The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions


1
Division of Hematology and Oncology, University of Illinois at Chicago, Chicago, IL 60612, USA
2
Division of Medical Oncology, Washington University School of Medicine, Saint Louis, MO 63110, USA
*
Authors to whom correspondence should be addressed.
Pharmaceuticals 2020, 13(11), 389; https://doi.org/10.3390/ph13110389
Submission received: 27 October 2020
/
Revised: 8 November 2020
/
Accepted: 12 November 2020
/
Published: 14 November 2020
(This article belongs to the Special Issue Malignant Glioma: Novel Therapeutic Strategies)
Abstract
:Background: Glioblastoma multiforme is a malignant intracranial neoplasm that constitutes a therapeutic challenge because of the associated high morbidity and mortality given the lack of effective approved medication and aggressive nature of the tumor. However, there has been extensive research recently to address the reasons implicated in the resistant nature of the tumor to pharmaceutical compounds, which have resulted in several clinical trials investigating promising treatment approaches. Methods: We reviewed literature published since 2010 from PUBMED and several annual meeting abstracts through 15 September 2020. Selected articles included those relevant to topics of glioblastoma tumor biology, original basic research, clinical trials, seminal reviews, and meta-analyses. We provide a discussion based on the collected evidence regarding the challenging factors encountered during treatment, and we highlighted the relevant trials of novel therapies including immunotherapy and targeted medication. Results: Selected literature revealed four main factors implicated in the low efficacy encountered with investigational treatments which included: (1) blood-brain barrier; (2) immunosuppressive microenvironment; (3) genetic heterogeneity; (4) external factors related to previous systemic treatment that can modulate tumor microenvironment. Investigational therapies discussed in this review were classified as immunotherapy and targeted therapy. Immunotherapy included: (1) immune checkpoint inhibitors; (2) adoptive cell transfer therapy; (3) therapeutic vaccines; (4) oncolytic virus therapy. Targeted therapy included tyrosine kinase inhibitors and other receptor inhibitors. Finally, we provide our perspective on future directions in treatment of glioblastoma. Conclusion: Despite the limited success in development of effective therapeutics in glioblastoma, many treatment approaches hold potential promise including immunotherapy and novel combinational drugs. Addressing the molecular landscape and resistant immunosuppressive nature of glioblastoma are imperative in further development of effective treatments.
1. Introduction
Glioblastoma multiforme is the most aggressive intracranial tumor; histologically considered as a malignant glioma arising from an astrocytic lineage (WHO classification: astrocytoma grade IV), and it represents the most common malignant primary brain tumor in adults [1,2]. Despite the low incidence rate of glioblastoma compared to other cancer types, it portends an extremely poor prognosis with a median overall survival (OS) of approximately 1 year, and a relative survival rate at 5 and 10 years approximately (5% and 3%, respectively) [3,4]. Glioblastoma is considered a privileged tumor because of its isolation from the surrounding structures, and the presence of the blood brain barrier (BBB) which impedes the passage of several immune cells and chemotherapeutics [5]. In addition, the heterogeneous genetic and molecular landscape of glioblastoma constitutes a challenge in the face of developing effective therapies, which is reflected by the lack of drug approval in the past decade [6]. The cornerstone of glioblastoma management relies on maximal surgical resection of the tumor with concurrent chemoradiation using the alkylating agent temozolomide (TMZ) followed by adjuvant TMZ for a total of 6 months according to the landmark trial by Stupp et al. [7]. The other treatment modality that has shown improved OS in glioblastoma patients is the addition of tumor treating field (TTF) to the current standard of care (Stupp protocol), which is a device worn by the patient on the scalp, and works by delivering alternating electrical fields that disrupt the microtubules in the mitotic spindle leading to tumor cell death [8]. However, this treatment has proved to be challenging for patients given the low compliance rates with its use and high cost [9]. The addition of bevacizumab which is a vascular endothelial growth factor (VEGF) inhibitor, in newly diagnosed and recurrent glioblastoma-selected patients demonstrated modest improvement in progression-free survival (PFS) and better quality of life, but was associated with a high rate of adverse events without an improvement in OS [10,11,12]. As the majority of glioblastoma ultimately recur during or after treatment, it is imperative to develop therapeutic drugs that could offer clinical benefit to these patients to improve survival, and decrease disease associated morbidity and complications. This unmet need has led to extensive preclinical research to further characterize the molecular and genetic alterations that control tumor growth, as well as the interaction between the immune system and glioblastoma in order to identify candidate targets for effective drug development. This comprehensive effort in the last decade has substantially expanded our understanding of glioblastoma and has been previously reviewed in depth [13,14]. Nevertheless, the limited drug approval for glioblastoma relative to the novel investigational therapies that have been tested, and the modest improvement in OS achieved in the past decade demonstrate the obligation for further research in the field of neuro-oncology to develop new therapies that can improve patient outcomes as outlined in Figure 1. Recently, there have been promising novel drugs including targeted molecular therapy and immunotherapy that have established a possible role in anti-tumor response in glioblastoma. Moreover, enhanced drug delivery across BBB and combination treatment modalities seem to hold a prospective role in the treatment of glioblastoma.
In this review, we discuss the distinct factors that are essential in understanding the challenges in the treatment of glioblastoma. Second, we offer a review of potential novel treatments including (immunotherapy and targeted therapy), and the rationale behind their use. Finally, we present our prospective on future directions in the pursuit of novel effective treatments for glioblastoma.
2. Exploiting the Blood-Brain Barrier and the Immune System against Glioblastoma: The Wrong Notion of the Immune Desert in Glioblastoma
Perhaps the most consequential discovery that has shifted the paradigm of cancer treatment and substantially led to improved outcomes in multiple cancer types is the discovery of the immune system role in preventing tumor growth and killing cancer cells which has coined the term “anti-tumor immunity” [15]. For example, checkpoint receptors are membrane-bound proteins that are found on malignant cells as well as immune cells such as T-cell subsets, natural killer (NK) cells, antigen presenting cells (APC) including macrophages and dendritic cells. These checkpoint receptors, are usually upregulated and highly expressed in the tumor microenvironment (TME) of several tumor types, and when bound to their respective ligands are responsible for negative regulation of immune cells leading to increased immune evasion by the tumor through the disruption of innate and adaptive anti-tumor function [16,17,18]. This has resulted in the development of several monoclonal antibodies directed at checkpoint receptors such as cytotoxic-associated lymphocyte antigen-4 (CTLA-4) and programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) [19]. This class of medication known as immune checkpoint inhibitor (ICI) has demonstrated a significant prolonged PFS and OS in several tumors including melanoma, lung, genitourinary, liver, and breast cancers [20,21,22,23,24,25,26,27]. Other checkpoint inhibitors are currently being investigated such as inhibitors of T-cell immunoglobulin and mucin-domain containing-3 (TIM3), lymphocyte activation gene-3 (LAG3), and indoleamine 2,3-dioxygenase-1 (IDO1) [28,29]. Another example that demonstrated the utility of immune derived cells in the treatment of cancer was the use of bioengineered chimeric antigen receptor loaded on autologous T-cells (CAR-T), which have shown efficacy and improved outcomes in relapsed acute lymphoblastic leukemia, refractory diffuse large-B cell lymphoma (DLBCL), and relapsed follicular lymphoma [30,31]. Moreover, the efficacy of oncolytic virus therapy such as talimogene laherparevec (TVEC) in melanoma and autologous dendritic vaccination therapy in prostate cancer have further extended the advent of immunotherapy in the treatment of cancer [32,33]. As such, several immunotherapies that recruit the immune system against cancer cells have been investigated in different tumor types including glioblastoma. To this end, glioblastoma has been regarded for a long time as an isolated tumor encased by BBB, which makes it devoid of active immune cells. This notion has been abandoned in the past decade after several preclinical and clinical studies demonstrated the role of the immune system in the TME of intracranial tumors including glioblastoma. Comprehensive analysis of the role of the immune system in glioblastoma is out of the scope of this review and has been summarized in detail elsewhere [14,34]. To better explain the current challenges in implementing effective immunotherapies for glioblastoma, it is imperative to understand the cross-talk between malignant cells of glioblastoma and immune cells and the immunosuppressive nature of the tumor. Several factors govern this interaction, and can be classified into the following: (1) factors related to BBB; (2) factors related to the immunosuppressive microenvironment; (3) factors related to tumor heterogeneity and genetic signature of glioblastoma subtypes; and (4) extrinsic factors related to the modulating effect of previous systemic treatment.
2.1. Factors Related to BBB
The blood-brain barrier (BBB) forms a dynamic interaction of endothelial cells, pericytes, microglia, astrocytes, and is responsible for the integrity of the central nervous system [35]. BBB plays an essential role in regulating the passage of water, ions, immune cells, nutrients, and therapeutic drugs through its complex physical barrier manifested by tight junctions and enzymatic–metabolic pathways that control transportation of different molecules. This can hinder the delivery of therapeutic drugs into the brain compartment as it is estimated that BBB restricts the penetration of 98% of small molecules and 100% of large molecules [36]. In glioblastoma, the integrity of the BBB is disrupted, which leads to alterations involving the vascular bed. The aggressive nature of glioblastoma leads to invasion of endothelial cells and breakage of tight junctions which results in a breach in the BBB [37,38]. Nevertheless, this disruption is not sufficient to allow increased concentration of therapeutic compounds in the tumor bed. Moreover, glioblastoma invasion into the surrounding structures is associated with angiogenesis because of hypoxia-induced VEGF formation, which though could potentially increase drug availability via the tumor vasculature, it also leads to pathological alterations in the BBB and suboptimal drug concentration in tumor foci outside the disrupted region [39]. The constellation of pathophysiological alterations in the vasculature bed associated with glioblastoma has coined the term blood-brain tumor barrier (BBTB) because of its unique features compared to BBB. To this end, analyzing the structure and changes associated with BBTB can aid in effective treatment development for glioblastoma and is an area for active research [40].
Of importance to immunotherapy, BBB plays an essential role in regulating immunosurveillance and restricting immune cell passage into the central nervous system (CNS), which could limit the efficacy of many novel drugs that work by recruiting the immune system to attack cancer cells. However, the insight gained from other CNS diseases on the ability of immune cells to cross BBB in certain autoimmune and inflammatory diseases, could help further research on exploiting this vulnerability in the administration of novel immunotherapies in glioblastoma [41,42,43].
