
Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges


Pole of Pharmacology and Therapeutics (FATH), Institut de Recherche Expérimentale et Clinique (IREC), UCLouvain, Avenue Hippocrate 57, B1.57.04, B-1200 Brussels, Belgium
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Organoids 2022, 1(1), 85-105; https://doi.org/10.3390/organoids1010008
Submission received: 20 May 2022
/
Revised: 9 June 2022
/
Accepted: 10 June 2022
/
Published: 13 June 2022
(This article belongs to the Special Issue Feature Papers in Organoids)
Abstract
:Organoid technologies represent a major breakthrough in biomedical research since they offer increasingly sophisticated models for studying biological mechanisms supporting human development and disease. Organoids are three-dimensional (3D) physiological in vitro systems that recapitulate the genetic, histological and functional features of the in vivo tissues of origin more accurately than classical cell culture methods. In the last decade, organoids have been derived from various healthy and diseased tissues and used for a wide range of applications in basic and translational research, including (cancer) tissue biology, development, regeneration, disease modeling, precision medicine, gene editing, biobanking and drug screening. Here, we report the current applications of organoid models to study (stem) cell metabolism in several pathophysiological contexts such as cancer and metabolic diseases. More precisely, we discuss the relevance and limitations of these 3D cultures to model and study metabolic (dys)functions associated with hepatic, renal or pancreatic disorders, as well as tumor development and progression. We also describe the use of organoids to understand the dynamic interaction between diet, microbiota and the intestinal epithelium. Finally, this review explores recent methodological improvements in organoid culture that may help to better integrate the influence of microenvironmental conditions in the study of tumor cell metabolic phenotypes.
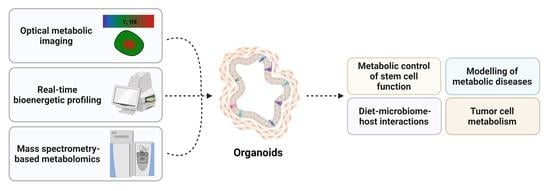
1. Introduction
Organoids are in vitro 3D structures in which cells spontaneously self-organize into progenitors and differentiated functional cell types, recapitulating complex aspects of the tissue of origin, from physiological processes to regeneration and disease [1]. They are primarily generated from primary tissues (single cells or tissue chunks) or stem cells such as adult stem cells (ASCs), induced pluripotent stem cells (iPSCs), or embryonic stem cells (ESCs). Since the seminal work of Sato and colleagues more than ten years ago [2] describing the establishment and growth of self-renewing intestinal epithelia that model the crypt–villus architecture, an extensive body of work has improved the methodology and expanded the range of tissues that can be studied [3,4]. Currently, ASC-derived organoids are established via two main methods, namely submerged culture (Figure 1a), which typically solely involves epithelial cells, and air–liquid interface (ALI) culture (Figure 1b), a more organotypic method that includes epithelial cells alongside integrated stromal and immune cells. In the former, organoids are embedded within solid gels of extracellular matrix (e.g., laminin-rich Matrigel, basement membrane extract (BME)) submerged beneath culture medium containing a tissue-specific combination of growth factors (e.g., ligands from the Wnt pathway, such as Wnt3a and/or R-spondin, epidermal and fibroblast growth factors (EGF, FGF) and the bone morphogenetic protein (BMP) inhibitor noggin) to allow ASCs to undergo long-term self-renewal and differentiation into all cell lineages. Moreover, Rho-associated protein kinase (ROCK) inhibitors have been shown to greatly increase the efficiency of organoid generation since they prevent the activation of programmed cell death, anoikis, as well as stress and injury responses upon cellular disaggregation during tissue processing. By comparison, the ALI method allows organoid initiation and long-term culturing upon the plating of mechanically dissociated tissue fragments into a type I collagen matrix on top of a permeable filter, with direct air exposure, thereby facilitating oxygen diffusion [5,6,7]. Such methodology has shown the ability to grow large multicellular organoids by preserving native tissue architecture, such as the epithelium, “en bloc” with endogenous immune and stromal elements, without reconstitution (i.e., indirect co-culture). Notably, ALI-based organoid cultures do not require the supplementation of exogenous growth factors, probably thanks to the production of essential endogenous niche factors by stromal cells. Finally, organoids can be derived from iPSCs through a series of differentiation steps, by culturing them with specific growth and signaling factors, which results in the generation of the desired tissue type (Figure 1c).
Advancements in 3D culture approaches have enabled organoids to be used to study various genomic, transcriptomic and proteomic alterations in many different pathophysiological situations. Although metabolic dysfunctions are now undoubtedly associated with a variety of diseases, studies describing the use of organoids to decipher cell metabolism are still scarce. In this review, we report the current applications and limitations of organoid cultures to model and study cell metabolism in metabolic diseases and cancer. We also describe the use of organoids to understand the dynamic interaction between circadian rhythms, diet, microbiota and the intestinal epithelium. Finally, this review explores recent methodological improvements that may help to better integrate the influence of microenvironmental conditions in the study of tumor cell metabolic phenotypes.
2. Organoid Models to Study the Metabolic Control of Stem Cell Function
Stem cells have the capacity for self-renewal and multipotent differentiation, which enables the regulation of tissue development and homeostasis. Several studies have indicated a role for metabolism in the function of some adult stem cell populations [8,9,10]. Based on complex microenvironmental cues, stem cells can indeed modify their metabolic preferences, in particular switching between glycolysis and mitochondrial oxidative metabolism, to fulfil bioenergetic and/or biosynthetic requirements during lineage specification, differentiation or maintenance [11]. The intestinal crypt is a perfect example to illustrate the influence of a microenvironmental niche on stem cell metabolic preferences and tissue homeostasis. Okkelman and colleagues have reported the application of live cell microscopy of oxygen, via the phosphorescence lifetime imaging microscopy (PLIM) method, to provide high-resolution real-time visualization of oxygen distribution in mouse intestinal organoids [12,13]. More precisely, the use of a cell-penetrating phosphorescent O2-sensitive probe revealed high heterogeneity in organoid oxygenation, with the existence of O2 microgradients, depending on the age of the culture and drug treatment, thereby indicating that integrating the metabolic heterogeneity is critical for proper data interpretation with organoids. In two other studies, the same authors also used fluorescence lifetime imaging microscopy (FLIM) to assess NAD(P)H levels as well as mitochondrial membrane potential and obtained a quantitative, multi-parameter, live readout of the balance between cell redox and energy production within the intestinal stem cell (ISC) niche [14,15] (Figure 2a). Other studies have allowed the measure of the real-time bioenergetic profile (i.e., oxygen consumption and extracellular acidification rates) of intestinal crypt organoids [16,17,18]. For instance, by using small intestinal organoids that recapitulate crypt structure and the interaction between Paneth cells and Lgr5+ (leucine-rich repeat-containing G protein-coupled receptor 5-positive) stem cells (i.e., crypt base columnar cells (CBCs)), Rodriguez-Colman and colleagues elegantly identified a role of metabolism in the maintenance of stem cell function [19]. The authors have described metabolic compartmentalization within organoids, with CBCs displaying high mitochondrial activity (as reflected by a high pyruvate/lactate ratio and increased mitochondrial membrane potential) while adjacent Paneth cells exhibit an enhanced glycolytic metabolism. Importantly, they have documented the establishment of a lactate-based metabolic symbiosis between the two cell types, with the lactate produced by Paneth cells serving as a respiratory substrate to sustain oxidative metabolism in Lgr5+ CBCs (Figure 2a). The interruption of this lactate shuttle with specific metabolic inhibitors in either cell type leads to decreased organoid reconstitution, thereby supporting the existence of a metabolic niche that provides optimal stem cell function in the intestinal crypt [19]. In proliferating Lgr5+ stem cells located at the base of the intestinal crypt, the inhibition of mitochondrial pyruvate import upon genetic deletion of MPC1 has been found to increase proliferation and expand the stem cell compartment [20]. A similar observation was made in intestinal organoids upon pharmacological inhibition of the mitochondrial pyruvate carrier (MPC) with UK-5099. In another study, jejunum-derived mouse organoids (enteroids) were used to assess the role of glutamine metabolism on ISC function [21]. The authors have shown that glutamine (as well as L-alanyl-L-glutamine, a stable glutamine dipeptide) supports intestinal epithelial homeostasis by promoting stem cell expansion, mTOR signal activation and crypt regeneration. Glutamine-deprived enteroids display gradual atrophy of crypt-like domains, with decreased epithelial proliferation via the activation of a reversible quiescent state in ISCs while maintaining Paneth and goblet cell differentiation. By using a similar model of jejunum-derived mouse enteroids, another study confirmed the important role of amino acid metabolism in the self-renewal and differentiation potential of ISCs [22]. The authors have indeed found that deprivation of the essential amino acid methionine markedly reduces the proportion of Lgr5+ stem cells and suppresses cell proliferation in enteroids, while enhancing expression of the enteroendocrine cell marker chromogranin A as well as markers of enterochromaffin, goblet and Paneth cells.
Several studies using intestinal organoids have also revealed a major contribution of lipid metabolism in the regulation of ISC activity. Indeed, high-fat or cholesterol-rich diets have been reported to alter the balance between self-renewal and differentiation in ISC and to endow organoid-initiating capacity to progenitors [23,24]. Fatty acid oxidation (FAO) has been found to support ISC renewal upon several dietary changes, such as short-term fasting [25] and high-fat diet [26], with hepatocyte nuclear factor 4 (HNF4) and peroxisome proliferator-activated receptor (PPAR) transcription factors as key regulators of the expression of FAO-related genes [26,27]. Pharmacological inhibition of carnitine palmitoyltransferase 1A (CPT1A), the mitochondrial acyl-CoA transporter, with etomoxir, reduced the diet-induced crypt organoid-forming capacities [25,26]. Importantly, since most intestinal tumors originate from ISCs [28,29,30], these studies have also shown that a high-fat diet, as well as excess dietary cholesterol, can promote intestinal tumor initiation, with this effect being delayed upon FAO inhibition [23,24,26,31]. Along the same lines, Sebastian and colleagues have elegantly shown, in a recent study, the presence of metabolic heterogeneity among intestinal epithelial cells by using adenoma-derived intestinal organoid models [32]. They reported that enhanced glycolytic metabolism, in the absence of SIRT6 deacetylase, drives intestinal tumorigenesis by increasing the number and activity of ISCs and by promoting their tumor-initiating potential. The authors have also identified a specific subpopulation of quiescent cells, with high pyruvate dehydrogenase kinase activity and increased stem cell potential, thereby providing new insights into the role of metabolism in ISC activity regulation and intestinal tumorigenesis.
