Differential Acute Effects of Selenomethionine and Sodium Selenite on the Severity of Colitis

Abstract
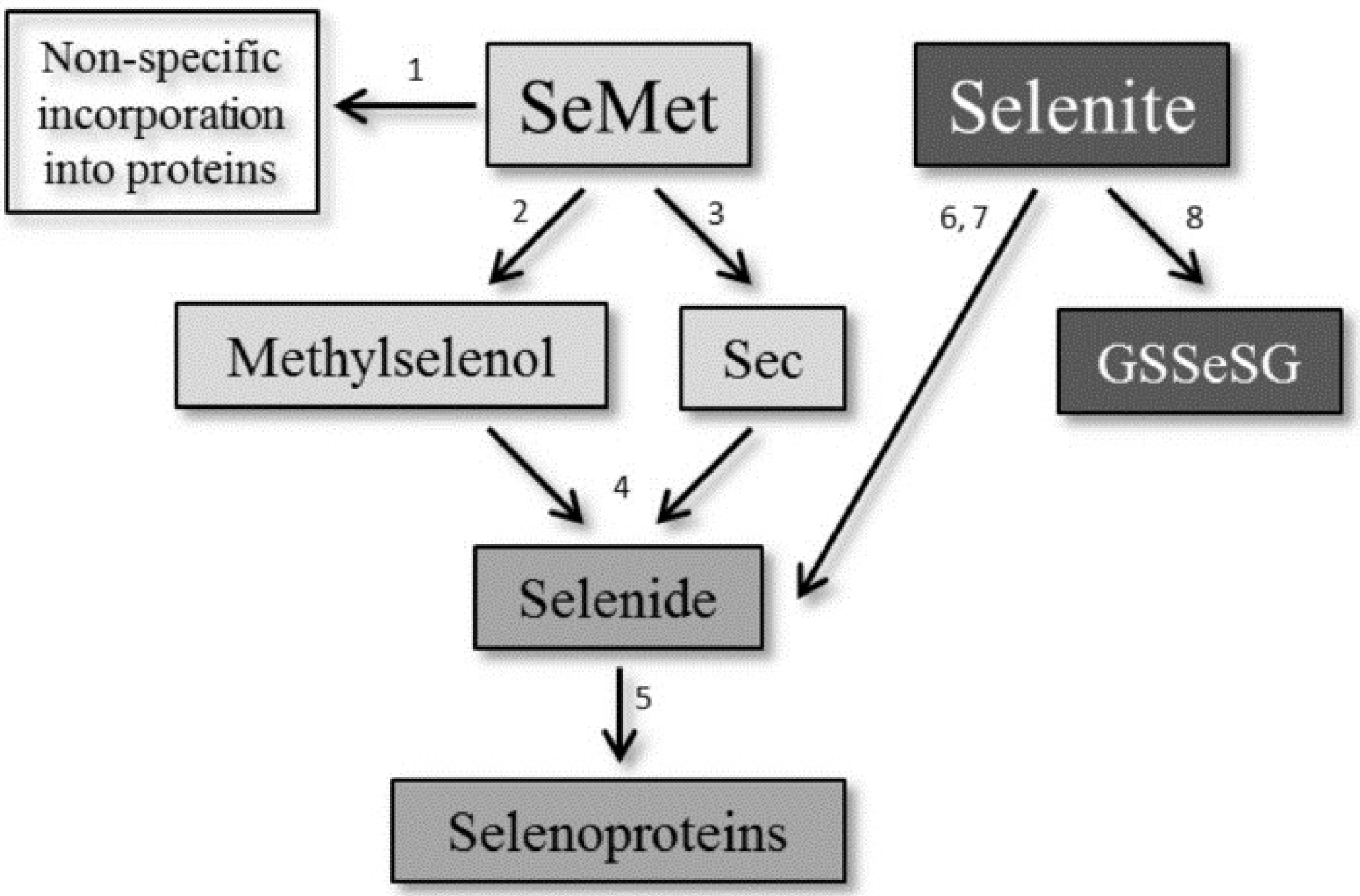
:1. Introduction

2. Experimental Section
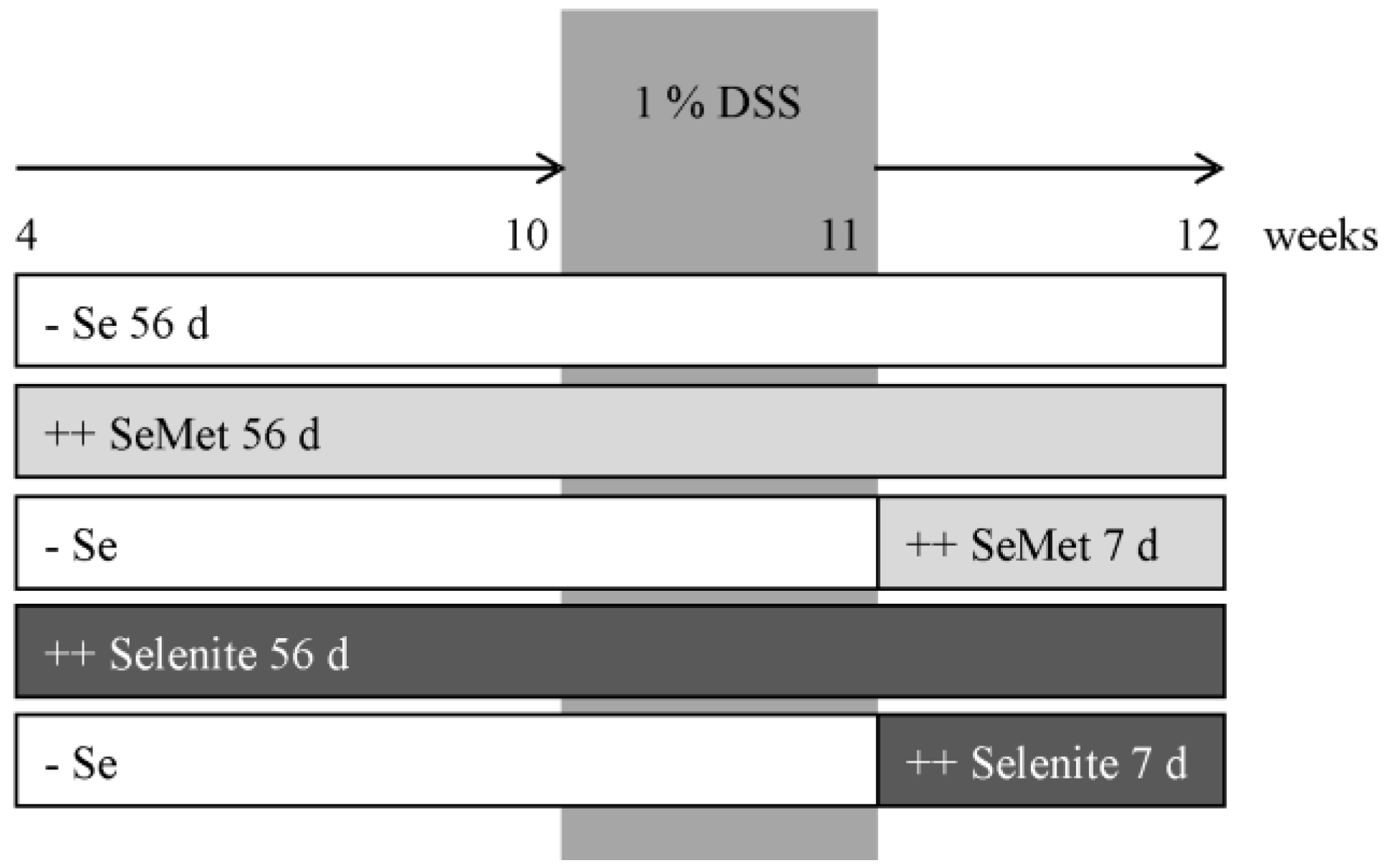
