxidants. Comparison with iron based Gif chemistry. Tetrahedron 1998, 54, 3367–3378. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Weng, L.; Smith, J.A. Binuclear manganese complexes as catalyst in selective and efficient oxidation of sulphides to sulfones. Tetrahedron Lett. 1998, 39, 7055–7058. [Google Scholar] [CrossRef]
- Braslau, R.; Anderson, M.O.; Rivera, F.; Jimenez, A.; Haddad, H.T.; Axon, J.R. Acyl hydrazines as precursor to acyl radical. Tetrahedron 2002, 58, 5513–5523. [Google Scholar] [CrossRef]
- Amos, R.I.J.; Gourlay, B.S.; Schiesser, C.H.; Smith, J.A.; Yates, B.F. A mechanistic study on the oxidation of hydrazides: Application to the tuberculosis drug isoniazid. Chem. Commun. 2008, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Amos, R.I.J.; Gourlay, B.S.; Yates, B.F.; Schiesser, C.H.; Lewis, T.W.; Smith, J.A. Mechanistic investigation of the oxidation of hydrazides: Implication for the activation of the TB drug isoniazid. Org. Biomol. Chem. 2013, 11, 170–176. [Google Scholar] [CrossRef]
- Zhao, X.B.; Yu, S.W.; Magliozzo, R.S. Characterization of the binding of isoniazid and analogues to Mycobacterium tuberculosis catalase-peroxidas. Biochemistry 2007, 46, 3161–3170. [Google Scholar] [CrossRef]
- Manjunatha, U.H.; Rao, S.P.S.; Kondreddi, R.R.; Noble, C.G.; Camacho, L.R.; Tan, B.H.; Ng, S.H.; Ng, P.S.; Ma, N.L.; Lakshminarayana, S.B.; et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015, 7, 269ra3. [Google Scholar] [CrossRef] [Green Version]
- Van de Waterbeemd, H.; Smith, D.A.; Beaumont, K.; Walker, D.K. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [Google Scholar] [CrossRef]
- Sangster, J. Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry; Wiley: New York, NY, USA, 1997; Volume 2. [Google Scholar]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a drug′s membrane permeability: A computational model validated with in vitro permeability assay data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 2000, 146, 1969–1975. [Google Scholar] [CrossRef] [Green Version]
- Dziadek, J.; Rajagopalan, M.; Parish, T.; Kurepina, N.; Greendyke, R.; Kreiswirth, B.N.; Madiraju, M.V. Mutations in the CCGTTCACA DnaA box of Mycobacterium tuberculosis oriC that abolish replication of oriC plasmids are tolerated on the chromosome. J. Bacteriol. 2002, 184, 3848–3855. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | C-N/H-N Distances (Å) | C-H-N Angle (°) |
|---|---|---|
| 14 | 3.33/2.39 | 142 |
| 15 | 3.49/2.75 | 124 |
| 21 | 3.50/2.65 | 134 |
| Compound | Mtb | katG | HaCaT | SI | |||
|---|---|---|---|---|---|---|---|
| MIC50 a (µM) | MIC99 b (µM) | MIC50 (µM) | MIC99 (µM) | CC50 c ± SD (µM) | Mtbd | katGe | |
| 4 (INH) | 0.073 | 0.36 | 1100 | 1500 | 725.80 ± 2.36 | 2016 | <1 |
| 8 | 44.70 | 100 | 150 | 300 | 86.64 ± 3.63 | <1 | <1 |
| 9 | 22.40 | 74.50 | 150 | 300 | 290.62 ± 1.45 | 4 | 1 |
| 10 | 29.80 | 74.50 | 150 | 300 | 160.97 ± 3.89 | 2 | <1 |
| 14 | 0.86 | 3.40 | 260 | 340 | 130.90 ± 3.46 | 38 | <1 |
| 15 | 10.30 | 17.20 | 510 | 680 | 305.45 ± 6.11 | 18 | <1 |
| 16 | 0.16 | 0.33 | 500 | >660 | 249.41 ± 0.56 | 756 | <1 |
| 20 | 0.082 | 1.60 | 160 | 240 | 81.50 ± 7.23 | 51 | <1 |
| 21 | 0.16 | 0.82 | 160 | 240 | 78.23 ± 0.71 | 95 | <1 |
| 22 | 0.16 | 1.60 | 160 | 330 | 80.19 ± 3.81 | 50 | <1 |
| 23 | 1.60 | 4.90 | 97.60 | 240 | 86.45 ± 4.59 | 18 | <1 |
| 24 | 0.81 | 3.25 | 81.30 | 160 | 105.41 ± 6.13 | 32 | <1 |
| 25 | 0.32 | 6.50 | 3.25 | 24.40 | 98.85 ± 1.48 | 15 | 4 |
| Compound | Partition/Distribution Coefficient | PAMPA a | |
|---|---|---|---|
| log P | log D7.4 | log Pe | |
| 4 (INH) | −1.00 ± 0.09 | - | −6.21 ± 0.10 |
| 8 | 0.67 ± 0.07 | - | −4.98 ± 0.12 |
| 9 | 1.11 ± 0.11 | - | −5.09 ± 0.07 |
| 10 | 0.99 ± 0.07 | - | −4.65 ± 0.09 |
| 14 | 2.03 ± 0.09 | - | −4.06 ± 0.09 |
| 15 | 2.28 ± 0.20 | - | −3.90 ± 0.17 |
| 16 | - | 0.67 ± 0.04 | −5.05 ± 0.30 |
| 20 | 1.26 ± 0.22 | - | −5.05 ± 0.09 |
| 21 | 1.66 ± 0.18 | - | −4.40 ± 0.10 |
| 22 | 1.30 ± 0.07 | - | −4.13 ± 0.07 |
| 23 | 0.64 ± 0.09 | - | −4.16 ± 0.05 |
| 24 | 0.66 ± 0.08 | - | −4.85 ± 0.06 |
| 25 | 1.15 ± 0.14 | - | −4.37 ± 0.04 |
Publisher′s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Różycka, D.; Korycka-Machała, M.; Żaczek, A.; Dziadek, J.; Gurda, D.; Orlicka-Płocka, M.; Wyszko, E.; Biniek-Antosiak, K.; Rypniewski, W.; Olejniczak, A.B. Novel Isoniazid-Carborane Hybrids Active In Vitro against Mycobacterium tuberculosis. Pharmaceuticals 2020, 13, 465. https://doi.org/10.3390/ph13120465
Różycka D, Korycka-Machała M, Żaczek A, Dziadek J, Gurda D, Orlicka-Płocka M, Wyszko E, Biniek-Antosiak K, Rypniewski W, Olejniczak AB. Novel Isoniazid-Carborane Hybrids Active In Vitro against Mycobacterium tuberculosis. Pharmaceuticals. 2020; 13(12):465. https://doi.org/10.3390/ph13120465
Chicago/Turabian StyleRóżycka, Daria, Małgorzata Korycka-Machała, Anna Żaczek, Jarosław Dziadek, Dorota Gurda, Marta Orlicka-Płocka, Eliza Wyszko, Katarzyna Biniek-Antosiak, Wojciech Rypniewski, and Agnieszka B. Olejniczak. 2020. "Novel Isoniazid-Carborane Hybrids Active In Vitro against Mycobacterium tuberculosis" Pharmaceuticals 13, no. 12: 465. https://doi.org/10.3390/ph13120465