Besides their applications in the understanding of ISC function, organoid models have been exploited to decipher stem cell metabolism in other tissues. Human pluripotent stem cell-derived cardiac organoids (hCOs) have been used to identify central regulators of the maturation process in the mammalian heart during postnatal life [33]. The use of hCOs in a miniaturized semiautomated cardiac organoid culture platform revealed that simulating the postnatal metabolic switch towards fatty acid utilization (at the detriment of carbohydrates) induced an increased expression of adult sarcomeric protein isoforms and cell cycle arrest. More precisely, the authors have found that a low-carbohydrate, low-insulin, palmitate-based medium represses key proliferation pathways, including β-catenin and Yes-associated protein 1 (YAP1) signaling, while inducing DNA damage response in hCOs, thereby supporting cardiac maturation. In another study, kidney organoids derived from iPSCs have been metabolically characterized to investigate the metabolic dynamics and function during kidney organoid differentiation [34]. Transcriptomics and untargeted metabolomics analyses validated a metabolic switch from glycolysis to mitochondrial oxidative phosphorylation (OXPHOS) during the iPSC differentiation process, also revealing a role for glycine, serine and threonine metabolism in the regulation of kidney organoid formation and lineage maturation (Figure 2b). More precisely, the authors described the contribution of serine metabolism in kidney organoid differentiation by regulating the production of S-adenosylmethionine and altering subsequent DNA methylation processes. Human pluripotent stem cell (hPSC)-derived retinal organoids have also allowed the characterization of metabolic changes accompanying photoreceptor differentiation [35]. By applying FLIM and hyperspectral imaging of organoids, the authors have revealed spatial and temporal metabolic changes during retinal organoid maturation, with increased glycolytic activity (detected with a higher free/bound NADH ratio) as well as retinol and retinoic acid accumulation in the organoid outer layer, coinciding with photoreceptor genesis. Finally, iPSC-derived human hepatic organoids have been generated and thoroughly characterized at the histological, transcriptional, metabolic and functional levels [36]. While the metabolic reprogramming of iPSCs from glycolysis to mitochondrial OXPHOS has been shown during organoid generation, transcriptomic analysis also revealed the increased expression of genes encoding proteins involved in many pathways of lipid metabolism, including the uptake and oxidation of exogenous fatty acids as well as cholesterol metabolism, upon differentiation. Altogether, these observations highlight the great potential of organoid models to reveal important roles of spatially and temporally controlled metabolic heterogeneity in the stem cell function and tissue homeostasis.
3. Organoids and Metabolic Diseases
Research in organoid models has opened up endless new possibilities for the study of metabolic diseases, including metabolic (dys)functions associated with hepatic, renal or pancreatic disorders, through the development of new models to assess functionality, pathogenicity and response to treatments [37,38].
3.1. Organoids to Model Liver Metabolic Diseases
Liver organoids, derived from either adult tissues (e.g., surgical resection, liver transplantation, needle biopsy) or iPSCs, are composed of multiple hepatic cell types, including hepatocytes, hepatic stellate cells and Kupffer cells. Several culture protocols have been reported to establish 3D liver organoids for the study of hepatocyte function, including glucose metabolism under normal and stress conditions [39], drug metabolism [40], as well as several hepatic metabolic diseases, including alpha-1 antitrypsin (A1AT) deficiency, citrullinemia type-1 (CTLN1), Wilson’s disease, Wolman’s disease and non-alcoholic fatty liver disease (NAFLD) [41,42,43] (Table 1).
3.1.1. Alpha-1 Antitrypsin Deficiency and Citrullinemia Type-1
A1AT deficiency is an inherited metabolic disorder caused by mutations in the SERPINA1 gene and characterized by low circulating levels of A1AT, a serine protease inhibitor known to protect the lung against proteolytic damage from neutrophil elastase. A1AT deficiency-related liver disease occurs due to the aberrant folding and subsequent intracellular retention of the mutant protein, thereby triggering endoplasmic reticulum (ER) stress and apoptosis in hepatocytes [53]. Huch and colleagues have reported that differentiation of cholangiocyte-derived liver organoids, established by using biopsies from patients with A1AT deficiency, could recapitulate in vitro fundamental characteristics of the disease, including the accumulation of A1AT protein aggregates, the reduction of A1AT secretion, the induction of ER stress and increased apoptosis [46]. Similar observations were recently reported when using liver organoids derived from patients with different A1AT deficiency-causing genotypes, thereby highlighting the potential of organoids for A1AT deficiency-related liver disease modeling [45].
Human iPSC-derived hepatic organoids have been used to model CTLN1, a urea cycle disorder caused by mutations in the gene encoding the argininosuccinate synthetase 1 (ASS1) enzyme that is essential for the conversion of excess ammonia into urea [47]. Indeed, the authors have shown that CTLN1 organoids exhibit an increased accumulation of ammonia in comparison to organoids derived from healthy controls while maintaining other important functions such as albumin secretion, glycogen storage and lipid uptake, as observed in patients. Importantly, they have reported that ammonia detoxification can be rescued in liver organoids upon overexpression of the wild-type ASS1 gene, thereby paving the way for gene therapy in CTLN1 patients.
3.1.2. Wilson’s and Wolman’s Diseases
Wilson’s disease is a rare genetic disorder caused by pathogenic loss-of-function variants in the ATP7B gene encoding a copper-dependent ATPase. It is characterized by disrupted copper homeostasis resulting in liver disease and/or neuropsychiatric symptoms. In dogs, mutations in the copper metabolism domain-containing 1 (COMMD1) gene have been associated with a defective biliary excretion of copper and with hepatic disorders similar to the clinical features observed in human patients with Wilson’s disease [54]. Nantasanti and colleagues have shown that canine liver organoids established from dogs with an autosomal recessive COMMD1 deficiency exhibit a higher intracellular accumulation of copper, compared to normal organoids, similar to the copper excretion defect in the in vivo situation [52]. Importantly, re-expression of a functional wild-type COMMD1 gene could restore copper excretion and improve liver organoid viability upon CuCl2 treatment. In a follow-up study, the same authors have provided preclinical proof of concept for organoid-based cell transplantation in vivo [51]. They have documented the use of autologous gene-corrected liver organoids for cell transplantation in a canine COMMD1-deficient model of copper storage disease. Although their results have shown low engraftment and repopulation rates, they have documented organoid cell survival up to two years post-transplantation.
Wolman’s disease is a severe form of lysosomal acid lipase (LAL) deficiency characterized by the accumulation of lipids in the tissues and organs of the body due to an impaired breakdown of triglycerides. Mutations in the LAL-encoding gene lead to a reduced or absent function of the enzyme, thereby triggering hepato(spleno)megaly and hepatic failure [55]. While the disease shares many characteristics of hepatic steatosis, life-threatening complications often develop during early childhood and it can be lethal in absence of treatment. Although Kanuma® (sebelipase alfa) is now approved as the first treatment for patients with LAL deficiency, new effective therapies are still urgently needed. By using iPSC-derived liver organoid models of steatohepatitis, Ouchi and colleagues confirmed a significant increase in lipid accumulation in organoids derived from patients with Wolman’s disease compared to healthy controls, a phenotype rescued by ectopic re-expression of the LAL enzyme [48]. These models were also used to test the farnesoid X receptor (FXR) agonist, obeticholic acid, for its efficiency in suppressing lipid accumulation in diseased organoids, thereby offering new therapeutic opportunities for patients with Wolman’s disease.
3.1.3. Non-Alcoholic Fatty Liver Disease
NAFLD encompasses a range of conditions caused by a build-up of fat in the liver and it ranges from hepatic steatosis to non-alcoholic steatohepatitis (NASH), a more serious form of the disease that includes liver inflammation and hepatocyte damage. NASH can progress to cirrhosis and liver failure as well as to the development of hepatocarcinoma. Current in vitro and in vivo models for NASH-related research, including primary human hepatocytes, hepatoma cell lines and animal models, have shown strong limitations in the identification and the study of new personalized therapies, thereby highlighting an urgent need for a predictive human model to assess the efficacy of anti-inflammatory and anti-fibrotic drugs. For this purpose, Ouchi and colleagues developed a multicellular iPSC-derived liver organoid model, composed of hepatocyte-, stellate- and Kupffer-like cells, which exhibit highly comparable transcriptomic signatures with the in vivo-derived tissues. Upon free fatty acid (FFA) treatment, organoids could recapitulate in vitro the key features of steatohepatitis, including steatosis, inflammation and fibrosis, in a sequential manner [48]. Remarkably, atomic force microscopy demonstrated that increased stiffness in organoids following FFA exposure was correlated with the severity of fibrosis. Similarly, another study reported lipid accumulation in liver organoids of human, mouse, cat and dog origin following treatment with FFA [50]. In a follow-up study, the same authors used feline liver organoid models to test drugs for their potential to reduce lipid accumulation and they identified T863 and AICAR (diacylglycerol O-acyltransferase 1 inhibitor and adenosine monophosphate-activated protein kinase activator, respectively), as two promising candidates for further clinical evaluation [49]. All these studies highlight the potential of organoids to model liver metabolic diseases and to offer new perspectives in personalized medicine and drug discovery.
3.2. Organoids to Model Pancreatic and Renal Metabolic Diseases
Organoid technology provides an opportunity to accelerate research on the pathophysiology of pancreatic and renal tissues through the development of 3D models that closely recapitulate key functional features of the organs.