To circumvent these challenges, several methods are under ongoing investigation to overcome the BBB and BBTB and achieve sufficient delivery of therapeutic compounds inside glioblastoma. One promising method is the delivery of drugs through nanoparticles which allow for the individual units loaded with drug to be carried to glioblastoma cells [44]. For example, nano-immunoconjugates (anti CTLA-4 and anti PD-1) on natural biopolymer scaffold were able to induce immune response in glioma cells and lead to a significant prolonged survival in mice with intracranial GL261 glioblastoma [45]. Similarly, another group of investigators showed that the delivery of small interfering RNAs (siRNA) against both epidermal growth factor receptor (EGFR) and PD-L1 through solid lipid nanoparticles in glioblastoma mouse models, was able to decrease the growth of glioblastoma and prolong mouse survival [46].
Convection-enhanced delivery (CED) is another promising method that has shown to encourage preclinical and clinical results [47]. This method relies on establishing a pressure gradient during interstitial infusion of the pharmaceutical compound to augment its distribution into the intracranial tumor to achieve enhanced local drug delivery. One recent example of the clinical benefit associated with CED is the work by Desjardins et al. that showed mild increased OS in patients with recurrent glioblastoma who received intertumoral recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) versus matching historical control (median OS 12.5 months vs. 11.3 months, respectively) [48]. Nevertheless, further research is essential to evaluate the safety and efficacy of CED to better identify the best drugs to use with this method as well as unifying treatment protocols and ascertaining the delivery of the infused molecule into tumor bed.
Moreover, it is important to note that some drugs can have an effect on glioblastoma despite not being able to penetrate BBB into the tumor. For example, bevacizumab, which is a VEGF inhibitor that has shown improved PFS and quality of life in glioblastoma patients, prevents angiogenesis within the vasculature compartment without the need of being present inside the tumor cells [10,11,12]. Another example is the use of ICI that can restore anti-tumor efficacy by blocking checkpoint receptors present not only in the tumor milieu, but also in the benign peripheral lymphatic system [49]. This warrants further investigation on novel treatments that could have a potential effect on glioblastoma without the need to penetrate BBB.
2.2. Factors Related to Glioblastoma Immunosuppressive Microenvironment
The long-standing historical notion of the CNS being considered an immune desert because it is devoid of effective resident immune cells has been challenged by rigorous research which demonstrated that the CNS has specific lymphatic system and is occupied by immune cells with unique characteristics compared to its counterparts in other organs [50].
The paramount advances in cancer treatment with the introduction of ICI as well as other immunotherapies have drastically shifted the focus in cancer treatment on exploiting pathways involved in host immune defense mechanisms to fight cancer cells “anti-tumor immunity.”
Although immunotherapies have shown significantly improved outcomes in different solid and hematological malignancies, the outcomes in glioblastoma have been mild to moderate at best. This stems from the fact that TME of glioblastoma employs an immunosuppressive effect despite the abundance of a variety of immune cell subsets [14,51]. For example, despite the presence of a functional lymphatic system in the CNS that provides the potential for tumor antigen to prime cognate T cells that could mediate anti-tumor immunity, glioblastoma exerts an inhibitory effect on the functionality of T cells through several mechanisms [52,53]. Moreover, glioblastoma can disrupt the function of non-resident T cells beyond the tumor bed and can affect potential effective peripheral T-cell populations and lead to their entrapment in the bone marrow because of the loss of spingosine-1-phosphate receptor 1 (S1P1) [54]. By contrast, glioblastoma allows the expansion of specific T-cell subsets such as T-regulatory (Treg) cells which are responsible for immune tolerance and promotion of tumor growth [55]. In addition, natural killer (NK) cells which are innate-like immune cells equipped with potent cytotoxic activity against tumor cells are suppressed in glioblastoma [56].
Additionally, tumor-associated macrophages (TAM) and microglia are essential subtypes of immune cells that constitute the majority of cellular mass in glioblastoma (30–50%) [57]. These cells are considered antigen presenting cells (APC) and are responsible for the presentation of tumor-associated antigens to be recognized by effector T cells [57]. TAMs constitute the majority of APC in glioblastoma while microglia account for an additional 15% of APCs [58,59]. Both TAMs and microglia are implicated in gliomagenesis in glioblastoma through several mechanisms including immune suppression and secretion of several soluble factors such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) that can further exacerbate immunosuppressive state [60,61,62]. Furthermore, antigen presentation processes are impaired in glioblastoma because of impaired upregulation of major histocompatibility complex (MHC) class II [63].
The other important factor that is associated with an immunosuppressive microenvironment in glioblastoma is the increased expression of checkpoint receptors such as PD-L1, IDO, TIM3 on T-cells and TAMs [64,65,66,67,68,69,70]. Similarly, glioblastoma favors the expression of several inhibitory molecules such as signal transducer and activator of transcription 3 (STAT3) and Fas ligand (FasL) which aggravate immune inhibitory signals and promote T-cell apoptosis, respectively [71,72].
For the abovementioned reasons, the negative impact of glioblastoma on the immune microenvironment which favors the presence of dysfunctional and deregulated immune cell subpopulations, constitute a major challenge on the implementation of effective immunotherapies in the treatment of glioblastoma. Therefore, efforts are focused on exploiting existing vulnerabilities in the TME of glioblastoma to reactivate the immune system against tumor cells. For example, TAMs display plasticity which refers to their ability of switching their phenotypic features leading to a change in their function. As such, this plasticity could be exploited to selectively express a phenotype that favors macrophage type-1 (M1) which is a pro-inflammatory cell that can boost host immunity versus macrophage type-2 (M2) which is considered pro-tumorigenic [73]. Similarly, the increased expression of checkpoint receptors in TME of glioblastoma can be targeted with the use of combinational ICI to rescue effector T-cells and to restore immune surveillance in glioblastoma.
2.3. Factors Related to Tumor Heterogeneity and Genetic Signature of Glioblastoma Subtypes
The common definition of glioblastoma by World Health Organization classification as a high-grade glioma-IV offers limited insight into the genetic heterogeneity of glioblastoma. Genomic characterization by The Cancer Genome Atlas (TCGA) research group and the comprehensive molecular analysis helped delineate three distinct tumor-intrinsic transcriptional glioblastoma subtypes including proneural, mesenchymal, and classical [74,75]. This classification relies on the predominant genetic signature associated with each subtype. For example, while the proneural subtype of glioblastoma is associated with amplification in platelet-derived growth factor receptor alpha (PDGFRA), the mesenchymal subtype can be associated with alteration in neurofibromatosis type 1 (NF1), and classical subtype is associated with amplification in EGFR [76]. This is of interest given the evidence that specific expression patterns due to amplification in PDFGRA and EGFR could serve as prognostic biomarkers [77,78]. In addition, some specific immune cell types such as TAM have been shown to have higher expression of their related genes in the mesenchymal subtype of glioblastoma [79].
The genetic heterogeneity appears to reflect distinct glioblastoma immune subsets based on the molecular signature [80]. For example, glioblastoma with wild-type isocitrate dehydrogenase (IDH) status is associated with a higher tumor infiltrative lymphocyte (TIL) and higher PD-L1 expression compared to IDH mutated glioblastoma [81]. Similarly, there is an increased PD-L1 expression on both resident T cells and circulating monocytes in glioblastoma with PTEN loss [82]. In addition, the glioblastoma mesenchymal subtype that is associated with NF-1 and RB-1 mutations can have significantly increased TIL [83]. This suggests that specific genetic alterations can be associated with an abundance of immune cells and increased expression of checkpoint receptors which could help select subgroups of glioblastoma patients who could derive a better response to immunotherapy. However, clinical correlation is still lacking with some evidence showing no predictive value of increased TIL or checkpoint receptors in glioblastoma subsets with improved response to ICI [84].
Furthermore, tumor mutation burden (TMB) status that estimates the frequency of mutations that are present per megabase of malignant cell DNA correlates with response observed in several solid malignancies during treatment with ICI. This is the result of increased neoantigen formation and presence of new mutant proteins that are immunogenic and can induce anti-tumor response [85]. As such, glioblastoma patients with high TMB could reflect a subpopulation that can have better response to immunotherapies such as ICI compared to glioblastoma patients with low TMB. This is of importance given that glioblastoma patients have low TMB at baseline which can limit anti-tumor immunity and explain weak response to immunotherapy [86]. Interestingly, there is a subset of patients with glioblastoma that can have hypermutated status with high TMB after treatment with TMZ and another subset of patients who have germline mutations leading to hypermutated status [87,88,89,90,91,92].
Therefore, further characterization of immune abundance based on gene expression of glioblastoma can prove to be helpful in identifying selected patients as immunotherapy continues to assume a critical role in the treatment of cancer. The correlation between glioblastoma genetic subtypes and tumor response to immunotherapy is an open area for research as deeper understanding is required to evaluate the effect of molecular variations on tumor cell survival and sensitivity or resistance to immunotherapy.
2.4. Extrinsic Factors Related to the Microenvironment Modulating Effect of Previous Glioblastoma Treatments
Several iatrogenic factors can affect and lead to alteration of glioblastoma on the genomic level as well as TME. As mentioned in the previous section, treatment with TMZ can increase hypermutated status, which is associated with higher neoantigen formation [87,88,89,90]. In addition, TMZ which is known to cause lymphopenia during treatment, has been shown to have an immunomodulatory effect which could enhance anti-tumor immune response [93]. In contrast, steroids which are commonly used in glioblastoma patients to treat tumor associated vasogenic edema and mass effect can lead to lymphodepletion and favor a more immunosuppressive environment which could hinder efficacy of novel drug effects such as immunotherapy [94,95]. Moreover, the use of glucocorticoids before or during treatment with ICI in different solid tumors is associated with a decreased response and worse OS [96]. This is of importance, as most patients with glioblastoma are treated with dexamethasone and are expected to have a degree of persistent immunosuppression that could counteract the efficacy of immunotherapies.
Additionally, bevacizumab which is used in the treatment of primary and recurrent glioblastoma can lead to diminished infiltrative immune suppressive cells, low PD-1/PD-L1 expression and increased cytotoxic T cells which can alter the efficacy of immunotherapies [97]. To this end, highlighting the influence of previous medications on TME of glioblastoma and subsequent treatments such as immunotherapy, is imperative when evaluating the efficacy of novel glioblastoma therapeutics [98].
