A Peculiar Case of Pneumonia due to Mycoplasma pneumoniae in a Child with Cystic Fibrosis and Sensibilization to Aspergillus fumigatus



Abstract
:1. Background
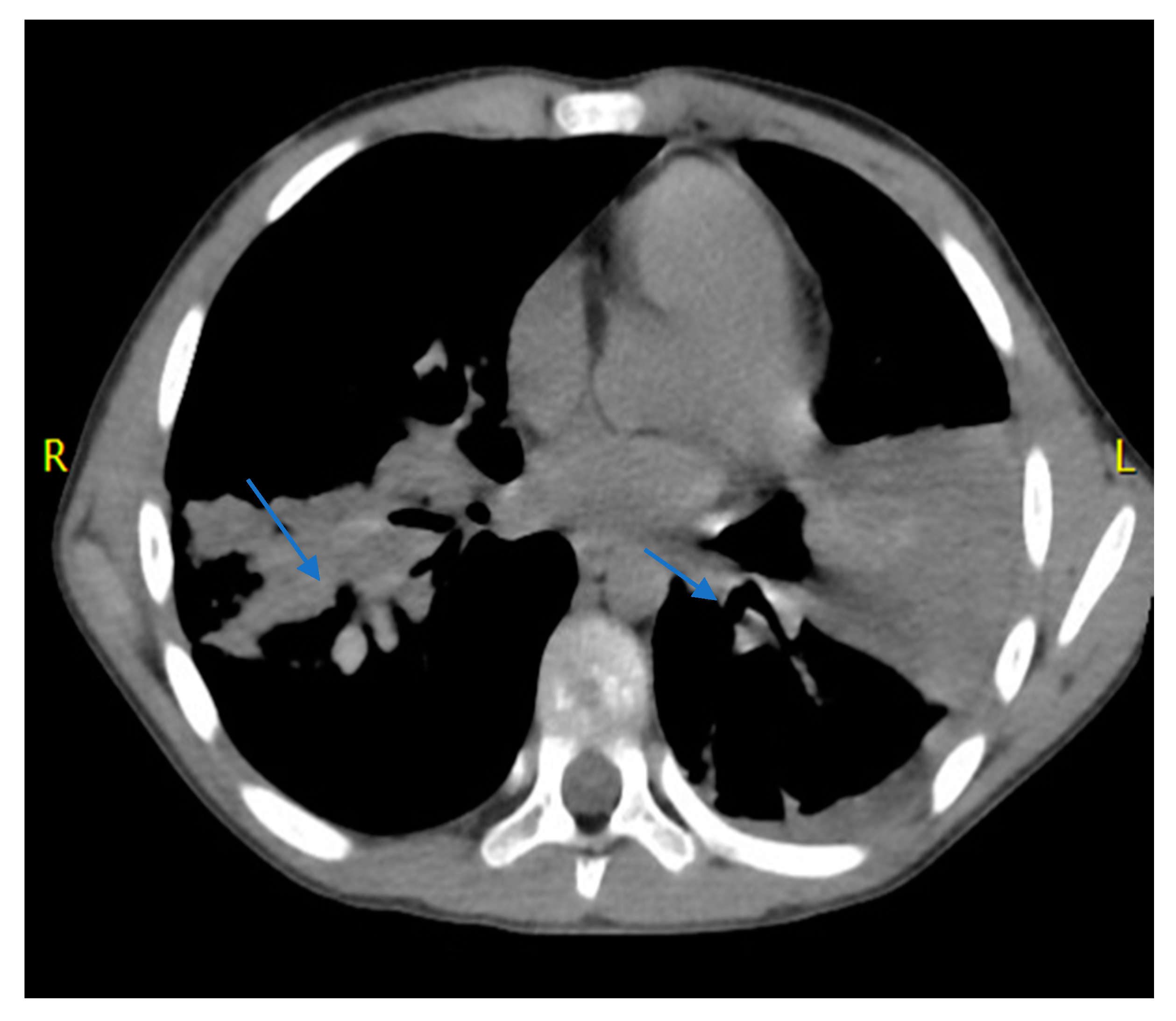
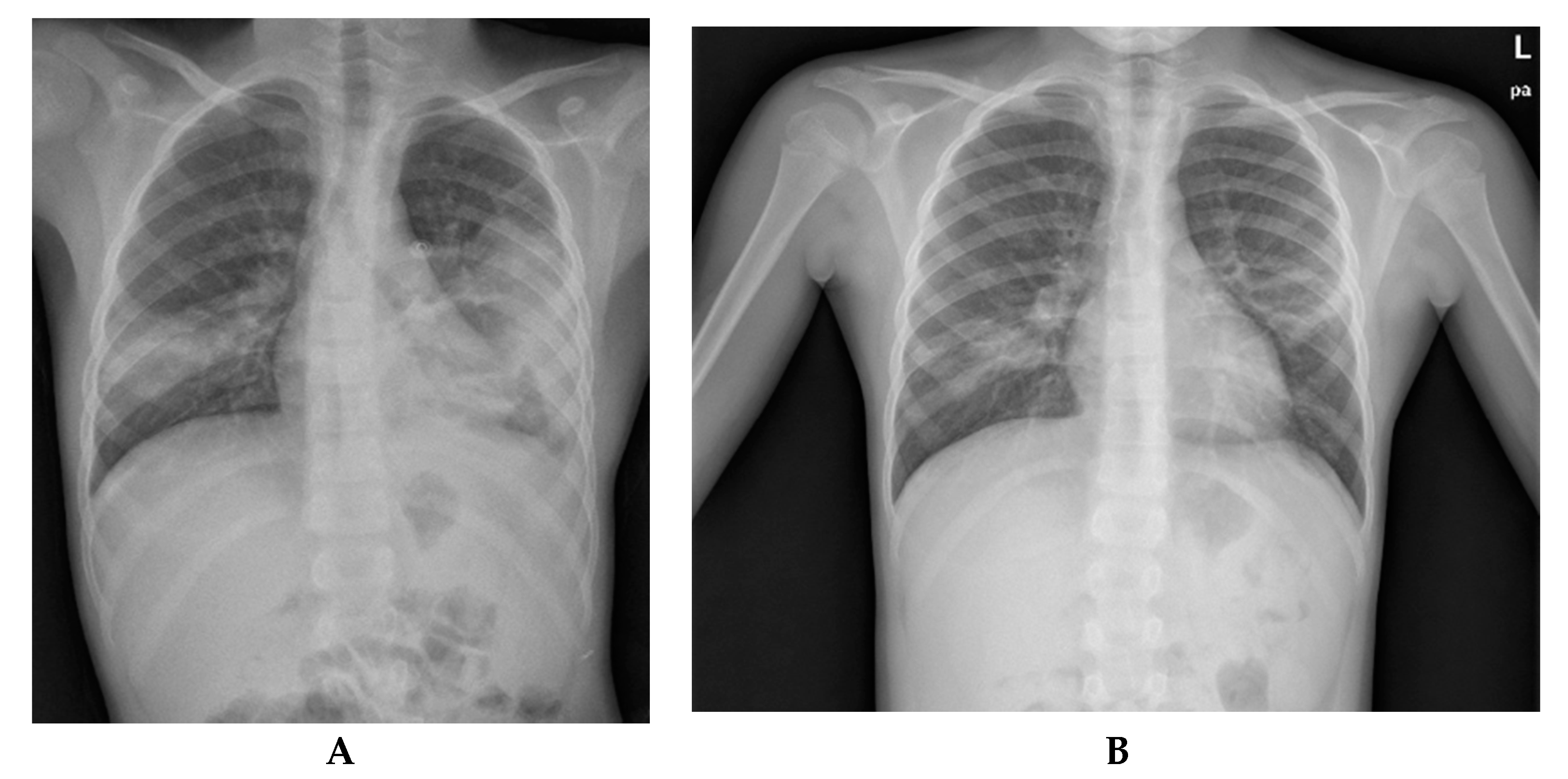
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rey, M.M.; Bonk, M.P.; Hadjiliadis, D. Cystic Fibrosis: Emerging understanding and therapies. Annu. Rev. Med. 2019, 70, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Colombo, C.; Ravaglia, R.; Faelli, N.; Tagliabue, C.; Corti, F.; Costantini, D.; Principi, N. Mycoplasma pneumoniae pericarditis and cardiac tamponade in a 7-year-old girl with cystic fibrosis. Infection 2006, 34, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Aksamit, T.R.; Chotirmall, S.H.; Dasenbrook, E.C.; Elborn, J.S.; LiPuma, J.J. Antibiotic management of lung infections in cystic fibrosis. II. Nontuberculous mycobacteria, anaerobic bacteria, and fungi. Ann. Am. Thorac. Soc. 2014, 11, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Sabino, R.; Ferreira, J.A.; Moss, R.B.; Valente, J.; Veríssimo, C.; Carolino, E. Molecular epidemiology of Aspergillus collected from cystic fibrosis patients. J. Cyst. Fibros. 2015, 14, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Breuer, O.; Schultz, A.; Garratt, L.W.; Turkovic, L.; Rosenow, T.; Murray, C.P.; Karpievitch, Y.V.; Akesson, L.; Dalton, S.; Sly, P.D.; et al. Aspergillus Infections and Progression of Structural Lung Disease in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019. Epub Nov 20. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Gresnigt, M.S.; Romani, L.; Netea, M.G.; Latgé, J.P. Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 2017, 15, 661–674. [Google Scholar] [CrossRef]
- Keown, K.; Abbott, S.; Kuzeljevic, B.; Rayment, J.H.; Chilvers, M.A.; Yang, C.L. An investigation into biomarkers for the diagnosis of ABPA and aspergillus disease in cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 1787–1793. [Google Scholar] [CrossRef]
- Janahi, I.A.; Rehman, A.; Al-Naimi, A.R. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. Ann. Thorac. Med. 2017, 12, 74–82. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; McElvaney, N.G. Fungi in the cystic fibrosis lung: Bystanders or pathogens? Int. J. Biochem. Cell Biol. 2014, 52, 161–173. [Google Scholar] [CrossRef]