3.2.1. Diabetes
Diabetes mellitus, characterized by hyperglycemia, is a group of chronic metabolic diseases affecting around 425 million people worldwide. While type 1 diabetes (T1D) is an autoimmune disease that results in the loss of insulin-producing β cells, leading to insulin deficiency, type 2 diabetes (T2D) is characterized by insulin resistance that may be combined with relatively reduced insulin secretion and it represents the most common form in adults. The pancreatic islet comprises several types of endocrine cells, including insulin-secreting β cells (~60%), glucagon-releasing α cells (~30%), somatostatin-releasing δ cells (~10%), pancreatic polypeptide-secreting PP cells and ghrelin-secreting ε cells [56]. By secreting hormones, islets are essential for controlling glucose homeostasis and islet transplantation has shown promise for the clinical management of patients with insulin-dependent diabetes. However, diabetes remains one of the most challenging health concerns since available drugs treat the symptoms but do not cure the disease. Human pancreatic islet organoids have recently emerged as promising models for diabetes to study the mechanisms supporting islet-related diseases, to test the potency and toxicity of novel therapeutic options and to serve as a considerable source of biological material for autologous islet transplantation, as extensively reviewed elsewhere [57,58,59,60]. Recent studies have optimized protocols to generate viable and functional islet organoids upon co-culturing of dissociated islet cells with human amniotic epithelial cells (hAECs) [61,62]. The authors have demonstrated that the incorporation of hAECs into islet organoids markedly improves engraftment, viability and graft function in a mouse T1D model, thereby offering new perspectives for the development of cell-based curative therapies for T1D patients. By using functionally mature iPSC-derived human islet-like organoids, another study has identified the non-canonical Wnt4 signaling as a main driver of the metabolic maturation necessary for efficient glucose-stimulated insulin secretion and has reported the potential of these models to re-establish glucose homeostasis in a diabetic mouse model [63]. Even more strikingly, the authors showed that ex vivo stimulation with interferon-γ induced endogenous expression of the immune checkpoint protein programmed death-ligand 1 (PD-L1) and suppressed T cell activation and graft rejection, thereby enabling the restoration of glucose homeostasis up to 50 days after transplantation. Altogether, these studies illustrate the great potential (but also current limitations) of organoid technology for diabetes research and therapy.
3.2.2. Kidney Diseases
Human and mouse iPSC-derived kidney organoids have been generated using different protocols [64,65,66,67]. An optimized method, based on the separate generation of nephron and ureteric bud progenitors before mixing in culture with embryo-derived stromal cells, has been shown to produce reassembled organoids in which the branching morphogenesis and inherent architecture of the embryonic kidney, including the peripheral progenitor niche and connection between nephrons and collecting ducts, were better recapitulated [68]. The generation and use of kidney organoids for biomedical research have been extensively reviewed by others in recent years [69,70,71,72]. Organoid cultures have successfully been applied to model several kidney diseases, including polycystic kidney disease [67,73,74], nephronophthisis-related ciliopathy [75], mucin 1 kidney disease [76] and podocytopathies [77,78]. A recent study also reported the use of human iPSC-derived kidney organoids combined with the CRISPR-Cas9 genome editing system to model Fabry disease, an X-linked lysosomal storage disease, upon the introduction of a mutation in the galactosidase alpha (GLA) gene [44]. The authors showed that GLA-mutant human kidney organoids phenocopied human Fabry nephropathy by inducing deformed podocytes and tubular cells with accumulation of globotriaosylceramide, similar to organoids expressing the wild-type enzyme. By using transmission electron microscopy and oil red O staining, they also documented the intracellular accumulation of lipid droplets, as well as damaged mitochondria, in kidney organoids expressing the mutant form of GLA. Gene expression profiling revealed a reduced metabolism of glutathione (GSH) in GLA-mutant kidney organoids and this was associated with increased oxidative stress and reactive oxygen species levels. Importantly, while enzyme replacement therapy with recombinant human α-Gal A attenuated oxidative stress as well as the structural and transcriptional changes in GLA-mutant human iPSC-derived kidney organoids, direct treatment with GSH could also reduce cell death and reverse the gene expression pattern (i.e., re-expression of podocyte and tubular markers), thereby positioning GSH as an efficient therapeutic option for Fabry disease. Finally, another recent study has described the establishment and use of hPSC-derived diabetic-like kidney organoids to unravel the molecular mechanisms underlying the higher susceptibility to SARS-CoV-2 infections in human diabetic patients [79]. The authors have observed that kidney organoids derived from diabetic patients display some metabolic alterations, including decreased mitochondrial biogenesis, increased oxidative metabolism and enhanced glycolysis, which result in increased susceptibility to SARS-CoV-2 infection. Indeed, the treatment of kidney organoids with dichloroacetate, an inhibitor of mitochondrial pyruvate dehydrogenase kinase, has been shown to increase mitochondrial oxidative metabolism (to the detriment of glycolysis) while reducing SARS-CoV-2 infection. These data provide new mechanistic insights to explain SARS-CoV-2 susceptibility in diabetic patients and open the door for the development and use of metabolism-interfering therapeutic strategies in the clinical management of COVID-19 patients.
4. Organoids for Modelling Diet–Microbiome–Host Interactions
Another field of application for organoid models is the study of the dynamic interaction between diet, microbiota and the host intestinal epithelium [80,81,82]. Complex regulatory networks and crosstalks occur between the host, its diet and its gut microbiota, and participate in the homeostasis of intestinal epithelial cells. Microorganisms and gut microbial metabolites have pivotal roles in a wide range of biological processes, including the regulation of lifespan, bioavailability and the biological activities of diet-, pharmaceuticals- and xenobiotics-derived compounds, thereby highlighting the need to extend our knowledge of diet–microbiome–host interactions. Intestinal organoids have rapidly proven to be valuable systems to overcome the limitations encountered with animal models or immortalized human cell lines grown as 2D monolayer cultures for such studies. They self-organize in vitro into 3D structures that closely recapitulate the tissue of origin [83] and they have been reported as relevant in vitro systems for concurrently studying nutrient transport, sensing and hormone secretion [84], drug uptake and metabolism [85], nutrient transport physiology during digestion [86], as well as dietary fat absorption [87]. By using intestinal organoids, Cai and colleagues have observed that, although several dietary constituents do not affect organoid growth, caffeic acid exhibits growth-inhibitory effects in a dose-dependent manner by reducing crypt-like structure formation [88]. However, contradictory effects were obtained with other compounds, such as monosodium glutamate and chlorogenic acid, thereby highlighting the need for future studies to carefully explore effects of phytochemicals on intestinal organoids. By using mouse gut organoids, Lukovac and colleagues documented that microbially produced short-chain fatty acids (SCFAs), including acetate, propionate and butyrate, regulate distinct gene expression profiles [89]. More precisely, they showed that the exposure of mature ileal organoids to supernatants collected from Akkermansia muciniphila affected several transcription factors and genes involved in fatty acid, cholesterol and bile acid metabolism, such as NR1H3, CPT1A and HMGCS1. Another study using intestinal organoids also revealed that butyrate treatment could improve tight junction integrity in intestinal epithelial cells, decrease apoptosis and mitigate graft-versus-host disease [90]. Moreover, Schilderink and colleagues showed that the butyrate-induced upregulation of ALDH1A1 and ALDH1A3 in human and mouse intestinal organoids contributed to retinoic acid production and the maintenance of gut homeostasis [91]. By exploiting the intestinal organoid technology, Bellono and colleagues also demonstrated the role of serotonergic enterochromaffin cells as chemosensors in the gut epithelium to detect and transduce environmental, metabolic, and homeostatic information from the gut directly to the nervous system [92]. Microbiota-derived SCFAs were shown to improve metabolic activation in human iPSC-derived liver organoids by promoting CYP3A4 expression and albumin secretion and identified troglitazone-induced hepatotoxicity [93]. Importantly, the latter effect could be reversed upon treatment with ketoconazole, a potent CYP3A4 inhibitor, highlighting the great potential of this culture system to evaluate CYP3A4-dependent drug toxicity. Finally, a recent study by Rosselot and colleagues elegantly showed that human intestinal organoids possess circadian rhythms and exhibit circadian phase-dependent necrotic cell death responses to Clostridium difficile toxin B [94]. Importantly, the authors showed that the source utilized for organoid generation was critical since iPSC-derived intestinal organoids did not display circadian rhythms while more mature structures such human intestinal enteroids exhibited robust circadian oscillation of key clock-controlled genes, thereby providing new insights for the use of organoid models in circadian medicine [95].
Altogether, these studies demonstrate that intestinal organoids recapitulate the gut microenvironment and, therefore, represent suitable models for studying the diet–microbiome–host interactions as well as circadian rhythms. This research field is still evolving and benefits from recent technical improvements, including the use of disrupted organoids [96] or microinjections [97], to accurately mimic the gut architecture, luminal accessibility and tissue polarity.