2.1. Animal Experiment

2.2. Scoring Systems
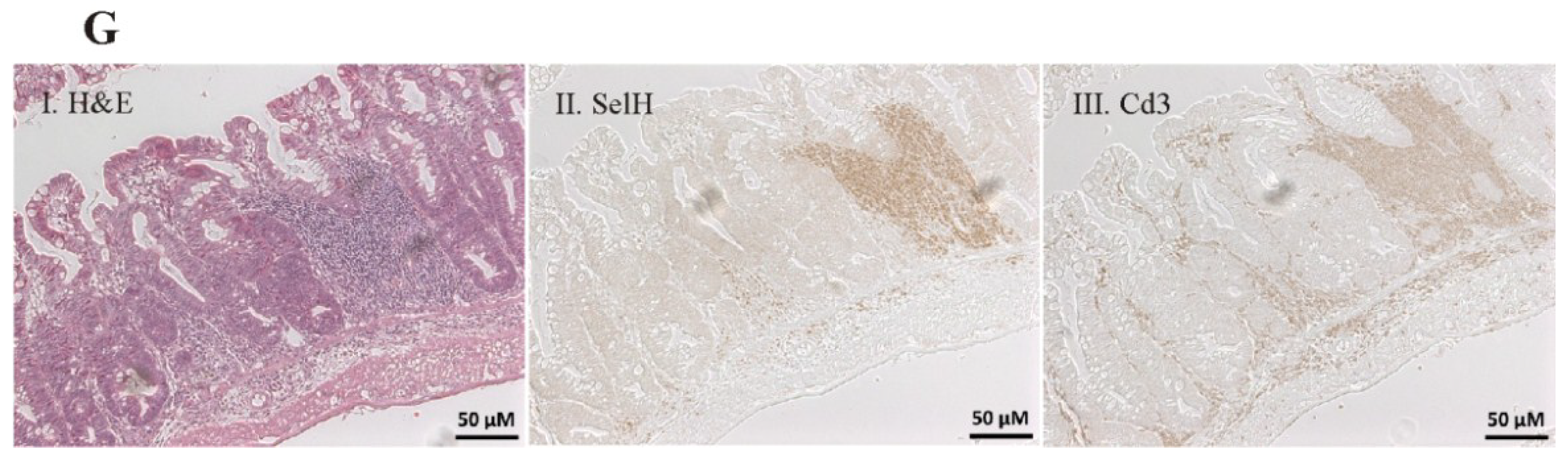
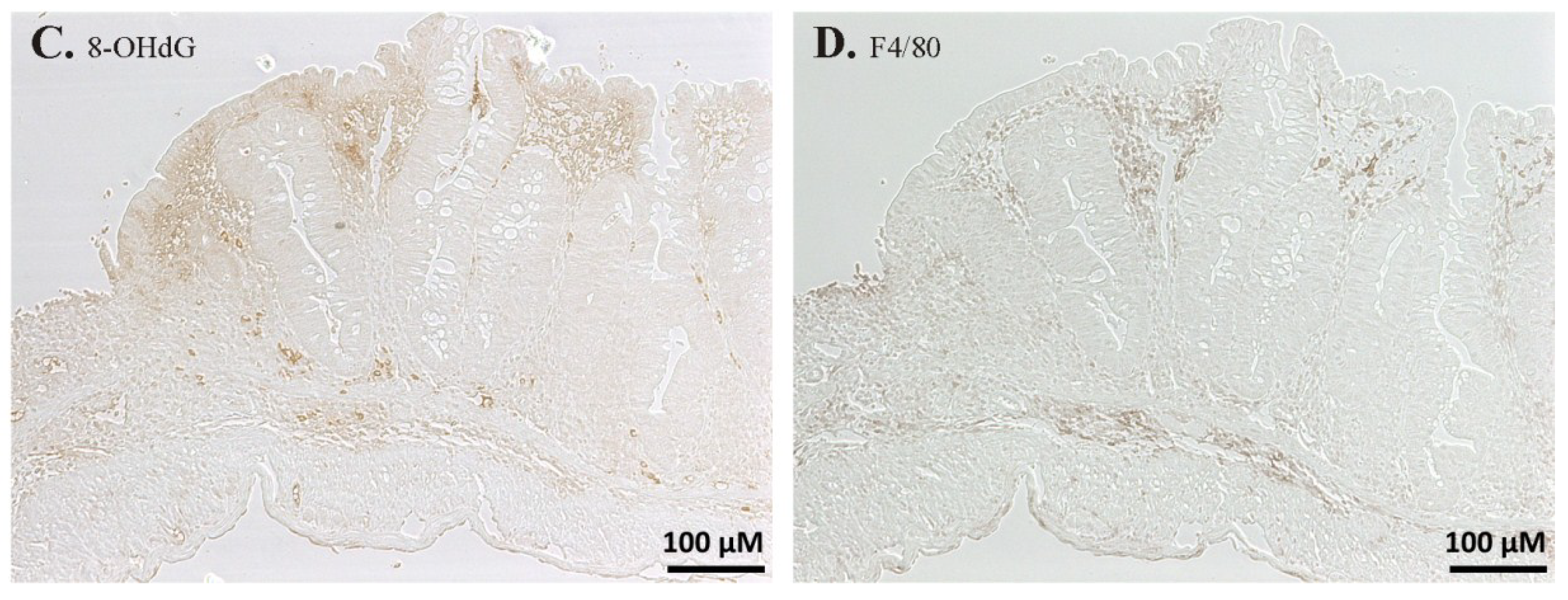
2.3. Immunohistochemistry and Histochemistry
2.4. Enzyme Activities
2.5. Western Blotting