3. Immunotherapy in the Treatment of Glioblastoma
Immunotherapy entails several pharmaceutical compounds that have different mechanism of action and can be categorized into: (1) Monoclonal antibodies (ICI) that target checkpoint protein receptors (CTLA-4, PD-1, PD-L1, TIM3, IDO, LAG3); (2) adoptive transfer of bioengineered immune cells which are designed to destroy malignant cells such as CAR-T cell therapy and CAR-natural killer therapy (CAR-NK); (3) cancer vaccines that use mutant tumor peptides or immune cell-based components to induce anti-tumor immunity; and (4) oncolytic viruses that preferentially destroy malignant cells through direct and indirect mechanisms by engaging effector immune cells against cancer cells.
3.1. Immune Checkpoint Inhibitors in Glioblastoma
Immune checkpoint inhibitors (ICIs) have revolutionized the management of cancer in the last decade since the approval of CTLA-4 inhibitor (ipilimumab) in 2011, which was followed by approval of other ICI (PD-1/PD-L1 inhibitors). As mentioned previously, ICI can reinvigorate anti-tumor immune response by blocking checkpoint receptor molecules which are responsible for an immune-inhibitory effect that leads to immune evasion of cancer cells. The clinical efficacy of this class of medication has been echoed by durable improved outcomes including prolonged OS and PFS in several solid tumors including melanoma, lung, urothelial, breast, and liver cancers. Following the successful implementation of ICI in several solid malignancies, further research was conducted to evaluate their efficacy in other tumor types including glioblastoma. As the main target of ICI is the checkpoint receptor in TME, the presence of these receptors appears to be essential to enhance the ability of these drugs to interrupt checkpoint protein interaction with the respective ligand receptor which eventually lead to reactivation of anti-tumor response [99]. This was reflected by several clinical trials which showed that the expression of a checkpoint receptor (PD-L1) correlated with improved outcomes in patients with metastatic lung and urothelial cancer who received PD-1/PD-L1 inhibitors (though this correlation can vary depending on the choice of ICI and tumor type) [100]. As such, a recent study demonstrated an increased expression of the checkpoint receptor PD-L1 in 90% of 135 patients who were diagnosed with glioblastoma [66]. This serves as a proof of concept of a potential role of ICI in treatment of glioblastoma because of the presence of targetable checkpoint receptors in TME. Consequentially, the presence of checkpoint receptors in glioblastoma appears to amplify immunosuppression and correlate with worse prognosis. For example, a study by Nduom et al. showed that expression of PD-L1 in 94 glioblastoma patients correlated with increased risk of death (hazards ratio [HR] = 1.54, p = 0.0343) [101]. Similarly, high expression of other checkpoint receptors such as CTLA-4, TIM3, and IDO was found to correlate with negative outcomes and decreased survival [70,102,103].
The enthusiasm to carry out the outstanding results achieved in cancer management with ICI, has not been as successful as anticipated in glioblastoma trials. This was demonstrated in a pivotal phase III randomized controlled trial (CheckMate-143), which compared the safety and efficacy of nivolumab (PD-1 inhibitor) versus bevacizumab in patients with recurrent glioblastoma who were previously treated with TMZ and radiotherapy at initial diagnosis [104]. This study demonstrated a lower objective response rate, lower PFS, similar median OS, and higher treatment-related adverse events in patients who received nivolumab compared to those who received bevacizumab [104]. Another phase-3 trial evaluating the addition of nivolumab to concurrent chemoradiation (TMZ and radiotherapy) in newly diagnosed MGMT-unmethylated glioblastoma failed to demonstrate improved outcomes [105]. Similarly, another PD-1 inhibitor (pembrolizumab) did not achieve improved PFS at 6 months or improved OS in a phase II study evaluating pembrolizumab versus pembrolizumab and bevacizumab in recurrent glioblastoma [106]. Despite the disappointing results of these clinical trials, they provided important clues which further helped in designing new trials testing different approaches of ICI treatment in glioblastoma.
One of the factors that is proposed to contribute to the decreased response of ICI is its inability to reach tumor cells. This is of importance as the BBB allows passage of limited small size compounds less than 400 Da, whereas nivolumab and pembrolizumab have a molecular weight of 146 kDA and 149 kDa respectively [107,108,109]. As such, there are several groups working on enhancing the delivery of these pharmaceuticals through the BBB into the TME. Some of this work focuses on nanoparticle delivery systems as described before [45], and other groups are investigating combining ICI with radiotherapy to increase the disruption of BBB (ClinicalTrials.gov Identifier: NCT02311582).
Another factor that appears to be fundamental for the efficacy of ICI in treatment of glioblastoma is the timing of administration. Most of the trials to date have examined efficacy of ICI either in the recurrent setting or in first line setting in combination with concurrent chemoradiation after surgical resection of the tumor. Therefore, it was tempting to evaluate the use of ICI prior to surgery as they have demonstrated improved outcomes in the neo-adjuvant setting in different tumors such as breast cancer and melanoma [110]. Several advantages can be achieved with neo-adjuvant administration of ICI including higher T-cell expansion in these patients compared to those who receive ICI after surgical resection of the tumor [111]. This is hypothesized to be the result of a large tumor bulk presence that contains potential high load of immunogenic antigens, which can elicit a more robust anti-tumor response in the presence of ICI. As such, the efficacy of ICI in the post-surgical setting could attenuate the anti-tumor immune response because of the unavailability of tumor antigens after surgical resection of the tumor. The advantage of neo-adjuvant ICI has been validated in recurrent glioblastoma in several studies. Cloughesy et al. randomized 35 patients with recurrent glioblastoma to receive either neo-adjuvant pembrolizumab (one single dose intravenously 2 weeks prior to surgery) and/or adjuvant ICI [112]. The study highlighted the role of neo-adjuvant ICI in upregulating T-cell populations and interferon-γ-related gene expression compared to subjects who did not receive neo-adjuvant pembrolizumab [112]. This also correlated with an observed median OS of approximately 14 months in patients who received neo-adjuvant pembrolizumab versus 7.5 months in those who did not receive ICI prior to surgical resection [112]. Similarly, another phase-II trial replicated similar findings in patients with recurrent glioblastoma who received neo-adjuvant nivolumab prior to surgical resection [113]. The administration of nivolumab before surgery was associated with a higher immune cell activity in the TME, higher tumor-infiltrating lymphocytes, and enhanced expression of chemokine transcripts, however, this trial did not result in prolonged OS [113]. To this end, the sequence of ICI administration in regards to surgery in glioblastoma appears to be imperative for the enhancement of the functional immune landscape leading to a substantial tumor response and seem to be promising, which led to several clinical trials that are ongoing to address this approach.
Furthermore, the potential role of ICI in the treatment of glioblastoma appears to be significant in specific subpopulations. For example, tumor mutation burden (TMB) and mismatch repair deficiency (MRD) reflect a state in which a tumor is loaded with a high number of mutated proteins and neoantigens given the defective DNA repair mechanisms. This can convert the tumor into a hotspot as a target for anti-tumor immunity mediated by ICI as higher number of immunogenic antigens are expected to elicit a strong immune response [114,115]. Despite the low frequency of both TMB and MRD in glioblastoma, some patients have been described to have defective DNA repair mechanisms leading to an immune permissive TME, which provides an advantage for ICI in priming an anti-glioblastoma immune response [116,117]. To illustrate this, Bouffet et al. reported a durable response with the use of nivolumab in two pediatric siblings with recurrent glioblastoma who had biallelic MRD [92]. Similarly, another report demonstrated a durable response to pembrolizumab in an adult patient with recurrent glioblastoma who had germline polymerase DNA epsilon (POLE) deficiency which correlated with a high tumor mutation load [91]. To this end, it is important to highlight that hypermutation status and high tumor mutation burden do not confer a better response when they develop after the administration of TMZ. This has been demonstrated in a study by Touat et al. that analyzed hypermutation and TMB status in 10,294 gliomas and found that hypermutation status post-TMZ was associated with lower abundance of tumor infiltrative lymphocytes and worse outcomes [118]. This highlights the difference between the hypermutation induced by germline deficiency in DNA repair mechanisms versus hypermutation induced by post-TMZ use. In addition, specific mutations in glioblastoma have not been reported to be associated with improved response when treated with ICI as demonstrated by Zhao et al. who reported increased and prolonged response to PD-1 inhibitors in glioblastoma patients who had B-raf murine sarcoma (BRAF) and protein tyrosine phosphatase non-receptor type 11 (PTPN11)-activating mutations [84]. This collective evidence points toward more individualized trial design and further investigation into genomic characteristics to identify potential patients who could be candidates for ICI therapy.
Current efforts are focused on combining ICI with different treatment modalities in glioblastoma such as the novel checkpoint inhibitors of LAG3 and TIM3 [119,120]. Interestingly, a variety of molecules and receptors have emerged recently as potential targets in combination with ICI such as TGF-β inhibitors, colony-stimulating factor-1 ligand (CSF-1) inhibitors, and CD47 blockade [121,122]. Other interesting approaches studying the effect of epigenetic in glioblastoma have been suggested such as the use of micro RNA (miRNA) modulation effect on the TME as a biomarker to predict response and resistance to ICI to guide treatment and has been reviewed [123]. The ongoing research on identifying selected patient population, identifying the right sequence of treatment, and combining ICI with other investigational therapeutics seem to hold a high promise in further advancement of glioblastoma care Figure 2.