5. Organoids and Tumor Metabolism
Tumors are complex and dynamic ecosystems in which the subclonal populations of cancer cells must adapt their metabolism to support disease progression [98]. Within the tumor microenvironment (TME), the limited access to oxygen and/or nutrients and the accumulation of protons in local compartments, together with the presence of tumor-associated stromal and immune cells generate metabolic heterogeneity [99,100]. Thus, according to their local TME, cancer (stem) cells can use a variety of substrates (e.g., glucose, amino acids, fatty acids) to fulfill their need in energy (ATP), biosynthetic precursors and reduced cofactors (NADH, NADPH) so that facilitating their survival, proliferation, metastasis and the development of resistance to anticancer therapies [101,102,103,104]. The TME-driven metabolic phenotype of cancer cells (and surrounding cells) evolves de facto with time and tumor development. The relevant pre-clinical models are therefore needed to explore the intimate relationship between TME and (cancer) cell metabolic preferences and to design novel strategies aiming to integrate and therapeutically exploit TME- and therapy-mediated metabolic addictions in cancer cells [105,106]. Major recent advances in 3D culture technology have led to the development of patient-derived tumor organoids (PDTO) for a variety of cancer types, including breast [107], bladder [108], colorectal [109], head and neck [110], liver [111], pancreatic [112], gastrointestinal [113], glioblastoma [114], retinoblastoma [115] and prostate cancers [116]. Importantly, they have been shown to recapitulate the histological and genetic features, as well as drug response, of the original tumor, thereby making them miniaturized in vitro avatars of patient tumors. Moreover, they retain the physicochemical characteristics of TME, such as hypoxic gradients [117,118,119], making them suitable tools for studying intratumoral metabolic heterogeneity. In the last decade, optical metabolic imaging (OMI) has been applied to quantify the fluorescence intensity and lifetime of NAD(P)H and FAD and detect distinct metabolic cell states within tumor organoids. Several studies have indeed shown that such technique allowed the identification of metabolic heterogeneity and the prediction of therapeutic response in organoid models of breast [120,121,122], colorectal [123], gastroenteropancreatic neuroendocrine [124], head and neck [125], and pancreatic cancers [126]. Besides fluorescence-based OMI, matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI) technology has been used in several studies to assess the distribution of anticancer drugs, as well as their metabolites, in a variety of PDTO models [127,128,129]. These data pave the way for the use of OMI and MALDI-MSI in PDTO as high-throughput platforms to identify optimal therapies for individual patients and to integrate cancer cell metabolism in the current genomics-driven cancer paradigm for precision oncology [130]. In addition, although metabolomics and 13C tracer analysis have been established as state-of-the-art techniques to determine the concentration of metabolites and the activity of metabolic pathways, respectively, there are very few examples of such studies in 3D organoid models. The metabolome of (tumor) organoids has been captured by using NMR [131] and targeted [132] or non-targeted [133,134,135] liquid chromatography (LC)-MS-based profiling. Neef and colleagues have recently performed LC-qTOF-MS-based metabolic and lipidomic profiling on colorectal cancer organoids to reveal changes upon treatment with 5-fluorouracil [136]. They observed major alterations in levels of metabolites involved in purine and pyrimidine metabolism, in accordance with the mechanism of action of the drug, thereby providing the first basis of evidence for assessing drug-induced metabolic response in 3D organoid models. However, since the influence of background signals derived from the basal membrane extract used for organoid culturing on metabolomics data processing is still not fully appreciated, studies reporting metabolomics analyses on organoid models remain scarce.
In most of the metabolic studies reported so far, organoids are solely composed of epithelial cancer cells embedded in an extracellular matrix (e.g., Matrigel) and cultured in a specific medium containing a defined combination of stem cell niche factors. Nevertheless, these models have demonstrated a great potential to reveal and study the epithelial–mesenchymal plasticity (EMP) of cancer cells [137,138,139] and they have been already used to screen drugs that can reverse the epithelial–mesenchymal transition [140]. This is of particular interest since many studies have reported a link between metabolism and EMP in development and disease, including cancer [141,142]. Still, the lack of stromal and immune cell components is actually a major limitation since tumor cells can either cooperate (i.e., metabolic symbiosis) or compete with these non-cancerous cell populations during tumor development [143]. For instance, while PDTO models of colorectal cancer were shown to recapitulate many of the genetic and transcriptomic features of donor tumors as well as response to anticancer drugs, they failed to reveal the high complexity of metabolism-related molecular alterations, as observed in primary tumors and partly in patient-derived tumor xenograft models [144]. Recent studies have reported new modalities of PDTO generation, such as the ALI method, to preserve “en bloc” cancer cells with tumor stroma, including cancer-associated fibroblasts (CAFs) and even functional native immune cells (T and B cells, myeloid cells, macrophages and NK cells) [145]. Alternatively, direct co-cultures of tumor organoids with autologous CAFs [146,147,148] or peripheral blood lymphocytes [149,150,151,152] have been described and may represent useful tools to study the metabolic communication within TME and better recapitulate the intratumoral metabolic heterogeneity (Figure 3).
Recently, by using a co-culture model of pancreatic cancer organoids with pancreatic stellate cells (PSCs) combined with OMI technology, Datta and colleagues reported the existence of a pyruvate-based metabolic symbiosis whereby fibroblasts facilitate oxidation reactions in cancer cells to support proliferation [153]. Another challenge in organoid cultures is the lack of functional vasculature, which may lead to the development of immature organoids and further prevent the study of anti-angiogenic drugs in these models. Co-culturing of human endothelial cells with mouse breast organoids has shown the development of a functional capillary vessel network, connected to the mice circulatory system, in an orthotopic model of human breast cancer [154]. Other studies have reported new experimental strategies to generate vascularized organoids [155,156], which may be further used for metabolic studies. Finally, although companies are developing synthetic scaffolds with the potential to offer xenogenic-free, chemically defined, highly tunable and reproducible alternatives [157], a major limitation of organoid cultures is still their reliance on an animal-derived extracellular matrix and defined medium conditions that might not represent nutrient levels encountered by cells within the tumor.
6. Conclusions
Organoid models are being rapidly integrated into various aspects of biomedical research and are constantly evolving due to improved derivation protocols and culture conditions. Applying organoids to the study of cell metabolism in a variety of pathophysiological contexts, such as metabolic diseases, cancer and diet–microbiome–host interactions, has the potential to greatly reduce the number of animal models used for equivalent purposes. Although the reduced cellular complexity in organoid models remains a major limitation for the study of metabolism in biological processes where multiple cell types interact in a spatial and highly dynamic way, recent improvements in culture methods and emerging technologies have the potential to allow metabolic characterization in organoids with single-cell and subcellular resolution (Table 2). We are thus convinced that all these new insights and novel technological approaches will help better assess metabolic (dys)function in a variety of diseases and will pave the way for the implementation of metabolic studies in organoids for disease detection, surveillance and treatment to improve outcomes and quality of life for patients with metabolic disorders.
Author Contributions
Conceptualization, C.C.; methodology; validation: E.R., V.V.d.B. and C.C.; writing—original draft preparation, E.R., V.V.d.B. and C.C.; writing—review and editing, E.R., V.V.d.B. and C.C.; supervision, C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
All figures have been created with https://biorender.com/ (accessed on 9 June 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.H.; Karlsson, K.; Kuo, C.J. Applications of Organoids for Cancer Biology and Precision Medicine. Nat. Cancer 2020, 1, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Katano, T.; Ootani, A.; Mizoshita, T.; Tanida, S.; Tsukamoto, H.; Ozeki, K.; Ebi, M.; Mori, Y.; Kataoka, H.; Kamiya, T.; et al. Establishment of a long-term three-dimensional primary culture of mouse glandular stomach epithelial cells within the stem cell niche. Biochem. Biophys. Res. Commun. 2013, 432, 558–563. [Google Scholar] [CrossRef]
- Flores, A.; Schell, J.; Krall, A.S.; Jelinek, D.; Miranda, M.; Grigorian, M.; Braas, D.; White, A.C.; Zhou, J.L.; Graham, N.A.; et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 2017, 19, 1017–1026. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Glasauer, A.; Hoover, P.; Yang, S.; Blatt, H.; Mullen, A.R.; Getsios, S.; Gottardi, C.J.; DeBerardinis, R.J.; Lavker, R.M.; et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal. 2013, 6, ra8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C. Stem Cell Metabolism in Cancer and Healthy Tissues: Pyruvate in the Limelight. Front. Pharmacol. 2017, 8, 958. [Google Scholar] [CrossRef] [Green Version]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Multi-Parametric Imaging of Hypoxia and Cell Cycle in Intestinal Organoid Culture. Adv. Exp. Med. Biol. 2017, 1035, 85–103. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Live cell imaging of mouse intestinal organoids reveals heterogeneity in their oxygenation. Biomaterials 2017, 146, 86–96. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Neto, N.; Papkovsky, D.B.; Monaghan, M.G.; Dmitriev, R.I. A deeper understanding of intestinal organoid metabolism revealed by combining fluorescence lifetime imaging microscopy (FLIM) and extracellular flux analyses. Redox. Biol. 2020, 30, 101420. [Google Scholar] [CrossRef]
- Okkelman, I.A.; Papkovsky, D.B.; Dmitriev, R.I. Estimation of the Mitochondrial Membrane Potential Using Fluorescence Lifetime Imaging Microscopy. Cytom. A 2020, 97, 471–482. [Google Scholar] [CrossRef]
- Bas, T.; Augenlicht, L.H. Real time analysis of metabolic profile in ex vivo mouse intestinal crypt organoid cultures. J. Vis. Exp. 2014, 93, e52026. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.Y.; Davidson, L.A.; Callaway, E.S.; Wright, G.A.; Safe, S.; Chapkin, R.S. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 309, G1–G9. [Google Scholar] [CrossRef] [Green Version]