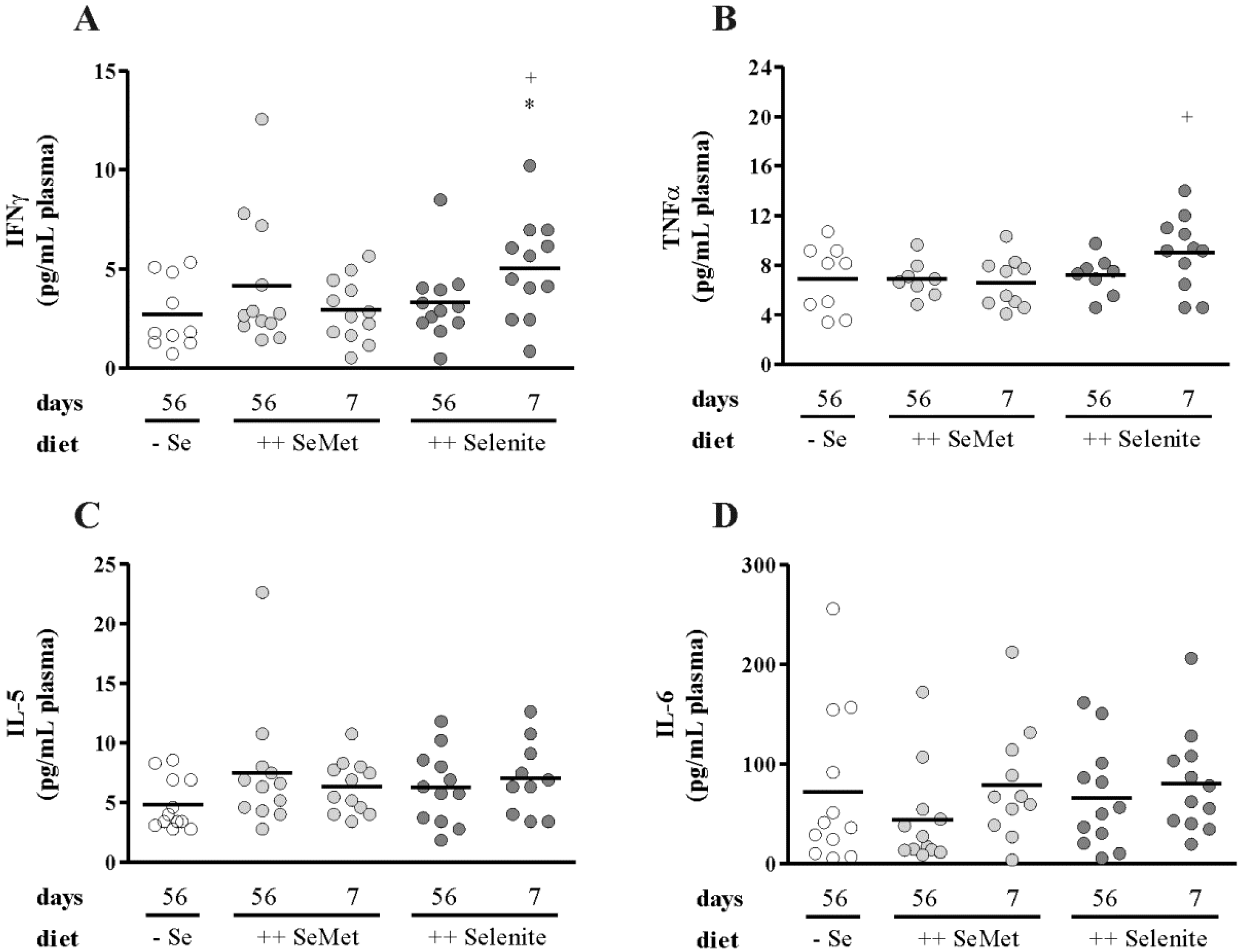
2.6. Cytokine Analysis
2.7. Quantitative Real-Time PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Species | |
|---|---|---|---|
| Cox-2 | fwd | TTCAAGACAGATCATAAGCGAG | mouse/human |
| rev | GTGGCATACATCATCAGACCA | ||