3.2. Adoptive Cell Therapy in Glioblastoma
Adoptive cell therapy in cancer treatment relies on redirecting immune cells to engage with specific tumor antigens based on bioengineered chimeric antigen receptors expressed in autologous T cells [124]. By targeting tumor antigens, bioengineered T-cells are able to initiate an augmented anti-tumor response through the activation of a variety of effector immune cells. This approach has shown promising results in hematological malignancies, which has generated enthusiasm toward applying this new approach in glioblastoma especially given that there was a signal of response observed in patients with secondary CNS lymphoma who received CAR-T therapy [125]. This therapy was initially investigated in glioblastoma to evaluate safety due to the known severe adverse event of immune effector-associated neurological syndrome (ICANS) which can increase toxicity and morbidities in glioblastoma patients who already have neurologic deficits. To this end, Brown et al. reported the safety of administering CAR IL13Rα2 expressed on cytotoxic T-lymphocyte clones in patients with recurrent glioblastoma [126]. The latter work has led to a report of a significant clinical response in a patient with recurrent glioblastoma who was treated with IL13BBζ–CAR T cells with sustained disease control that lasted 7.5 months but later progressed to four different new locations [127]. Other investigators explored targeting different glioblastoma-related tumor cell receptors such as epidermal growth factor receptor variant-III (EGFRvIII) and human epidermal receptor-2 (HER2) as they are commonly expressed on glioblastoma cells. These studies demonstrated safety of EGFRvIII-CAR and HER2- CAR-T infusions and an active anti-tumor effect, however, this was yet to demonstrate a clinical benefit with an improved survival [128,129,130]. Another approach of adoptive cell therapy that has just entered early clinical investigation involves the administration of bioengineered NK cells [131]. The advantage of using NK cells compared to CAR-T cell therapy relies on the fact that NK cells are leveraged to retain high cytotoxic effect against tumor cells irrespective of specific antigen expression which could lead to an increased efficacy against antigenically heterogeneous tumor cells [131]. Nevertheless, the immature results of CAR-cell-based therapy in glioblastoma require further evidence and investigation to support safety and efficacy in glioblastoma. Among the important factors to address during the ongoing research on CAR-cell therapy in glioblastoma include: finding a common targetable antigen that is universally expressed on tumor cells and is involved in driving tumorigenesis, expanding the repertoire of the bioengineered cells to target more than one antigen such as using bi-specific CAR-T-cell therapy and acceptable safety [132].
3.3. Therapeutic Vaccine Therapy in Glioblastoma
Therapeutic cancer vaccination relies on the method of inducing an anti-tumor immune response by selecting tumor-associated antigens or mutated peptides that are specific for cancer cells and are not universally expressed in healthy tissue. These candidate antigens are then either enhanced ex vivo or incubated in specific immune cells like dendritic cells and then reintroduced into the patient. By doing so, immunogenic tumor-associated antigens are hypothesized to initiate a tumor-specific immune response that can contribute to regression of malignant cells [133]. The platform of therapeutic vaccines in glioblastoma utilizes different approaches and can be classified into: (a) Tumor antigen associated vaccines that use commonly expressed oncogene-derived proteins on tumor cell surface such as the use of EGFRvIII and IDH1(R132H) to elicit a specific immune response; (b) vaccines that use heat shock protein family to illicit an immune response against a variety of different tumor antigens; (c) cell-based vaccine using autologous or allogenic dendritic cell (DC) which serves in initiating anti-tumor response through enhanced antigen presentation and; (d) personalized vaccines using immunogenomic methods to detect patient-specific novel peptides which are considered to be immunogenic neoantigen to enhance the response against malignant cells.
Vaccine-based therapies using EGFRvIII have generated a lot of enthusiasm as extracellular mutations of EGFRvIII are commonly expressed in glioblastoma (approximately 20%) and are responsible for constitutive EGFR pathway activation [134]. Early phase-II clinical trial conducted by Schuster et al. demonstrated safety and increased immunogenicity of EGFRvIII peptide sequence conjugated to keyhole limpet hemocyanin when administered with Stupp protocol in newly diagnosed glioblastoma [135]. However, despite the early encouraging results, a phase-III randomized controlled trial failed to demonstrate survival advantage of the candidate vaccine [136]. Interestingly, a signal of clinical benefit was observed in the previous trial when a post-hoc analysis showed improved OS in patients with bulky glioblastoma who received the vaccines although this was not the primary end point of the study [136]. In addition, a recent randomized phase-II trial demonstrated improved PFS in patients with EGFRvIII-positive recurrent glioblastoma when they received EGFRvIII vaccine in combination with bevacizumab versus those receiving placebo-bevacizumab [137]. To this end, EGFRvIII vaccines could assume a future role in the treatment of glioblastoma based on an enhanced clinical signal observed in specific subpopulations or when the vaccine is used in combination with other treatment modalities, although further confirmatory large randomized trials will be needed. Another, potential mutated protein that is commonly encountered in glioblastoma and has been shown to induce immunogenicity is IDH1 R132H [138]. Early results demonstrated safety and immunogenicity of a vaccine targeting this mutation in a German phase I clinical trial in 32 patients with newly diagnosed IDH1 R132H mutant malignant astrocytoma [139].
While vaccines against EGFRvIII and IDH1 R132H target specifically malignant cells that express the mutant protein, vaccination using heat shock protein family can leverage a broader response targeting a variety of tumor antigens in glioblastoma which could mitigate the heterogeneous nature of the tumor as a resistance mechanism. This is hypothesized to be the result of the function of heat shock protein family in post translational protein processes and presentation of antigens on MHC class I [140]. Several groups have demonstrated relative safety and immunogenicity using heat shock protein family vaccines in newly diagnosed and recurrent glioblastoma but data is still immature to support their clinical utility [141,142,143].
The rationale of using DC vaccines stems from their unique characteristics as highly potent cells that have the capacity to present antigens. The presentation of antigens by DC leads to sensitization of naïve T cells and priming of an anti-tumor immune response. The potential benefit of dendritic cell vaccination in newly diagnosed and recurrent glioblastoma has been illustrated in a systematic review that included a total of 403 patients and showed improved OS in patients with glioblastoma who received DC vaccines compared to historical average survival [144]. A randomized phase-III trial by Liau et al. have further corroborated the evidence of a benefit with the addition of autologous tumor lysate-pulsed DC vaccine (DCVax-L) to Stupp protocol in patients with newly diagnosed glioblastoma which has led to the approval of this vaccine in Switzerland [145].
Finally, personalized vaccines using newly detected neoantigens is another promising avenue in glioblastoma research. This personalized vaccine approach is based on the fact that neoantigens are composed of immunogenic-mutated peptides that are the product of genomic disruption in glioblastoma cells and when expressed in the TME can lead to an anti-tumor response [146]. For example, the use of personalized vaccine composed of DCVax followed by a neoantigen-based synthetic long peptide vaccine demonstrated immunogenicity of such an approach in a patient with newly diagnosed glioblastoma [147]. Of interest, two recent clinical trials have further demonstrated the immunogenic potential and clinical efficacy using whole-exome sequencing or microarray analysis in identifying candidate neoantigen peptide vaccines that were shown to generate anti-tumor response [148,149].
Key challenges remain when developing therapeutic glioblastoma vaccines including heterogeneity of glioblastoma as well as the immunosuppressive environment, which constitute a major hurdle given their direct dependency on antigen-specific derived immune response. The ongoing work evaluating variable vaccines in glioblastoma treatment should offer further insight.
3.4. Oncolytic Virus Therapy
Oncolytic viral therapy in glioblastoma involves the intratumoral administration of genetically modified viruses that usually have neural tropism and an ability to selectively replicate inside malignant infected cells. The amplified replication of lytic viruses leads to the destruction of the target cell and further propagation of viral progeny in addition to intracellular molecules, which can induce and augment anti-tumor immune response [150,151]. Some tumor-selective lytic viruses have shown ability to replicate in glioma cells and are currently being studied in early phase clinical trials including herpes simplex virus (HSV), adenovirus, poliovirus, gamma-retrovirus, and zika virus [150,152,153]. The method of administration is by intraparenchymal or intracranial arterial convection-enhanced delivery which allows for the direct intratumoral administration of the oncolytic virus. Although the majority of the results of this therapeutic avenue remains confined to early phase clinical trials, the results to date have demonstrated accepted safety and efficacy of this approach. For example, Ji et al. reported the results of administration of intra-arterial cerebral infusion of adenovirus mutant thymidine kinase in 53 patients with recurrent glioblastoma and showed improved PFS and OS compared to control group [154]. This study demonstrated acceptable safety compared to control groups with the exception of one patient who developed vasospasm 10 days after oncolytic viral administration [154]. Similarly, Cloughesy et al. demonstrated tolerability of using retroviral replicating virus (Toca511) followed by administration of Toca FC which led to the preferential killing of infected cancer cells through delivery of a yeast cytosine deaminase, which in turn converts 5-fluorocytosine to 5-fluorouracil (a chemotherapeutic compound that inhibits tumor cell growth and leads to cytotoxicity) [155]. In this phase I, open-label trial, 45 patients with recurrent glioblastoma received the viral therapy. The trials demonstrated the safety of this approach and an OS survival benefit although it was not the primary endpoint of this study [155]. As mentioned previously, Desjardins et al. demonstrated safety and significant OS improvement in patients with recurrent glioblastoma who received a recombinant oncolytic polio-rhinovirus (PVSRIPO) with an OS rate of 21% at 24 months which was maintained at 36 months [48]. The promising efficacy of oncolytic viral therapy in glioblastoma treatment led to FDA breakthrough designation for PVSRIPO in 2016. Recently, the overall survival rate at 36 months from Desjardins group (OS rate of 20%) was replicated by Lang et al. who administered an oncolytic-replicating adenovirus (DNX-2401) in 25 patients with recurrent glioblastoma [156]. Interestingly, it was observed that there was a decrease in the exhaustion of TIM3 and increased infiltration by CD8+ T cells of tumor specimens after the treatment with the oncolytic vaccine which has led to another clinical trial evaluating the combination of this vaccine with PD-1 inhibitor (NCT02798406). This cumulative evidence opens a new avenue for the immunotherapies in glioblastoma although it is essential to confirm the previous findings in large phase 3 randomized trials.
4. Targeted Therapies in Glioblastoma
The advent of molecular targeted therapy has revamped cancer treatment since their introduction in early 2000. Their efficacy relies on targeting specific molecular alterations or their protein products which govern malignant cell growth and enhance the invasive nature of the tumor. Most of the targeted therapies are tyrosine kinase inhibitors (TKIs) that can prevent kinase phosphorylation which are implicated in cancer cell proliferation and angiogenesis [157]. Conversely, drugs that can block receptors in the TME involved in tumor growth such as VEGF inhibitors are considered targeted therapies. The successful use of these targeted therapies in multiple solid and hematological malignancies has persuaded the investigation of their efficacy in glioblastoma. To this point, glioblastoma can be associated with a variety of mutations which has been described by The Cancer Genome Atlas research network [134]. This has resulted in several investigations which have been conducted to evaluate the outcomes in glioblastoma patients treated with specific molecular targets. Variable drugs have been investigated and were directed against mutations involving EGFR, fibroblast growth factor receptor (FGFR), MET, BRAF-MAPK, PI3K-AKT mTOR pathway, VEGF, and integrins Figure 3.