- Ludikhuize, M.C.; Meerlo, M.; Burgering, B.M.T.; Rodriguez Colman, M.J. Protocol to profile the bioenergetics of organoids using Seahorse. STAR Protoc. 2021, 2, 100386. [Google Scholar] [CrossRef]
- Rodriguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Moore, S.R.; Guedes, M.M.; Costa, T.B.; Vallance, J.; Maier, E.A.; Betz, K.J.; Aihara, E.; Mahe, M.M.; Lima, A.A.; Oria, R.B.; et al. Glutamine and alanyl-glutamine promote crypt expansion and mTOR signaling in murine enteroids. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G831–G839. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Iwatsuki, K.; Hanyu, H.; Maruyama, N.; Aihara, E.; Tadaishi, M.; Shimizu, M.; Kobayashi-Hattori, K. Effect of essential amino acids on enteroids: Methionine deprivation suppresses proliferation and affects differentiation in enteroid stem cells. Biochem. Biophys. Res. Commun. 2017, 488, 171–176. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martin, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220.e4. [Google Scholar] [CrossRef] [Green Version]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.W.; Cao, A.Q.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769–778.e4. [Google Scholar] [CrossRef] [Green Version]
- Mana, M.D.; Hussey, A.M.; Tzouanas, C.N.; Imada, S.; Barrera Millan, Y.; Bahceci, D.; Saiz, D.R.; Webb, A.T.; Lewis, C.A.; Carmeliet, P.; et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 2021, 35, 109212. [Google Scholar] [CrossRef]
- Chen, L.; Vasoya, R.P.; Toke, N.H.; Parthasarathy, A.; Luo, S.; Chiles, E.; Flores, J.; Gao, N.; Bonder, E.M.; Su, X.; et al. HNF4 Regulates Fatty Acid Oxidation and Is Required for Renewal of Intestinal Stem Cells in Mice. Gastroenterology 2020, 158, 985–999.e9. [Google Scholar] [CrossRef]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Beyaz, S.; Chung, C.; Mou, H.; Bauer-Rowe, K.E.; Xifaras, M.E.; Ergin, I.; Dohnalova, L.; Biton, M.; Shekhar, K.; Eskiocak, O.; et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 2021, 28, 1922–1935.e25. [Google Scholar] [CrossRef]
- Sebastian, C.; Ferrer, C.; Serra, M.; Choi, J.E.; Ducano, N.; Mira, A.; Shah, M.S.; Stopka, S.A.; Perciaccante, A.J.; Isella, C.; et al. A non-dividing cell population with high pyruvate dehydrogenase kinase activity regulates metabolic heterogeneity and tumorigenesis in the intestine. Nat. Commun. 2022, 13, 1503. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–E8381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Xiong, Y.; Zhang, S.; Sui, Y.; Yu, C.; Liu, P.; Li, H.; Guo, W.; Gao, Y.; Przepiorski, A.; et al. The Dynamics of Metabolic Characterization in iPSC-Derived Kidney Organoid Differentiation via a Comparative Omics Approach. Front. Genet. 2021, 12, 632810. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.W.; Arnesano, C.; Harutyunyan, N.; Khuu, T.; Martinez, J.C.; Pollack, H.A.; Koos, D.S.; Lee, T.C.; Fraser, S.E.; Moats, R.A.; et al. Structural and Functional Characterization of Human Stem-Cell-Derived Retinal Organoids by Live Imaging. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3311–3318. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Bustos, J.F.; Alvarado Gonzalez, J.C.; de Abreu, D.A.R.; Liebisch-Rey, H.; Silva, A.; Ortiz, D.; Ramirez, L.B.; Ortega, J.; Celis Regalado, L.G. Modeling Metabolic Diseases with Organoids: A Review. J. Biomed. Res. Environ. Sci. 2021, 2, 272–279. [Google Scholar] [CrossRef]
- Rauth, S.; Karmakar, S.; Batra, S.K.; Ponnusamy, M.P. Recent advances in organoid development and applications in disease modeling. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188527. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, G.; Shen, C.; Uygun, K.; Yarmush, M.L.; Meng, Q. A novel 3D liver organoid system for elucidation of hepatic glucose metabolism. Biotechnol. Bioeng. 2012, 109, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Kim, H.K.; Jee, J.; Hahn, S.; Jeong, S.; Yoo, J. Development of organoid-based drug metabolism model. Toxicol. Appl. Pharmacol. 2019, 385, 114790. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, dmm039347. [Google Scholar] [CrossRef] [Green Version]
- Nantasanti, S.; de Bruin, A.; Rothuizen, J.; Penning, L.C.; Schotanus, B.A. Concise Review: Organoids Are a Powerful Tool for the Study of Liver Disease and Personalized Treatment Design in Humans and Animals. Stem Cells Transl. Med. 2016, 5, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.M.; Kim, Y.K. Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef]
- Gomez-Mariano, G.; Matamala, N.; Martinez, S.; Justo, I.; Marcacuzco, A.; Jimenez, C.; Monzon, S.; Cuesta, I.; Garfia, C.; Martinez, M.T.; et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol. Int. 2020, 14, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Akbari, S.; Sevinc, G.G.; Ersoy, N.; Basak, O.; Kaplan, K.; Sevinc, K.; Ozel, E.; Sengun, B.; Enustun, E.; Ozcimen, B.; et al. Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Rep. 2019, 13, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Haaker, M.W.; Kruitwagen, H.S.; Vaandrager, A.B.; Houweling, M.; Penning, L.C.; Molenaar, M.R.; van Wolferen, M.E.; Oosterhoff, L.A.; Spee, B.; Helms, J.B. Identification of potential drugs for treatment of hepatic lipidosis in cats using an in vitro feline liver organoid system. J. Vet. Intern. Med. 2020, 34, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Kruitwagen, H.S.; Oosterhoff, L.A.; Vernooij, I.; Schrall, I.M.; van Wolferen, M.E.; Bannink, F.; Roesch, C.; van Uden, L.; Molenaar, M.R.; Helms, J.B.; et al. Long-Term Adult Feline Liver Organoid Cultures for Disease Modeling of Hepatic Steatosis. Stem Cell Rep. 2017, 8, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Kruitwagen, H.S.; Oosterhoff, L.A.; van Wolferen, M.E.; Chen, C.; Nantasanti Assawarachan, S.; Schneeberger, K.; Kummeling, A.; van Straten, G.; Akkerdaas, I.C.; Vinke, C.R.; et al. Long-Term Survival of Transplanted Autologous Canine Liver Organoids in a COMMD1-Deficient Dog Model of Metabolic Liver Disease. Cells 2020, 9, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nantasanti, S.; Spee, B.; Kruitwagen, H.S.; Chen, C.; Geijsen, N.; Oosterhoff, L.A.; van Wolferen, M.E.; Pelaez, N.; Fieten, H.; Wubbolts, R.W.; et al. Disease Modeling and Gene Therapy of Copper Storage Disease in Canine Hepatic Organoids. Stem Cell Rep. 2015, 5, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoller, J.K.; Aboussouan, L.S. Alpha1-antitrypsin deficiency. Lancet 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- Favier, R.P.; Spee, B.; Schotanus, B.A.; van den Ingh, T.S.; Fieten, H.; Brinkhof, B.; Viebahn, C.S.; Penning, L.C.; Rothuizen, J. COMMD1-deficient dogs accumulate copper in hepatocytes and provide a good model for chronic hepatitis and fibrosis. PLoS ONE 2012, 7, e42158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, Z.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Coric, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency—An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68. [Google Scholar] [CrossRef]
- Tsakmaki, A.; Fonseca Pedro, P.; Bewick, G.A. Diabetes through a 3D lens: Organoid models. Diabetologia 2020, 63, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Bittenglova, K.; Habart, D.; Saudek, F.; Koblas, T. The Potential of Pancreatic Organoids for Diabetes Research and Therapy. Islets 2021, 13, 85–105. [Google Scholar] [CrossRef]
- Jiang, L.; Shen, Y.; Liu, Y.; Zhang, L.; Jiang, W. Making human pancreatic islet organoids: Progresses on the cell origins, biomaterials and three-dimensional technologies. Theranostics 2022, 12, 1537–1556. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Z.; Song, E.; Xu, T. Islet organoid as a promising model for diabetes. Protein Cell 2022, 13, 239–257. [Google Scholar] [CrossRef]
- Wassmer, C.H.; Lebreton, F.; Bellofatto, K.; Perez, L.; Cottet-Dumoulin, D.; Andres, A.; Bosco, D.; Berney, T.; Othenin-Girard, V.; De Tejada, B.M.; et al. Bio-Engineering of Pre-Vascularized Islet Organoids for the Treatment of Type 1 Diabetes. Transpl. Int. 2021, 35, 10214. [Google Scholar] [CrossRef]
- Lebreton, F.; Lavallard, V.; Bellofatto, K.; Bonnet, R.; Wassmer, C.H.; Perez, L.; Kalandadze, V.; Follenzi, A.; Boulvain, M.; Kerr-Conte, J.; et al. Insulin-producing organoids engineered from islet and amniotic epithelial cells to treat diabetes. Nat. Commun. 2019, 10, 4491. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; de Sousa Lopes, S.M.C.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; Nishinakamura, R. Higher-Order Kidney Organogenesis from Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 730–746.e6. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Cardilla, A.; Ngeow, J.; Gong, X.; Xia, Y. Studying Kidney Diseases Using Organoid Models. Front. Cell Dev. Biol. 2022, 10, 845401. [Google Scholar] [CrossRef]
- Romero-Guevara, R.; Ioannides, A.; Xinaris, C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front. Physiol. 2020, 11, 563981. [Google Scholar] [CrossRef]
- Islam, M.; Nishinakamura, R. How to rebuild the kidney: Recent advances in kidney organoids. J. Biochem. 2019, 166, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Nishinakamura, R. Human kidney organoids: Progress and remaining challenges. Nat. Rev. Nephrol. 2019, 15, 613–624. [Google Scholar] [CrossRef]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, T.A.; Howden, S.E.; Lawlor, K.; Phipson, B.; Maksimovic, J.; Hale, L.; Wilson, S.; Quinlan, C.; Ho, G.; Holman, K.; et al. Patient-iPSC-Derived Kidney Organoids Show Functional Validation of a Ciliopathic Renal Phenotype and Reveal Underlying Pathogenetic Mechanisms. Am. J. Hum. Genet. 2018, 102, 816–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvela-Levitt, M.; Kost-Alimova, M.; Emani, M.; Kohnert, E.; Thompson, R.; Sidhom, E.H.; Rivadeneira, A.; Sahakian, N.; Roignot, J.; Papagregoriou, G.; et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 2019, 178, 521–535.e3. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Islam, M.; Sharmin, S.; Naganuma, H.; Yoshimura, Y.; Haque, F.; Era, T.; Nakazato, H.; Nakanishi, K.; Sakuma, T.; et al. Organoids from Nephrotic Disease-Derived iPSCs Identify Impaired NEPHRIN Localization and Slit Diaphragm Formation in Kidney Podocytes. Stem Cell Rep. 2018, 11, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, L.J.; Howden, S.E.; Phipson, B.; Lonsdale, A.; Er, P.X.; Ghobrial, I.; Hosawi, S.; Wilson, S.; Lawlor, K.T.; Khan, S.; et al. 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 2018, 9, 5167. [Google Scholar] [CrossRef] [Green Version]
- Garreta, E.; Prado, P.; Stanifer, M.L.; Monteil, V.; Marco, A.; Ullate-Agote, A.; Moya-Rull, D.; Vilas-Zornoza, A.; Tarantino, C.; Romero, J.P.; et al. A diabetic milieu increases ACE2 expression and cellular susceptibility to SARS-CoV-2 infections in human kidney organoids and patient cells. Cell Metab. 2022, 34, 857–873.e9. [Google Scholar] [CrossRef]
- Cassotta, M.; Forbes-Hernandez, T.Y.; Calderon Iglesias, R.; Ruiz, R.; Elexpuru Zabaleta, M.; Giampieri, F.; Battino, M. Links between Nutrition, Infectious Diseases, and Microbiota: Emerging Technologies and Opportunities for Human-Focused Research. Nutrients 2020, 12, 1827. [Google Scholar] [CrossRef]