| Gpx1 | fwd | CACACCAGGAGAATGGCAAGA | mouse |
| rev | GAACTTCTCAAAGTTCCAGGCA | ||
| Ifnγ | fwd | GCCAAGTTTGAGGTCAACAACCC | mouse |
| rev | CCGAATCAGCAGCGACTCCT | ||
| Rpl13a | fwd | GTTCGGCTGAAGCCTACCAG | mouse |
| rev | TTCCGTAACCTCAAGATCTGCT | ||
| Selh | fwd | CCTTATTCCACCAACGCGCCA | mouse |
| rev | GCGTCAGCTCGTACAATGCTC | ||
| Sepw1 | fwd | ATGCCTGGACATTTGTGGCGA | mouse |
| rev | GCAGCTTTGATGGCGGTCAC | ||
| Sepp1 | fwd | CTCATCTATGACAGATGTGGCCGT | mouse |
| rev | AAGACTCGTGAGATTGCAGTTTCC | ||
| Tnfα | fwd | CCACGTCGTAGCAAACCACC | mouse |
| rev | TACAACCCATCGGCTGGCAC | ||
2.8. Statistical Analysis
3. Results
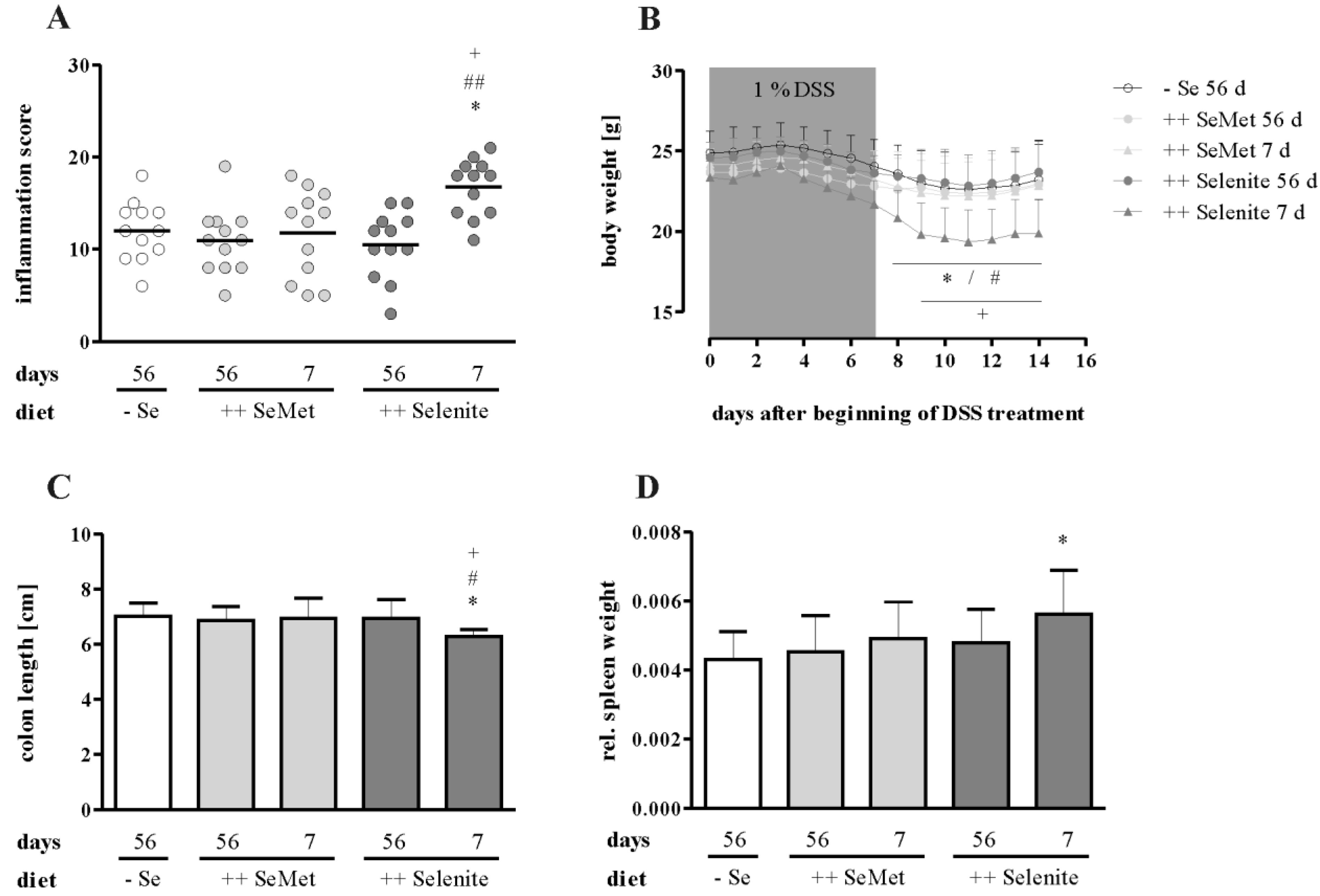