Frequency of EGFR amplification is estimated to reach 40%, and in-frame deletions (EGFRvIII) in approximately 25% [134]. The alterations in EGFR can lead to constitutive activation of cell proliferation and contribute to prognosis which makes it a plausible target in specific tumors such as breast and lung cancer. However, it appears that EGFR-associated alterations in glioblastoma lack prognostic significance, which is multifactorial because of the difference in structural mutation location (extracellular in glioblastoma versus intracellular in lung cancer) as well as the presence of regulatory downstream circuits that can lead to cell growth independent of EGFR status [158,159]. As such, majority of clinical trials failed to demonstrate improved PFS or OS in recurrent patients treated with different EGFR inhibitors including erlotinib, gefitinib, afatinib, and dacomitinib [160,161,162,163]. The failure of the previous therapeutics has shifted the focus toward different strategies using EGFR receptors as binding sites on the tumor cells for the delivery of cytotoxic payload. Lassman et al. demonstrated safety in 60 patients with recurrent glioblastoma who received the antibody–drug conjugate depatuxizumab mafodotin (depatux-m) [164]. However, a more recent phase-II trial using depatux-m in combination with TMZ at progression did not improve OS significantly compared to the control arm [165].
The presence of FGFR gene fusion was investigated as a target for therapy despite its low frequency (3.5%) in glioblastoma which is exclusive to IDH1/2 wild-type tumors [166]. In the previous study, two patients with FGFR fusion gene were treated with erdafitinib (FGFR inhibitor), and showed promising tumor response but both eventually had progressive disease [166]. Similarly, targeting c-MET overexpression (frequency of 13% in glioblastoma) and amplification (5%) with two phase II trials in recurrent glioblastoma, failed to demonstrate improved outcomes [167,168,169].
Targeting the PI3K-AKT-mTOR pathway which is universally altered in glioblastoma has also been unsuccessful and showed no improved outcomes in both newly diagnosed and recurrent glioblastoma using PI3K inhibitors and mTOR inhibitors [170,171,172]. Interestingly, despite the low frequency of BRAFV600E mutation in glioblastoma (up to 3%), some subtypes of glioblastoma such as epithelioid glioblastoma show high frequency reaching 50% [173,174]. Based on the efficacy of targeting mutations in the MAPK pathway including BRAFV600E in melanoma, several anecdotal reports described rapid and durable anti-tumor response using inhibitors of this pathways such as BRAF and MEK inhibitors [175,176]. A study by Kaley et al. has further corroborated the efficacy of BRAF inhibitors in glioblastoma patients [177]. Nevertheless, utility and access to these medications remain scarce given that most of the patients eventually develop progressive disease while on treatment and because of the restricted number of eligible patients with such mutation.
Finally, other study groups attempted targeting receptors in the TME of glioblastoma which are not necessarily expressed on tumor cells such as VEGF and integrins. The trials using the anti-VEGF (bevacizumab) later led to its approval in the treatment of glioblastoma as described earlier in the introduction. Several trials evaluated the efficacy of several integrin inhibitors but failed to show any benefit of the addition of these targeted therapies and have been summarized in detail by Le Rhun et al. [178].
Collectively, the failure of most targeted therapy trials to date in treatment of glioblastoma has resulted in the decline in the interest of targeted molecular therapy with the exception of novel molecular targets that are under active research [178]. Supplementary research remains essential to identify pathways and molecular alterations that control tumor survival and resistant nature of glioblastoma in order to develop further effective therapeutics.
5. Future Perspective of Glioblastoma Research
Despite the increased armamentarium of effective cancer therapeutics across multiple tumor types, management of glioblastoma continues to prove the unmet need for further research to develop new effective medication, which is echoed by the high number of clinical trials that are under way (470 clinical trials registered at clinicaltrials.gov as of October 2020). Phase 3 randomized clinical trials are crucial for evaluating the role of novel therapies in glioblastoma despite the promising results of early phase clinical trials.
Addressing the immunological landscape and the crosstalk between intracranial glioblastoma and the extracranial immune components appear to be of a high priority given that most of the concentration revolves around exploiting the immune system against glioblastoma. Achieving clarity on the utility of immunotherapy (ICI) up-front in the neo-adjuvant setting is promising but requires further validation in large prospective randomized trials. In addition, several proposed approaches have been suggested to overcome the immunosuppressive TME in glioblastoma and are currently under investigation (summarized in Table 1).
Furthermore, combinatorial therapies with immunotherapies is currently being explored and appear to be promising. For example, inclusion of poly-ADP ribose polymerase (PARP) inhibitors which induces synthetic lethality and leads to overload with defective DNA repair mechanisms appears to be promising. Preclinical studies suggest that PARP inhibition with drugs such as olaparib can increase neoantigen formation, lead to PD-L1 upregulation, boost type I Interferon signaling, and reprogram TME to facilitate anti-tumor response when using ICI [179]. In addition, PARP inhibition is thought to cause catastrophic DNA damage which may also lead to resensitized tumor cells to TMZ in MRD glioblastoma [180]. Similarly, Hanna et al. demonstrated in a phase I trial the radio-sensitizing effect and the safety of olaparib administration with TMZ prior to surgery in patients with recurrent glioblastoma [181].
Ultimately, as immunotherapy continues to dominate the research arena in glioblastoma therapeutic development, it would be important to stratify patients based on their previous glucocorticoid use as most of the patients require to be on steroids which is implicated in suppression of the immune system and can counteract the effect of drugs targeting the immune system.
6. Conclusions
The lack of drug approvals for the treatment of glioblastoma and the mild to modest improvement in survival in the past decade represent a growing need for well-designed research to develop effective therapeutics. The ongoing effort should consider the factors that are implicated in the resistance observed with these novel investigational therapeutics to further leverage our understanding and optimize our treatment approach. Immunotherapy holds a promise in glioblastoma despite the immunosuppressive nature of the tumor. Combination immunotherapy with other treatment modalities, investigational novel compounds, and innovative delivery modalities such as antibody–drug conjugates, convection-enhanced delivery, and nanoparticles are currently under ongoing research which will address their safety and efficacy in glioblastoma. Further characterization of patients’ tumor molecular profiles can help identify subpopulations who could derive a benefit from currently available treatments.
Author Contributions
Conceptualization, K.K., T.M.J. and G.A.; writing—original draft preparation, K.K.; writing—review K.K. Editing K.K., T.M.J., G.A. Figure and table creation, K.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Some research content reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number K12 CA167540. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Figure 2 and Figure 3 were created by K.K. using licensed Biorender software.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109, Erratum in 2007, 114, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. cbtrus statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20 (Suppl. 4), iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Sminia, P.; Westerman, B.A. Blood-brain barrier crossing and breakthroughs in glioblastoma therapy. Br. J. Clin. Pharmacol. 2016, 81, 1018–1020. [Google Scholar] [CrossRef] [Green Version]
- Parker, N.R.; Ekhong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Bent, M.J.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs. maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316, Erratum in 2018, 319, 1824. [Google Scholar] [CrossRef] [Green Version]
- Fabian, D.; Eibl, M.D.P.G.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.K.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the addition of Tumor-Treating Fields (TTF): A review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and bevacizumab in progressive glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, J.-F.; Denizot, F.; Luciani, M.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nat. Cell Biol. 1987, 328, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Ardolino, L.; Joshua, A. Immune checkpoint inhibitors in malignancy. Aust. Prescr. 2019, 42, 62–67. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; Von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Galsky, M.D.; E Rosenberg, J.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76, Erratum in 2017, 390, 848. [Google Scholar] [CrossRef] [Green Version]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.; Hahn, N.M.; De Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Seock-Ah IMpassion130 Trial Investigators; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gautier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; Riddell, S.R.; et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 2019, 134, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Hambardzumyan, D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar] [CrossRef]
- Dhermain, F.G.; Hau, P.; Lanfermann, H.; Jacobs, A.H.; van den Bent, M.J. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet Neurol. 2010, 9, 906–920. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellisdavies, G.C.R.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Platt, M.P.; Agalliu, D.; Cutforth, T. Hello from the other side: How autoantibodies circumvent the blood-brain barrier in autoimmune encephalitis. Front. Immunol. 2017, 8, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majerova, P.; Michalicova, A.; Cente, M.; Hanes, J.; Vegh, J.; Kittel, Á.; Kosikova, N.; Cigankova, V.; Mihaljevic, S.; Jadhav, S.; et al. Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS ONE 2019, 14, e0217216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, L.; Coll, C.; Erthal, L.C.S.; de la Torre, C.; Serrano, D.R.; Martínez-Máñez, R.; Santos-Martínez, M.J.O.; Ruiz-Hernández, E. Drug delivery nanosystems for the localized treatment of glioblastoma multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850, Erratum in 2020, 11, 701. [Google Scholar] [CrossRef]
- Erel-Akbaba, G.; Carvalho, L.A.; Tian, T.; Zinter, M.; Akbaba, H.; Obeid, P.J.; Chiocca, E.A.; Weissleder, R.; Kantarci, A.G.; Tannous, B.A. Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [Google Scholar] [CrossRef]
- Shi, M.; Sanche, L. Convection-enhanced delivery in malignant gliomas: A review of toxicity and efficacy. J. Oncol. 2019, 2019, 9342796. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Ii, J.E.H.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Toki, M.I.; Kumar, D.; Ahmed, F.S.; Rimm, D.L.; Xu, M.L. Benign lymph node microenvironment is associated with response to immunotherapy. Precis. Clin. Med. 2020, 3, 44–53. [Google Scholar] [CrossRef]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Gomez, G.G.; Kruse, C.A. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther. Mol. Biol. 2006, 10, 133–146. [Google Scholar]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341, Erratum in 2016, 533, 278. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; DeChant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468, Erratum in 2019, 25, 529. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Wiendl, H.; Mitsdoerffer, M.; Hofmeister, V.; Wischhusen, J.; Bornemann, A.; Meyermann, R.; Weiss, E.H.; Melms, A.; Weller, M. A functional role of HLA-G expression in human gliomas: An alternative strategy of immune escape. J. Immunol. 2002, 168, 4772–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Balça-Silva, J.; Da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/astrocytes-glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, S.; Strommer, K.; Frei, K.; Siepl, C.; De Tribolet, N.; Heid, I.; Fontana, A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J. Immunol. 1989, 143, 3222–3229. [Google Scholar] [PubMed]
- Huettner, C.; Czub, S.; Kerkau, S.; Roggendorf, W.; Tonn, J.-C. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer. Res. 1997, 17, 3217–3224. [Google Scholar] [PubMed]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, T.; Li, G.; Nagpal, S. History and current state of immunotherapy in glioma and brain metastasis. Ther. Adv. Med. Oncol. 2017, 9, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Garber, S.T.; Hashimoto, Y.; Weathers, S.-P.; Xiu, J.; Gatalica, Z.; Verhaak, R.G.W.; Zhou, S.; Fuller, G.N.; Khasraw, M.; De Groot, J.; et al. Immune checkpoint blockade as a potential therapeutic target: Surveying CNS malignancies. Neuro-Oncology 2016, 18, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301, Erratum in 2015, 21, 662. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y.; et al. Molecular and clinical characterization of TIM-3 in glioma through 1,024 samples. OncoImmunology 2017, 6, e1328339. [Google Scholar] [CrossRef]