- Rubert, J.; Schweiger, P.J.; Mattivi, F.; Tuohy, K.; Jensen, K.B.; Lunardi, A. Intestinal Organoids: A Tool for Modelling Diet-Microbiome-Host Interactions. Trends Endocrinol. Metab. 2020, 31, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Nigro, G.; Hanson, M.; Fevre, C.; Lecuit, M.; Sansonetti, P.J. Intestinal Organoids as a Novel Tool to Study Microbes-Epithelium Interactions. Methods Mol. Biol. 2019, 1576, 183–194. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793.e6. [Google Scholar] [CrossRef] [Green Version]
- Zietek, T.; Rath, E.; Haller, D.; Daniel, H. Intestinal organoids for assessing nutrient transport, sensing and incretin secretion. Sci. Rep. 2015, 5, 16831. [Google Scholar] [CrossRef] [Green Version]
- Zietek, T.; Giesbertz, P.; Ewers, M.; Reichart, F.; Weinmuller, M.; Urbauer, E.; Haller, D.; Demir, I.E.; Ceyhan, G.O.; Kessler, H.; et al. Organoids to Study Intestinal Nutrient Transport, Drug Uptake and Metabolism—Update to the Human Model and Expansion of Applications. Front. Bioeng. Biotechnol. 2020, 8, 577656. [Google Scholar] [CrossRef]
- Foulke-Abel, J.; In, J.; Yin, J.; Zachos, N.C.; Kovbasnjuk, O.; Estes, M.K.; de Jonge, H.; Donowitz, M. Human Enteroids as a Model of Upper Small Intestinal Ion Transport Physiology and Pathophysiology. Gastroenterology 2016, 150, 638–649.e8. [Google Scholar] [CrossRef] [Green Version]
- Jattan, J.; Rodia, C.; Li, D.; Diakhate, A.; Dong, H.; Bataille, A.; Shroyer, N.F.; Kohan, A.B. Using primary murine intestinal enteroids to study dietary TAG absorption, lipoprotein synthesis, and the role of apoC-III in the intestine. J. Lipid Res. 2017, 58, 853–865. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Qi, Y.; Jergens, A.; Wannemuehler, M.; Barrett, T.A.; Wang, Q. Effects of six common dietary nutrients on murine intestinal organoid growth. PLoS ONE 2018, 13, e0191517. [Google Scholar] [CrossRef] [Green Version]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [Green Version]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; Seppen, J.; Muncan, V.; van den Brink, G.R.; Lambers, T.T.; van Tol, E.A.; de Jonge, W.J. The SCFA butyrate stimulates the epithelial production of retinoic acid via inhibition of epithelial HDAC. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G1138–G1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017, 170, 185–198.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, S.J.; Lee, J.; Chung, K.S.; Son, M.Y.; Son, M.J. Effect of Microbial Short-Chain Fatty Acids on CYP3A4-Mediated Metabolic Activation of Human Pluripotent Stem Cell-Derived Liver Organoids. Cells 2021, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Rosselot, A.E.; Park, M.; Kim, M.; Matsu-Ura, T.; Wu, G.; Flores, D.E.; Subramanian, K.R.; Lee, S.; Sundaram, N.; Broda, T.R.; et al. Ontogeny and function of the circadian clock in intestinal organoids. EMBO J. 2022, 41, e106973. [Google Scholar] [CrossRef]
- Yamada, R.G.; Ueda, H.R. The circadian clock ticks in organoids. EMBO J. 2022, 41, e110157. [Google Scholar] [CrossRef]
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2018, 25, 1657–1670. [Google Scholar] [CrossRef] [Green Version]
- Williamson, I.A.; Arnold, J.W.; Samsa, L.A.; Gaynor, L.; DiSalvo, M.; Cocchiaro, J.L.; Carroll, I.; Azcarate-Peril, M.A.; Rawls, J.F.; Allbritton, N.L.; et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 301–319. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, 6487. [Google Scholar] [CrossRef]
- Goncalves, A.C.; Richiardone, E.; Jorge, J.; Polonia, B.; Xavier, C.P.R.; Salaroglio, I.C.; Riganti, C.; Vasconcelos, M.H.; Corbet, C.; Sarmento-Ribeiro, A.B. Impact of cancer metabolism on therapy resistance—Clinical implications. Drug Resist. Updat. 2021, 59, 100797. [Google Scholar] [CrossRef]
- Van den Bossche, V.; Zaryouh, H.; Vara-Messler, M.; Vignau, J.; Machiels, J.P.; Wouters, A.; Schmitz, S.; Corbet, C. Microenvironment-driven intratumoral heterogeneity in head and neck cancers: Clinical challenges and opportunities for precision medicine. Drug Resist. Updat. 2022, 60, 100806. [Google Scholar] [CrossRef]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Emerging roles of lipid metabolism in cancer progression. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 254–260. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Metabolic and mind shifts: From glucose to glutamine and acetate addictions in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Vander Linden, C.; Corbet, C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 2019, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Vander Linden, C.; Corbet, C.; Bastien, E.; Martherus, R.; Guilbaud, C.; Petit, L.; Wauthier, L.; Loriot, A.; De Smet, C.; Feron, O. Therapy-induced DNA methylation inactivates MCT1 and renders tumor cells vulnerable to MCT4 inhibition. Cell Rep. 2021, 35, 109202. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lohmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Retinoblastoma from human stem cell-derived retinal organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [Green Version]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 4862. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.J.; Cook, R.S.; Sanders, M.E.; Aurisicchio, L.; Ciliberto, G.; Arteaga, C.L.; Skala, M.C. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 2014, 74, 5184–5194. [Google Scholar] [CrossRef] [Green Version]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-Derived Cancer Organoid Cultures to Predict Sensitivity to Chemotherapy and Radiation. Clin. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Gillette, A.A.; Babiarz, C.P.; VanDommelen, A.R.; Pasch, C.A.; Clipson, L.; Matkowskyj, K.A.; Deming, D.A.; Skala, M.C. Autofluorescence Imaging of Treatment Response in Neuroendocrine Tumor Organoids. Cancers 2021, 13, 1873. [Google Scholar] [CrossRef]
- Shah, A.T.; Heaster, T.M.; Skala, M.C. Metabolic Imaging of Head and Neck Cancer Organoids. PLoS ONE 2017, 12, e0170415. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef]
- David, B.P.; Dubrovskyi, O.; Speltz, T.E.; Wolff, J.J.; Frasor, J.; Sanchez, L.M.; Moore, T.W. Using Tumor Explants for Imaging Mass Spectrometry Visualization of Unlabeled Peptides and Small Molecules. ACS Med. Chem. Lett. 2018, 9, 768–772. [Google Scholar] [CrossRef]
- Liu, X.; Flinders, C.; Mumenthaler, S.M.; Hummon, A.B. MALDI Mass Spectrometry Imaging for Evaluation of Therapeutics in Colorectal Tumor Organoids. J. Am. Soc. Mass. Spectrom. 2018, 29, 516–526. [Google Scholar] [CrossRef]
- Wang, Y.; Hummon, A.B. MS imaging of multicellular tumor spheroids and organoids as an emerging tool for personalized medicine and drug discovery. J. Biol. Chem. 2021, 297, 101139. [Google Scholar] [CrossRef]
- Vander Linden, C.; Corbet, C. Reconciling environment-mediated metabolic heterogeneity with the oncogene-driven cancer paradigm in precision oncology. Semin. Cell Dev. Biol. 2020, 98, 202–210. [Google Scholar] [CrossRef]
- Weygand, J.; Carter, S.E.; Salzillo, T.C.; Moussalli, M.; Dai, B.; Dutta, P.; Zuo, X.; Fleming, J.B.; Shureiqi, I.; Bhattacharya, P. Can an organoid recapitulate the metabolome of its parent tissue? A pilot NMR spectroscopy study. J. Cancer Prev. Curr. Res. 2017, 8, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, H.; Ogiso, H.; Okazaki, T.; Kiyokawa, E. Comparative lipid analysis in the normal and cancerous organoids of MDCK cells. J. Biochem. 2016, 159, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.; Mukha, D.; Maor, I.I.; Sedov, E.; Koren, E.; Yosefzon, Y.; Shlomi, T.; Fuchs, Y. Blimp1+ cells generate functional mouse sebaceous gland organoids in vitro. Nat. Commun. 2019, 10, 2348. [Google Scholar] [CrossRef] [Green Version]
- Lindeboom, R.G.; van Voorthuijsen, L.; Oost, K.C.; Rodriguez-Colman, M.J.; Luna-Velez, M.V.; Furlan, C.; Baraille, F.; Jansen, P.W.; Ribeiro, A.; Burgering, B.M.; et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol. Syst. Biol. 2018, 14, e8227. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.K.; Janssen, N.; Winter, S.; Wallisch, S.K.; Hofmann, U.; Dahlke, M.H.; Schwab, M.; Murdter, T.E.; Haag, M. Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites 2020, 10, 494. [Google Scholar] [CrossRef]
- Hahn, S.; Nam, M.O.; Noh, J.H.; Lee, D.H.; Han, H.W.; Kim, D.H.; Hahm, K.B.; Hong, S.P.; Yoo, J.H.; Yoo, J. Organoid-based epithelial to mesenchymal transition (OEMT) model: From an intestinal fibrosis perspective. Sci. Rep. 2017, 7, 2435. [Google Scholar] [CrossRef] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bansal, A.; Dunbar, K.B.; Chang, Y.; Zhang, J.; Balaji, U.; Gu, J.; Zhang, X.; Podgaetz, E.; Pan, Z.; et al. A human Barrett’s esophagus organoid system reveals epithelial-mesenchymal plasticity induced by acid and bile salts. Am. J. Physiol.-Gastrointest. Liver Physiol. 2022, 322, G598–G614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Powell, R.T.; Yuan, X.; Bae, G.; Roarty, K.P.; Stossi, F.; Strempfl, M.; Toneff, M.J.; Johnson, H.L.; Mani, S.A.; et al. Morphological screening of mesenchymal mammary tumor organoids to identify drugs that reverse epithelial-mesenchymal transition. Nat. Commun. 2021, 12, 4262. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e6. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Jiang, E.; Shang, Z. 3D Co-culture of Cancer-Associated Fibroblast with Oral Cancer Organoids. J. Dent. Res. 2021, 100, 201–208. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [Green Version]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Sivanand, S.; Lau, A.N.; Florek, L.V.; Barbeau, A.M.; Wyckoff, J.; Skala, M.C.; Vander Heiden, M.G. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. Sci. Adv. 2022, 8, eabg6383. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Perianez, R.; Molina-Privado, I.; Rojo, F.; Guijarro-Munoz, I.; Alonso-Camino, V.; Zazo, S.; Compte, M.; Alvarez-Cienfuegos, A.; Cuesta, A.M.; Sanchez-Martin, D.; et al. Basement membrane-rich organoids with functional human blood vessels are permissive niches for human breast cancer metastasis. PLoS ONE 2013, 8, e72957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Methods for organoid culturing. (a) Submerged culture: upon harvesting and mechanical/enzymatic dissociation of tissues to obtain a single cell suspension, ASCs are embedded into a laminin-rich extracellular matrix (e.g., Matrigel or BME) and overlaid with a culture medium containing stemness-promoting factors to generate epithelial organoids. (b) Air–liquid interface (ALI) culture: alternatively, minced adult tissues can be cultivated in a type I collagen matrix within an inner Transwell dish. The top of the collagen gel is exposed to air, allowing a sufficient oxygen supply for cells. ALI method enables the generation of organoids comprising epithelial cells with their surrounding stroma, including fibroblasts and immune cells. (c) Induced pluripotent stem cell (iPSC) culture: iPSC-derived organoids are generated by a stepwise differentiation protocol resulting in the generation of the desired tissue type, often with the accompanying stroma. iPSC-derived organoids are then initiated by using submerged culture.