- Patel, A.R.; Singh, S.; Khawaja, I. Diagnosing allergic bronchopulmonary Aspergillosis: A review. Cureus 2019, 11, e4550. [Google Scholar] [CrossRef] [Green Version]
- Hinson, K.F.; Moon, A.J.; Plummer, N.S. Broncho-pulmonary aspergillosis: A review and a report of eight new cases. Thorax 1952, 7, 317–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, N.; Pant, C.; Bhagat, R.; Shah, A. Central bronchiectasis in allergic bronchopulmonary aspergillosis: Comparative evaluation of computed tomography of the thorax with bronchography. Eur. Respir. J. 1994, 7, 1290–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, S.; Pennoni, G.; Mencarini, V.; Palladino, N.; Peccini, L.; Principi, N. Antimicrobial Treatment of Staphylococcus aureus in patients with cystic fibrosis. Front. Pharmacol. 2019, 10, 849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Cheng, W.; He, X.; Liu, Y. The co-colonization prevalence of Pseudomonas aeruginosa and Aspergillus fumigatus in cystic fibrosis: A systematic review and meta-analysis. Microb. Pathog. 2018, 125, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Emerging role of Mycoplasma pneumoniae and Chlamydia pneumoniae in paediatric respiratory-tract infections. Lancet Infect Dis. 2001, 1, 334–344. [Google Scholar] [CrossRef]
- Principi, N.; Esposito, S. Macrolide-resistant Mycoplasma pneumoniae: Its role in respiratory infection. J. Antimicrob. Chemother. 2013, 68, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Goyal, R.; White, C.S.; Templeton, P.A.; Britt, E.J.; Rubin, L.J. High attenuation mucous plugs in allergic bronchopulmonary aspergillosis: CT appearance. J. Comput. Assist. Tomogr. 1992, 16, 649–650. [Google Scholar]
- Stevens, D.A.; Moss, R.B.; Kurup, V.P.; Knutsen, A.P.; Greenberger, P.; Judson, M.A. Allergic bronchopulmonary aspergillosis in cystic fibrosis—State of the art: Cystic Fibrosis Foundation Consensus Conference. Clin. Infect. Dis. 2003, 37, S225–S264. [Google Scholar] [CrossRef]