- Chang, N.; Ahn, S.H.; Kong, D.-S.; Lee, H.W.; Nam, D.-H. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Didenko, V.V.; Ngo, H.N.; Minchew, C.; Baskin, D.S. Apoptosis of T lymphocytes invading glioblastomas multiforme: A possible tumor defense mechanism. J. Neurosurg. 2002, 96, 580–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068, Erratum in 2013, 494, 506. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6, Erratum in 2018, 33, 152. [Google Scholar] [CrossRef] [Green Version]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef]
- Cantanhede, I.; Oliveira, J.R.M. PDGF family expression in glioblastoma multiforme: Data compilation from ivy glioblastoma atlas project database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [Green Version]
- Engler, J.R.; Robinson, A.E.; Smirnov, I.; Hodgson, J.G.; Berger, M.S.; Gupta, N.; James, C.D.; Molinaro, A.; Phillips, J.J. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE 2012, 7, e43339. [Google Scholar] [CrossRef] [Green Version]
- Luoto, S.; Hermelo, I.; Vuorinen, E.M.; Hannus, P.; Kesseli, J.; Nykter, M.; Granberg, K.J. Computational characterization of suppressive immune microenvironments in glioblastoma. Cancer Res. 2018, 78, 5574–5585. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro-Oncology 2017, 19, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; A Crane, C.; Parney, I.F.; Barry, J.J.; Cachola, K.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.D.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Sa, J.K.; Choi, S.W.; Zhao, J.; Lee, Y.; Zhang, J.; Kong, D.; Choi, J.W.; Seol, H.J.; Lee, J.; Iavarone, A.; et al. Hypermutagenesis in untreated adult gliomas due to inherited mismatch mutations. Int. J. Cancer 2019, 144, 3023–3030. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, I.-H.; Cho, H.J.; Park, C.-K.; Jung, Y.-S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.-S.; et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Mahlokozera, T.; Vellimana, A.K.; Li, T.; Mao, D.D.; Zohny, Z.S.; Kim, D.H.; Tran, D.D.; Marcus, D.S.; Fouke, S.J.; Campian, J.L.; et al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro-Oncology 2018, 20, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachi, A.; Dastmalchi, F.; A Mitchell, D.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Chitadze, G.; Flüh, C.; Quabius, E.S.; Freitag-Wolf, S.; Peters, C.; Lettau, M.; Bhat, J.; Wesch, D.; Oberg, H.-H.; Luecke, S.; et al. In-depth immunophenotyping of patients with glioblastoma multiforme: Impact of steroid treatment. OncoImmunology 2017, 6, e1358839. [Google Scholar] [CrossRef]
- Grossman, S.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S.; NABTT CNS Consortium. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, F.; Signorelli, D.; Ghidini, M.; Ghidini, A.; Pizzutilo, E.G.; Ruggieri, L.; Cabiddu, M.; Borgonovo, K.; Dognini, G.; Brighenti, M.; et al. Association of steroids use with survival in patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Cancers 2020, 12, 546. [Google Scholar] [CrossRef] [Green Version]
- Tamura, R.; Tanaka, T.; Ohara, K.; Miyake, K.; Morimoto, Y.; Yamamoto, Y.; Kanai, R.; Akasaki, Y.; Murayama, Y.; Tamiya, T.; et al. Persistent restoration to the immunosupportive tumor microenvironment in glioblastoma by bevacizumab. Cancer Sci. 2019, 110, 499–508. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Morimoto, Y.; Kuranari, Y.; Yamamoto, Y.; Takei, J.; Murayama, Y.; Yoshida, K.; Sasaki, H. Alterations of the tumor microenvironment in glioblastoma following radiation and temozolomide with or without bevacizumab. Ann. Transl. Med. 2020, 8, 297. [Google Scholar] [CrossRef]
- Zhang, C.; Fan, Y.; Che, X.; Zhang, M.; Li, Z.; Li, C.; Wang, S.; Wen, T.; Hou, K.; Shao, X.; et al. Anti-PD-1 Therapy Response Predicted by the Combination of Exosomal PD-L1 and CD28. Front. Oncol. 2020, 10, 760. [Google Scholar] [CrossRef]
- Zappasodi, R.; Wolchok, J.; Merghoub, T. Strategies for predicting response to checkpoint inhibitors. Curr. Hematol. Malign Rep. 2018, 13, 383–395, Erratum in 2019, 14, 62. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, M.; Pistillo, M.P.; Carosio, R.; Morabito, A.; Banelli, B. Immune checkpoints and innovative therapies in glioblastoma. Front. Oncol. 2018, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic antitumor effect with indoleamine 2, 3-dioxygenase inhibition and temozolomide in a murine glioma model. J. Neurosurg. 2016, 124, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. Phase-3 CheckMate-498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo nivolumab Plus Radiation in Patients with Newly Diagnosed MGMT Unmethylated Glioblastoma Multiforme. 9 May 2019. Available online: https://news.bms.com/news/corporate-financial/2019/Bristol-Myers-Squibb-Announces-Phase-3-CheckMate--498-Study-Did-Not-Meet-Primary-Endpoint-of-Overall-Survival-with-Opdivo-nivolumab-Plus-Radiation-in-Patients-with-Newly-Diagnosed-MGMT-Unmethylated-Glioblastoma-Multiforme/default.aspx (accessed on 1 October 2020).
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; De Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2006. [Google Scholar] [CrossRef]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9 (Suppl. 1), S3. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125554lbl.pdf (accessed on 13 November 2020).
- Available online: https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf (accessed on 10 September 2020).
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef]
- Versluis, J.M.; Long, G.V.; Blank, C.U. Learning from clinical trials of neoadjuvant checkpoint blockade. Nat. Med. 2020, 26, 475–484. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.; Hugo, W.; Lee, A.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; de Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Yang, Z.; Wei, S.; Deng, Y.; Wang, Z.; Liu, L. Clinical significance of tumour mutation burden in immunotherapy across multiple cancer types: An individual meta-analysis. Jpn. J. Clin. Oncol. 2020, 50, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A., Jr. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya-Matsuoka, C.; Metrus, N.R.; Shaw, K.R.; Penas-Prado, M.; Weathers, S.-P.; Loghin, M.E.; Alfaro-Munoz, K.; Yuan, Y.; O’Brien, B.J.; Harrison, R.A.; et al. The natural course of hypermutator gliomas. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2014. [Google Scholar]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination therapy with Anti-PD-1, Anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Weenink, B.; French, P.J.; Smitt, P.S.; Debets, R.; Geurts, M. Immunotherapy in glioblastoma: Current shortcomings and future perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [Green Version]
- Gholamin, S.; Mitra, S.S.; Feroze, A.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Osuchowska, I.; Maciejewski, R.; Roliński, J.; Grajkowska, W.A.; Grochowski, C. Micro RNA Molecules as Modulators of Treatment Resistance, Immune Checkpoints Controllers and Sensitive Biomarkers in Glioblastoma Multiforme. Int. J. Mol. Sci. 2020, 21, 1507. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280ps7. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Dietrich, J.; Martinez-Lage, M.; Leick, M.; Choi, B.D.; DeFilipp, Z.; Chen, Y.-B.; Abramson, J.; Crombie, J.; Armand, P.; et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood 2019, 134, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.R.; Wright, C.; et al. Bioactivity and safety of IL13Rα2-Redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Schlom, J.; Hodge, J.W.; Palena, C.; Tsang, K.-Y.; Jochems, C.; Greiner, J.W.; Farsaci, B.; Madan, R.A.; Heery, C.R.; Gulley, J.L. Therapeutic cancer vaccines. Adv. Cancer Res. 2014, 121, 67–124. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477, Erratum in 2014, 157, 753. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; A Goldlust, S.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with Bevacizumab for patients with relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a double-blind randomized phase II trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Platten, M.; Schilling, D.; Bunse, L.; Wick, A.; Bunse, T.; Riehl, D.; Green, E.; Sanghvi, K.; Karapanagiotou-Schenkel, I.; Harting, I.; et al. ATIM-33. NOA-16: A first-in-man multicenter phase i clinical trial of the german neurooncology working group evaluating a mutation-specific peptide vaccine targeting idh1r132h in patients with newly diagnosed malignant astrocytomas. Neuro-Oncology 2018, 20 (Suppl. 6), vi8–vi9. [Google Scholar] [CrossRef] [Green Version]
- Ampie, L.; Choy, W.; Lamano, J.B.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Heat shock protein vaccines against glioblastoma: From bench to bedside. J. Neurooncol. 2015, 123, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3, e99145. [Google Scholar] [CrossRef] [Green Version]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142, Erratum in 2018, 16, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Johanns, T.M.; Miller, C.A.; Liu, C.J.; Perrin, R.J.; Bender, D.; Kobayashi, D.K.; Campian, J.L.; Chicoine, M.R.; Dacey, R.G.; Huang, J.; et al. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. OncoImmunology 2019, 8, e1561106. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245, Erratum in 2019, 566, E13. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Iorgulescu, J.B.; Reardon, D.A.; Chiocca, E.A.; Wu, C.J. Immunotherapy for glioblastoma: Going viral. Nat. Med. 2018, 24, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Paget, M.; Tshilenge, K.-T.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Restriction of replication of oncolytic herpes simplex virus with a deletion of γ34.5 in glioblastoma stem-like cells. J. Virol. 2018, 92, e00246:1–e00246:18. [Google Scholar] [CrossRef] [Green Version]
- Ji, N.; Weng, D.; Liu, C.; Gu, Z.; Chen, S.; Guo, Y.; Fan, Z.; Wang, X.; Chen, J.; Zhao, Y.; et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 2016, 7, 4369–4378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Carter, B.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lai, A.; et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.A.; Sabel, M.C.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.M.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 2010, 12, 95–103. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Sepúlveda-Sánchez, J.M.; Vaz, M. Ángeles; Balañá, C.; Gil-Gil, M.; Reynés, G.; Gallego, Óscar; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.B.; Bent, M.J.V.D.; Gan, H.K.; A Reardon, D.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro-Oncology 2019, 21, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, M.J.V.D.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.-S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, Y.; Kim, S.-I.; Park, C.-K.; Paek, S.H.; Lee, S.-T.; Park, S.-H. C-MET overexpression and amplification in gliomas. Int. J. Clin. Exp. Pathol. 2015, 8, 14932–14938. [Google Scholar]
- Wen, P.Y.; Schiff, D.; Cloughesy, T.F.; Raizer, J.J.; Laterra, J.; Smitt, M.; Wolf, M.; Oliner, K.S.; Anderson, A.; Zhu, M.; et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro-Oncology 2011, 13, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, double-blind, placebo-controlled, multicenter phase II study of onartuzumab plus bevacizumab versus placebo plus bevacizumab in patients with recurrent glioblastoma: Efficacy, safety, and hepatocyte growth factor and O6-Methylguanine-DNA methyltransferase biomarker analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: An open-label, multicenter, multi-arm, phase II trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; McGraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncology 2015, 17, 1261–1269. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; Bent, M.J.V.D.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [Green Version]
- Behling, F.; Barrantes-Freer, A.; Skardelly, M.; Nieser, M.; Christians, A.; Stockhammer, F.; Rohde, V.; Tatagiba, M.; Hartmann, C.; Stadelmann, C.; et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn. Pathol. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, A.; Chavez, L.; Sharma, T.; Ryzhova, M.; Schrimpf, D.; Stichel, D.; Capper, D.; Sturm, D.; Kool, M.; Habel, A.; et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018, 28, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanns, T.M.; Ferguson, C.J.; Grierson, P.M.; Dahiya, S.; Ansstas, G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with braf-mutated high-grade glioma. J. Natl. Compr. Cancer Netw. 2018, 16, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnirsky, M.; Feun, L.G.; Gultekin, S.H.; De La Fuente, M.I. Prolonged complete response with combined dabrafenib and trametinib after BRAF inhibitor failure in BRAF-mutant glioblastoma. JCO Precis Oncol. 2020, 4, PO.19.00272. [Google Scholar] [CrossRef]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.-D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-mutant gliomas: Results from the VE-BASKET study. J. Clin. Oncol. 2018, 36, 3477–3484. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; Bent, M.V.D.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.-F.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, noaa104, Epub ahead of print. [Google Scholar] [CrossRef]
Figure 1.