Figure 1.
Methods for organoid culturing. (a) Submerged culture: upon harvesting and mechanical/enzymatic dissociation of tissues to obtain a single cell suspension, ASCs are embedded into a laminin-rich extracellular matrix (e.g., Matrigel or BME) and overlaid with a culture medium containing stemness-promoting factors to generate epithelial organoids. (b) Air–liquid interface (ALI) culture: alternatively, minced adult tissues can be cultivated in a type I collagen matrix within an inner Transwell dish. The top of the collagen gel is exposed to air, allowing a sufficient oxygen supply for cells. ALI method enables the generation of organoids comprising epithelial cells with their surrounding stroma, including fibroblasts and immune cells. (c) Induced pluripotent stem cell (iPSC) culture: iPSC-derived organoids are generated by a stepwise differentiation protocol resulting in the generation of the desired tissue type, often with the accompanying stroma. iPSC-derived organoids are then initiated by using submerged culture.

Figure 2.
Organoids for studying metabolic control of stem cell function. (a) Several methodological approaches, including phosphorescence (PLIM) and fluorescence (FLIM) lifetime imaging and real-time bioenergetic profiling, have revealed a metabolic heterogeneity within intestinal organoids, in particular, with the existence of a lactate-based metabolic symbiosis between Paneth cells and Lgr5+ crypt base columnar cells. (b) Metabolic characterization of iPSC-derived kidney organoids has shown a metabolic switch from glycolysis to mitochondrial OXPHOS and tricarboxylic acid (TCA) cycle and the specific role of serine metabolism during kidney organoid differentiation. DNMT: DNA methyltransferase, ROS: reactive oxygen species, SAM: S-adenosylmethionine, TMRM: tetramethylrhodamine methyl ester.
Figure 2.
Organoids for studying metabolic control of stem cell function. (a) Several methodological approaches, including phosphorescence (PLIM) and fluorescence (FLIM) lifetime imaging and real-time bioenergetic profiling, have revealed a metabolic heterogeneity within intestinal organoids, in particular, with the existence of a lactate-based metabolic symbiosis between Paneth cells and Lgr5+ crypt base columnar cells. (b) Metabolic characterization of iPSC-derived kidney organoids has shown a metabolic switch from glycolysis to mitochondrial OXPHOS and tricarboxylic acid (TCA) cycle and the specific role of serine metabolism during kidney organoid differentiation. DNMT: DNA methyltransferase, ROS: reactive oxygen species, SAM: S-adenosylmethionine, TMRM: tetramethylrhodamine methyl ester.

Figure 3.
Organoid culture methods to model TME conditions. Stromal and immune cell components of the TME can be integrated within organoid cultures via two types of approach. The submerged culture enables the reconstitution of the TME upon direct co-culture of tumor epithelial organoids with stromal and/or immune cells obtained from autologous peripheral blood or tumor. Alternatively, the use of air–liquid interface culture or microfluidic culture devices allows the preservation of the native TME, including functional immune cells and cancer-associated fibroblasts (CAFs), from mechanically minced or enzymatically digested tumor fractions. NK cell: natural killer cell.
Figure 3.
Organoid culture methods to model TME conditions. Stromal and immune cell components of the TME can be integrated within organoid cultures via two types of approach. The submerged culture enables the reconstitution of the TME upon direct co-culture of tumor epithelial organoids with stromal and/or immune cells obtained from autologous peripheral blood or tumor. Alternatively, the use of air–liquid interface culture or microfluidic culture devices allows the preservation of the native TME, including functional immune cells and cancer-associated fibroblasts (CAFs), from mechanically minced or enzymatically digested tumor fractions. NK cell: natural killer cell.


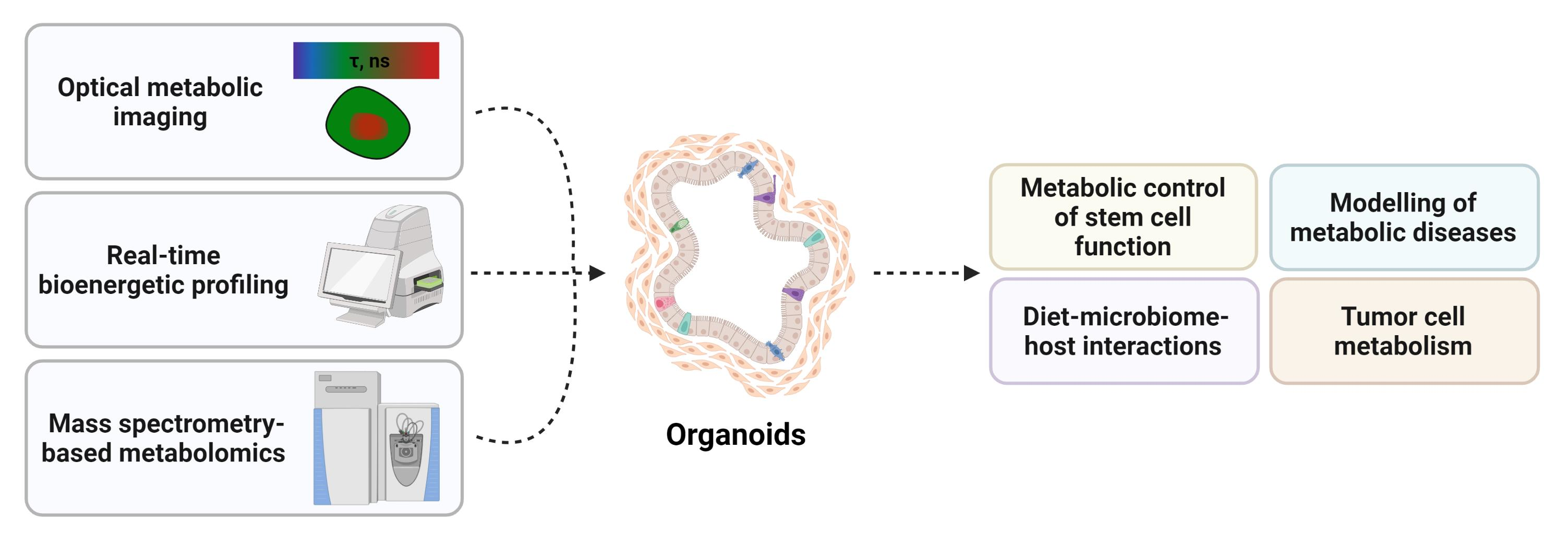{kind=link}
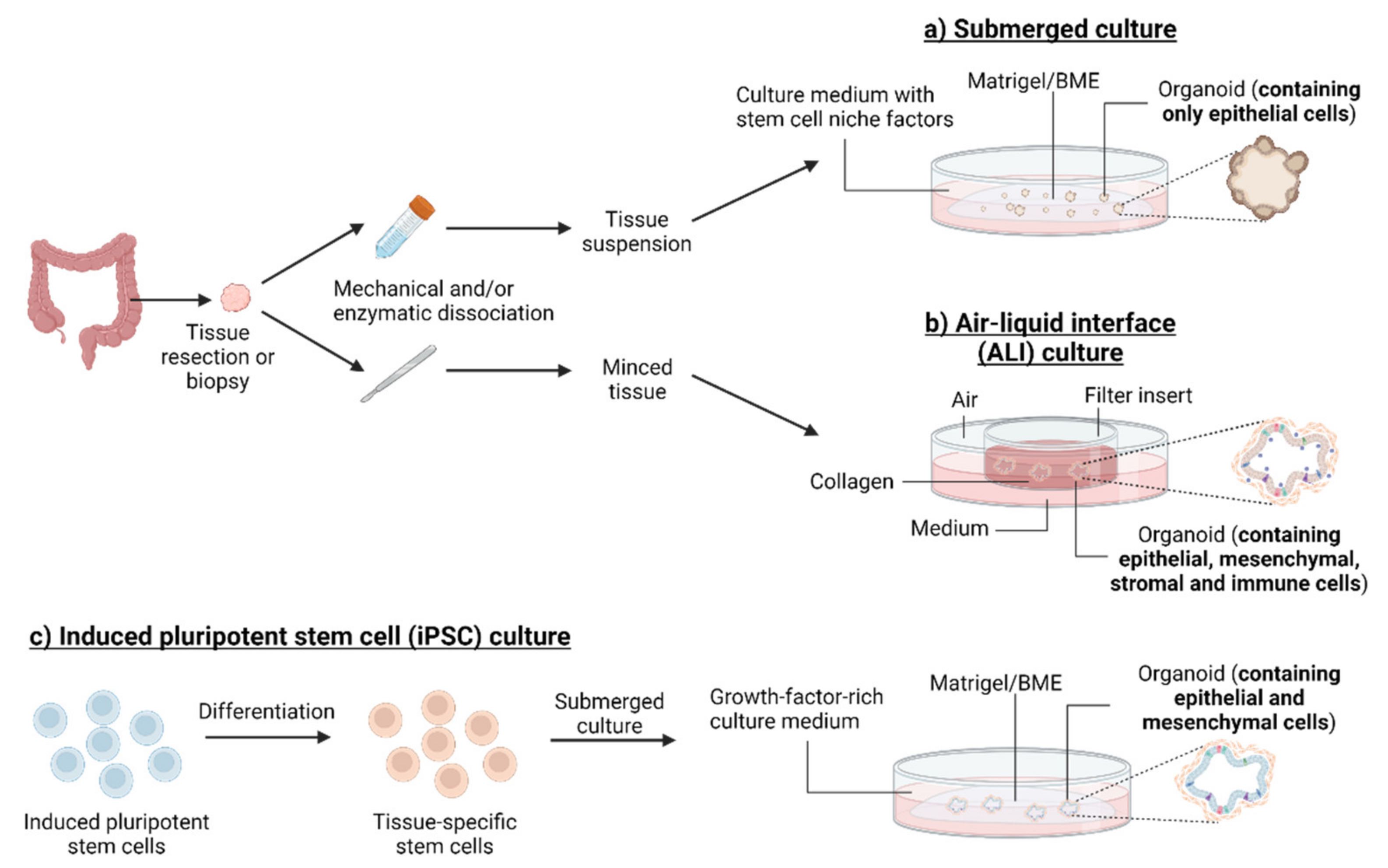{kind=link}
{kind=link}
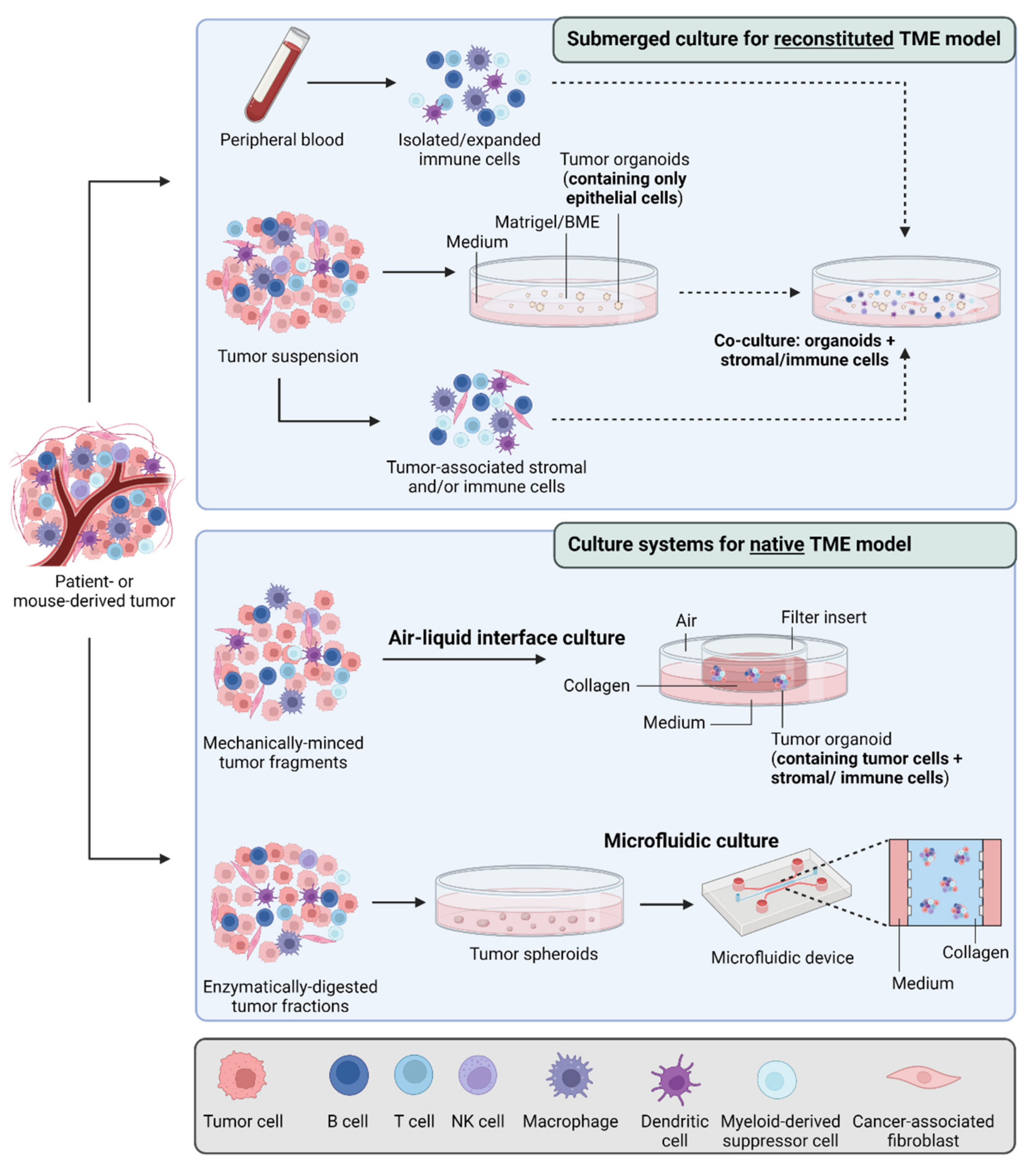{kind=link}
Table 1.
Organoid-based models of kidney and liver metabolic diseases.