- Mahdavinia, M.; Grammer, L.C. Management of allergic bronchopulmonary aspergillosis: A review and update. Ther. Adv. Respir. Dis. 2012, 6, 173–187. [Google Scholar] [CrossRef]
- Hilliard, T.; Edwards, S.; Buchdahl, R.; Francis, J.; Rosenthal, M.; Balfour-Lynn, I. Voriconazole therapy in children with cystic fibrosis. J. Cyst. Fibros. 2005, 4, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shoseyov, D.; Brownlee, K.G.; Conway, S.P.; Kerem, E. Aspergillus bronchitis in cystic fibrosis. Chest 2006, 130, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Launay, M.; Roux, A.; Beaumont, L.; Douvry, B.; Lecuyer, L.; Douez, E. Posaconazole Tablets in Real-Life Lung Transplantation: Impact on Exposure, Drug-Drug Interactions, and Drug Management in Lung Transplant Patients, Including Those with Cystic Fibrosis. Antimicrob. Agents Chemother. 2018, 62, e02061-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Blood Exams | Admission | After 6 Days | After 16 Days (Beginning of ABPA Therapy) | After 2 Weeks of ABPA Therapy |
|---|---|---|---|---|
| Leucocytes, cells/mm3 | 17,130 | 19,870 | 17,080 | 18,060 |
| Neutrophils, % | 75.4 | 66.6 | 59.3 | 68.4 |
| Eosinophils, % | 2.7 | 8.0 | 19.3 | 5.1 |
| CRP, mg/dL | 11.19 | 16.80 | 4.26 | 0.90 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peccini, L.; Pennoni, S.; Mencarini, V.; Saponara, M.; Palladino, N.; Principi, N.; Pennoni, G.; Esposito, S. A Peculiar Case of Pneumonia due to Mycoplasma pneumoniae in a Child with Cystic Fibrosis and Sensibilization to Aspergillus fumigatus. Pathogens 2020, 9, 15. https://doi.org/10.3390/pathogens9010015
Peccini L, Pennoni S, Mencarini V, Saponara M, Palladino N, Principi N, Pennoni G, Esposito S. A Peculiar Case of Pneumonia due to Mycoplasma pneumoniae in a Child with Cystic Fibrosis and Sensibilization to Aspergillus fumigatus. Pathogens. 2020; 9(1):15. https://doi.org/10.3390/pathogens9010015
Chicago/Turabian StylePeccini, Laura, Serena Pennoni, Valeria Mencarini, Marco Saponara, Nicola Palladino, Nicola Principi, Guido Pennoni, and Susanna Esposito. 2020. "A Peculiar Case of Pneumonia due to Mycoplasma pneumoniae in a Child with Cystic Fibrosis and Sensibilization to Aspergillus fumigatus" Pathogens 9, no. 1: 15. https://doi.org/10.3390/pathogens9010015