Timeline of treatment approval for glioblastoma and landmark investigational non-approved drugs. The lower panel demonstrates Kaplan–Meier curves of three pivotal randomized controlled trials in newly diagnosed glioblastoma which led to the approval of standard of care systemic treatment including concurrent radiation and temozolomide (Stupp protocol) [7], addition of bevacizumab to Stupp protocol [10,11], and use of tumor-treating fields with Stupp protocol [8]. Despite the improvement of OS with the addition of temzolomide, bevacizumab and TTF, the overall survival remains poor (as noted by the continued drop in slope of the curves). There are no approved treatments shown to improve the survival in recurrent glioblastoma (not shown in this figure) [12]. Kaplan–Meier curves reprinted by permission from the Massachusetts Medical Society (NEJM) and American Medical Association (JAMA). N Engl J Med. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K et al., copyright © 2005. N Engl J Med. A randomized trial of bevacizumab for newly diagnosed glioblastoma. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA et al., copyright © 2014. JAMA. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B et al., copyright © 2017. Abbreviations: TMZ: temozolomide, mOS: median overall survival, mPFS: median progression free survival, RT: radiation therapy, SOC: standard of care, TTF: tumor-treating fields.
Figure 1.
Timeline of treatment approval for glioblastoma and landmark investigational non-approved drugs. The lower panel demonstrates Kaplan–Meier curves of three pivotal randomized controlled trials in newly diagnosed glioblastoma which led to the approval of standard of care systemic treatment including concurrent radiation and temozolomide (Stupp protocol) [7], addition of bevacizumab to Stupp protocol [10,11], and use of tumor-treating fields with Stupp protocol [8]. Despite the improvement of OS with the addition of temzolomide, bevacizumab and TTF, the overall survival remains poor (as noted by the continued drop in slope of the curves). There are no approved treatments shown to improve the survival in recurrent glioblastoma (not shown in this figure) [12]. Kaplan–Meier curves reprinted by permission from the Massachusetts Medical Society (NEJM) and American Medical Association (JAMA). N Engl J Med. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K et al., copyright © 2005. N Engl J Med. A randomized trial of bevacizumab for newly diagnosed glioblastoma. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA et al., copyright © 2014. JAMA. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B et al., copyright © 2017. Abbreviations: TMZ: temozolomide, mOS: median overall survival, mPFS: median progression free survival, RT: radiation therapy, SOC: standard of care, TTF: tumor-treating fields.

Figure 2.
Proposed approaches for the improvement of glioblastoma treatment with immune checkpoint inhibitors (ICI) include timing of administration and method of delivery, patient selection, and combinatorial therapies. Panel (A) demonstrates the promising approach of administering ICI in the neo-adjuvant setting prior to surgical resection of glioblastoma and followed by adjuvant ICI [112,113]. Convection enhanced delivery of nanoparticle ICI is another method that could improve the availability and action of ICI in glioblastoma [44]. Panel (B) shows that proper patient selection for treatment with ICI could be of a clinical benefit such as patients with high tumor mutation burden due to germline deficiency affecting DNA repair mechanisms and in patients with specific mutations that could attenuate the immunosuppressive effect in the microenvironment of the tumor [84,91,92]. Panel (C) illustrates ongoing investigational combination of ICI with other novel therapies.
Figure 2.
Proposed approaches for the improvement of glioblastoma treatment with immune checkpoint inhibitors (ICI) include timing of administration and method of delivery, patient selection, and combinatorial therapies. Panel (A) demonstrates the promising approach of administering ICI in the neo-adjuvant setting prior to surgical resection of glioblastoma and followed by adjuvant ICI [112,113]. Convection enhanced delivery of nanoparticle ICI is another method that could improve the availability and action of ICI in glioblastoma [44]. Panel (B) shows that proper patient selection for treatment with ICI could be of a clinical benefit such as patients with high tumor mutation burden due to germline deficiency affecting DNA repair mechanisms and in patients with specific mutations that could attenuate the immunosuppressive effect in the microenvironment of the tumor [84,91,92]. Panel (C) illustrates ongoing investigational combination of ICI with other novel therapies.

Figure 3.
Targeting pathways and receptors involved in cell proliferation, aggressiveness, invasion, and angiogenesis in glioblastoma. Some of the pharmaceuticals which have been trialed to target these specific receptors or pathways are illustrated.
Figure 3.
Targeting pathways and receptors involved in cell proliferation, aggressiveness, invasion, and angiogenesis in glioblastoma. Some of the pharmaceuticals which have been trialed to target these specific receptors or pathways are illustrated.