| Disease | Species | Organoid Source and Derivation | Refs |
|---|---|---|---|
| Kidney metabolic diseases | |||
| Fabry disease | Human | iPSCs (fibroblast-derived) | [44] |
| Liver metabolic diseases | |||
| Alpha-1 antitrypsin deficiency | Human | Adult tissue (surgical resection; liver transplantation; biopsy) | [45,46] |
| Citrullinemia type 1 | Human | iPSCs (fibroblast-derived) | [47] |
| Steatosis, steatohepatitis | Human Cat | iPSCs (fibroblast-derived) Adult tissue (post-mortem) | [48] [49,50] |
| Wilson’s disease | Dog | Adult tissue (surgical resection, needle biopsy, fine needle aspiration) | [51,52] |
| Wolman’s disease | Human | iPSCs (fibroblast-derived) | [48] |
Table 2.
Methods to study metabolism in organoids. Here, the main methods reported to study cell metabolism in organoid models are listed. Their applications, advantages and disadvantages are indicated. ECAR: extracellular acidification rate; FAD: flavin adenine dinucleotide; NADH: nicotinamide adenine dinucleotide; OCR: oxygen consumption rate; TMRM: tetramethylrhodamine, methyl ester.
Table 2.
Methods to study metabolism in organoids. Here, the main methods reported to study cell metabolism in organoid models are listed. Their applications, advantages and disadvantages are indicated. ECAR: extracellular acidification rate; FAD: flavin adenine dinucleotide; NADH: nicotinamide adenine dinucleotide; OCR: oxygen consumption rate; TMRM: tetramethylrhodamine, methyl ester.
| Methods | Applications | Advantages | Disadvantages | Refs |
|---|---|---|---|---|
| Extracellular flux analysis (Seahorse XF analyzer) | Evaluation of mitochondrial respiration (OCR) and glycolysis (ECAR) | Real-time and simultaneous measurements Up to 4 injections for nutritional and/or pharmacological modulation Label-free assay system, highly sensitive microplate format | No spatial resolution Technically challenging (e.g., need for accurate plating, optimal organoid density) Use of saturating concentrations of substrates and drugs | [16,17,18,19,20] |
| Fluorescence lifetime imaging microscopy (FLIM)/Optical metabolic imaging (OMI) | Live cell microscopy of endogenous (NAD(P)H, FAD) or exogenous chromophores (TMRM for mitochondrial membrane potential) | Non-invasive, cell-specific and direct analysis of metabolism within organoid models Compatible with other imaging methods (e.g., PLIM) for multiparametric quantitative analysis | Complex interpretation of fluorescence data (due to double or multi-exponential decays for most of fluorescent reporters) Shorter lifetimes than PLIM | [14,15,120,121,122,123,124,125,126,153] |
| Mass spectrometry-based metabolomics | Absolute or relative quantification of extra- and/or intracellular metabolites within organoids | Suitable for targeted and untargeted profiling of several metabolite classes (e.g., lipids, polar metabolites) | No spatial resolution No information on metabolic flux (steady-state conditions) Background signal from animal-derived extracellular matrix | [133,134,135] |
| Phosphorescence lifetime imaging microscopy (PLIM) | Live cell microscopy of oxygen (with dedicated cell-penetrating phosphorescent O2-sensitive probes) | Suitable for single-cell analysis of inter- and intra-organoid variability of oxygenation levels Direct, reversible and non-chemical process Compatible with other imaging methods (e.g., FLIM) for multiparametric quantitative analysis High sensitivity and high stability of the signal | Emission intensity dependent on the probe distribution within organoids Limited number of applications (depending on probe availability) | [12,13,14] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Richiardone, E.; Van den Bossche, V.; Corbet, C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids 2022, 1, 85-105. https://doi.org/10.3390/organoids1010008
AMA Style
Richiardone E, Van den Bossche V, Corbet C. Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges. Organoids. 2022; 1(1):85-105. https://doi.org/10.3390/organoids1010008
Chicago/Turabian StyleRichiardone, Elena, Valentin Van den Bossche, and Cyril Corbet. 2022. "Metabolic Studies in Organoids: Current Applications, Opportunities and Challenges" Organoids 1, no. 1: 85-105. https://doi.org/10.3390/organoids1010008