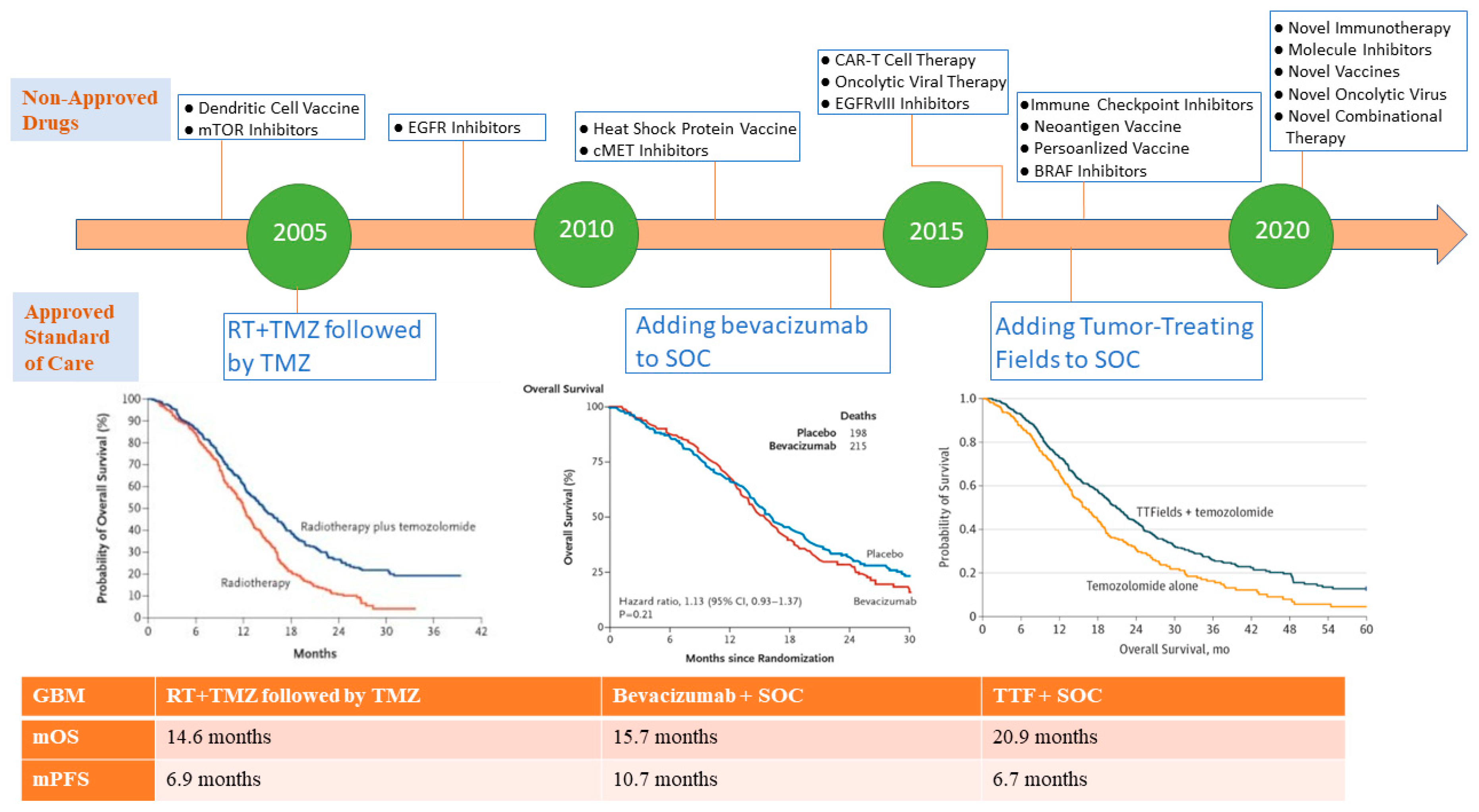{kind=link}
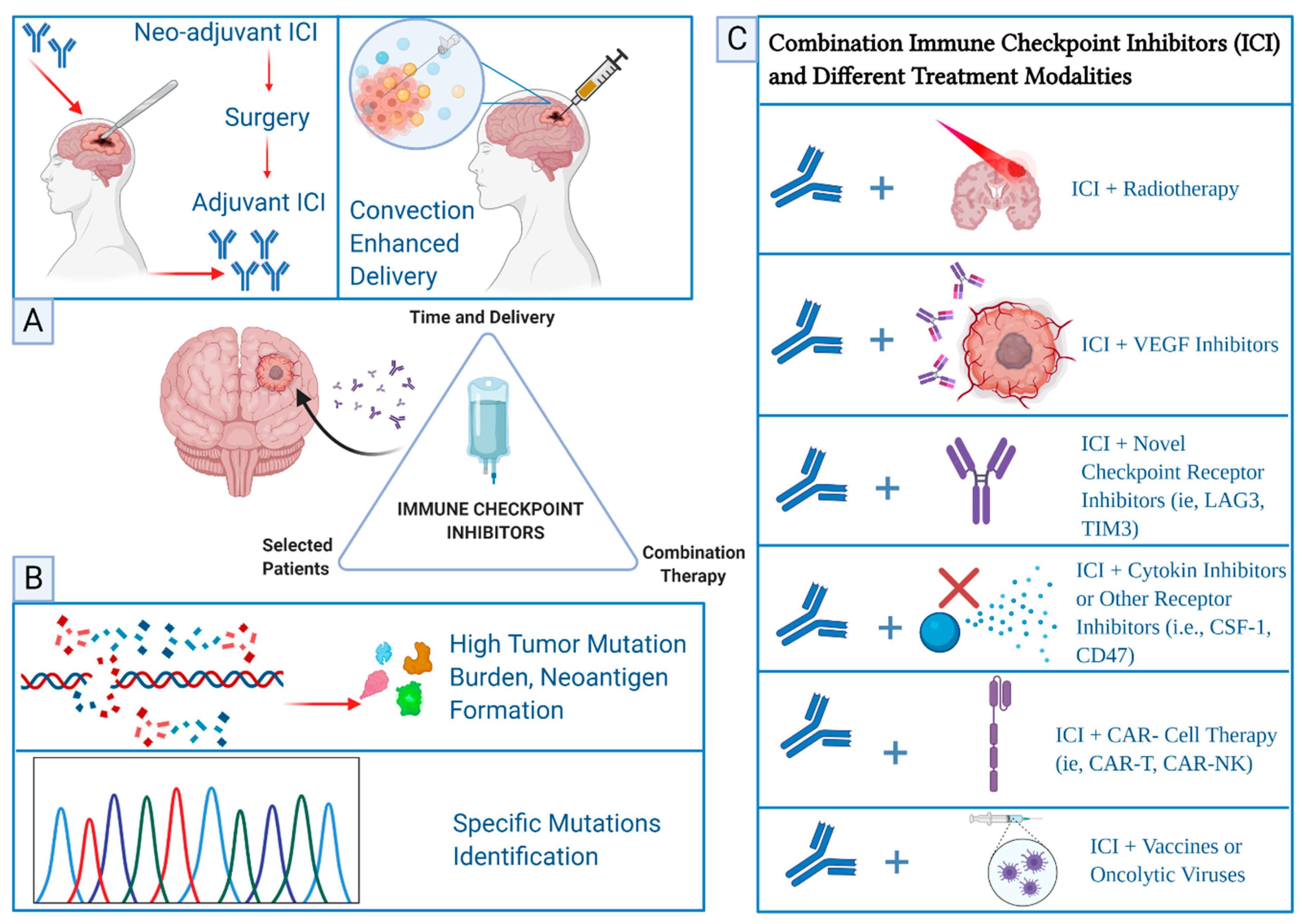{kind=link}
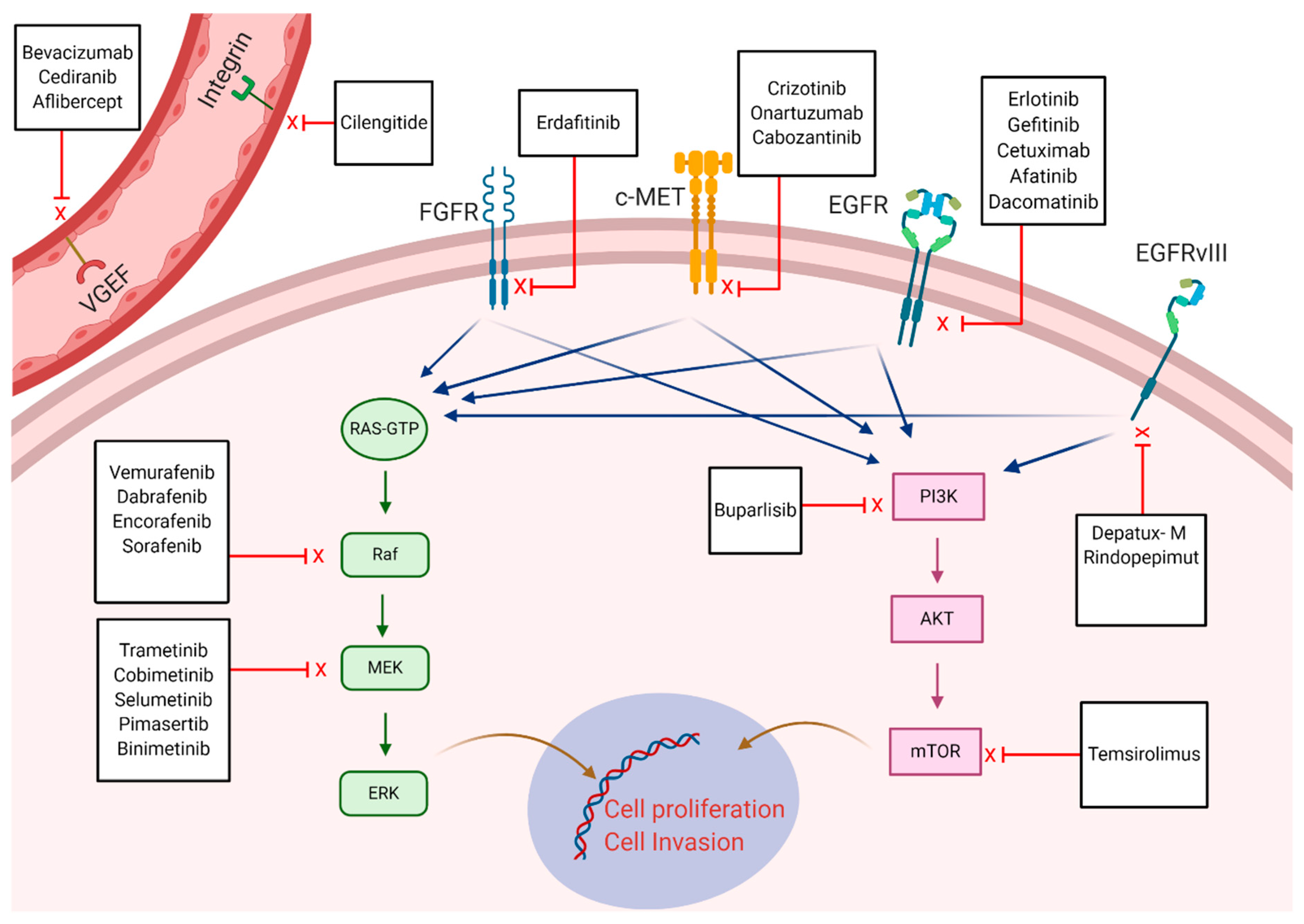{kind=link}
Table 1.
Factors implicated in the immunosuppressive environment of glioblastoma and possible solutions to enhance response.
Table 1.
Factors implicated in the immunosuppressive environment of glioblastoma and possible solutions to enhance response.
| Factors Related to Glioblastoma | Possible Solutions | Reference |
|---|---|---|
| Restricted drug delivery due to blood brain barrier | Convection enhanced delivery (CED) and nano particles. Disruption of blood brain barrier with radiation therapy combined with immune checkpoint inhibitors Adoptive cell therapy | [44,45] [121] [127,128] |
| Effector T-cell suppression and peripheral T-cell entrapment | Use of immune checkpoint inhibitors, therapeutic vaccines or adoptive cell transfer therapy | [52,53,54,55] |
| Increased expression of checkpoint receptors | Use of combination checkpoint inhibitors targeting several checkpoint receptors | [64,65,66,67,68,69,70,121] |
| Suppression of natural killer cells | Use of checkpoint inhibitors Natural killer cell adoptive transfer | [131] |
| Abundance of tumor associated macrophages (TAM) (protumorigenic phenotype) | Boosting host immunity through exploiting plasticity in TAM to express inflammatory phenotype M1 | [73] |
| Increased secretion of TGF-β | Use of checkpoint inhibitors with TGF-β inhibitors | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Khaddour, K.; Johanns, T.M.; Ansstas, G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals 2020, 13, 389. https://doi.org/10.3390/ph13110389
AMA Style
Khaddour K, Johanns TM, Ansstas G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals. 2020; 13(11):389. https://doi.org/10.3390/ph13110389
Chicago/Turabian StyleKhaddour, Karam, Tanner M. Johanns, and George Ansstas. 2020. "The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions" Pharmaceuticals 13, no. 11: 389. https://doi.org/10.3390/ph13110389
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.