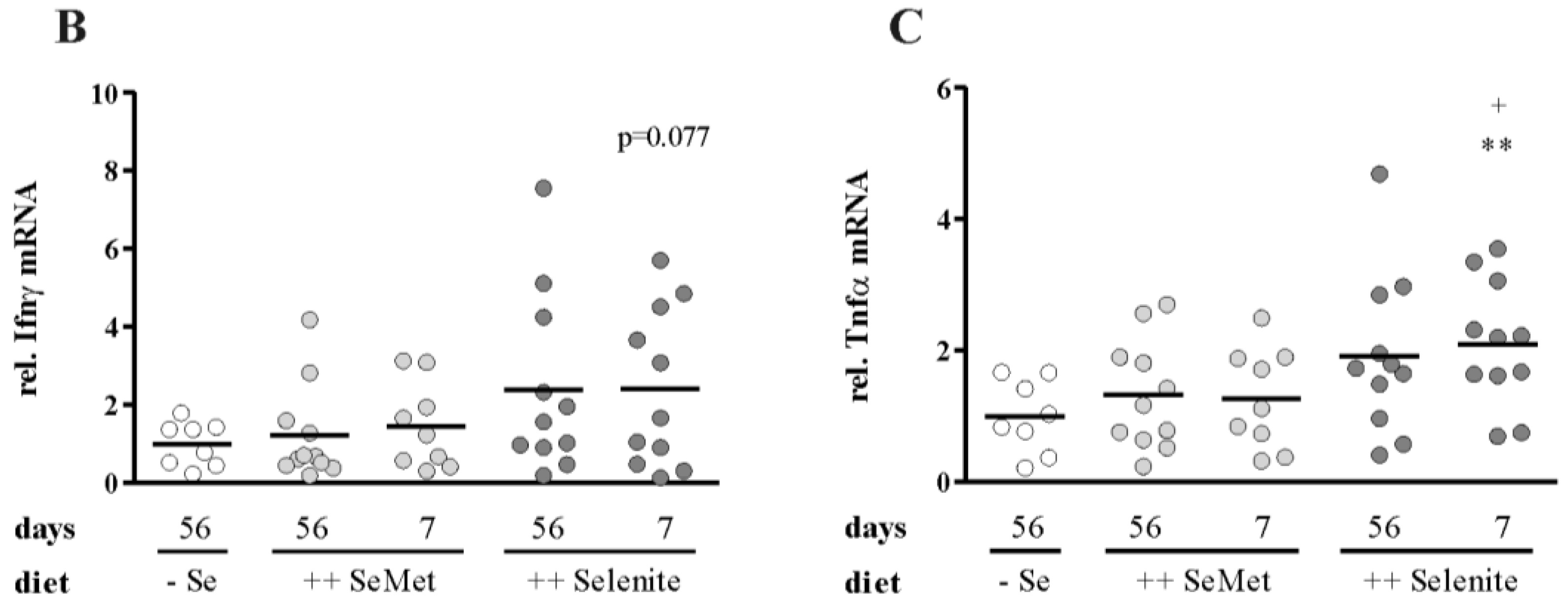
3.1. Selenium Status at Colitis Onset Did not Influence Inflammation Severity
3.2. Selenite Supplementation during Acute Inflammation Increased Parameters of Colitis




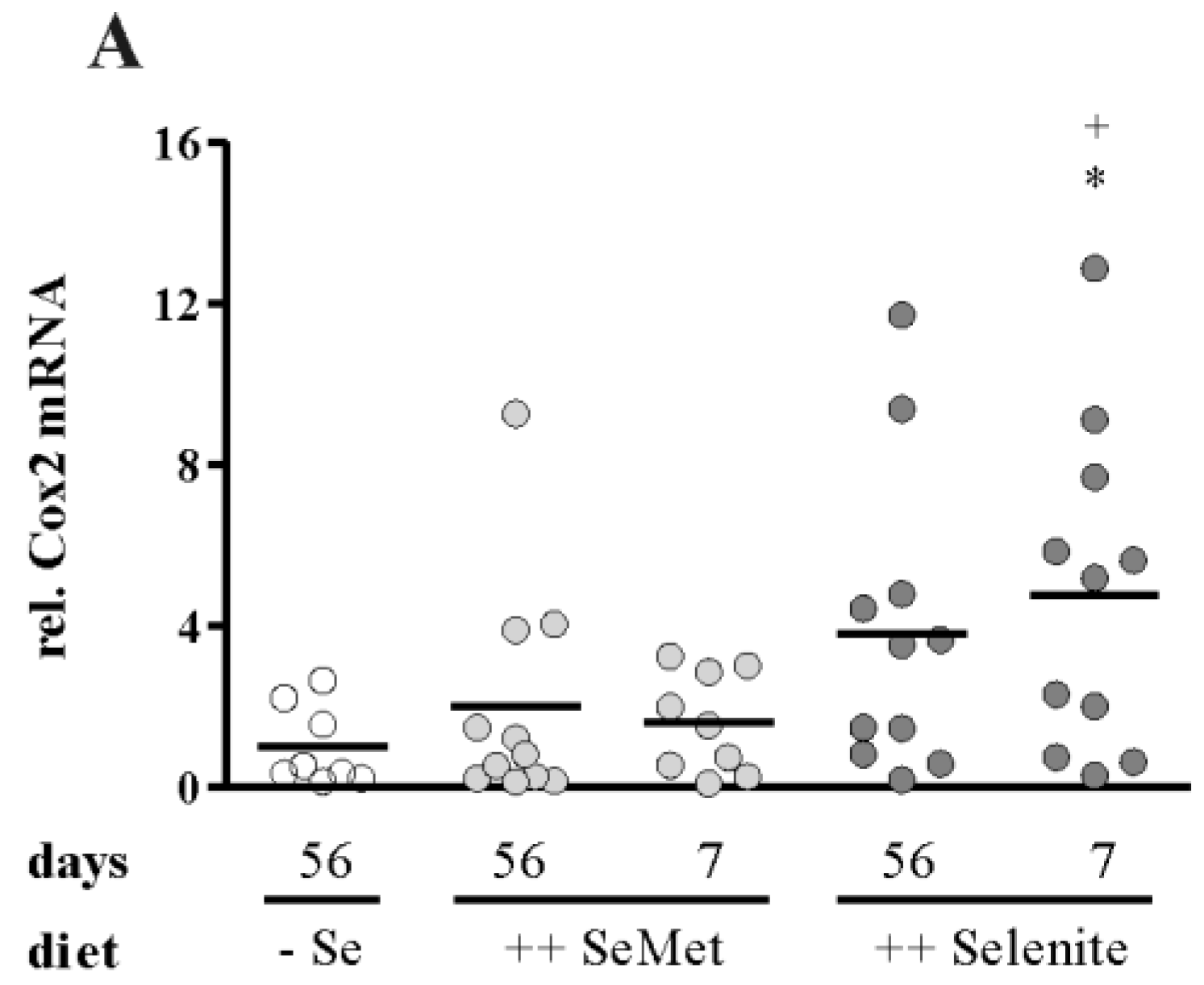
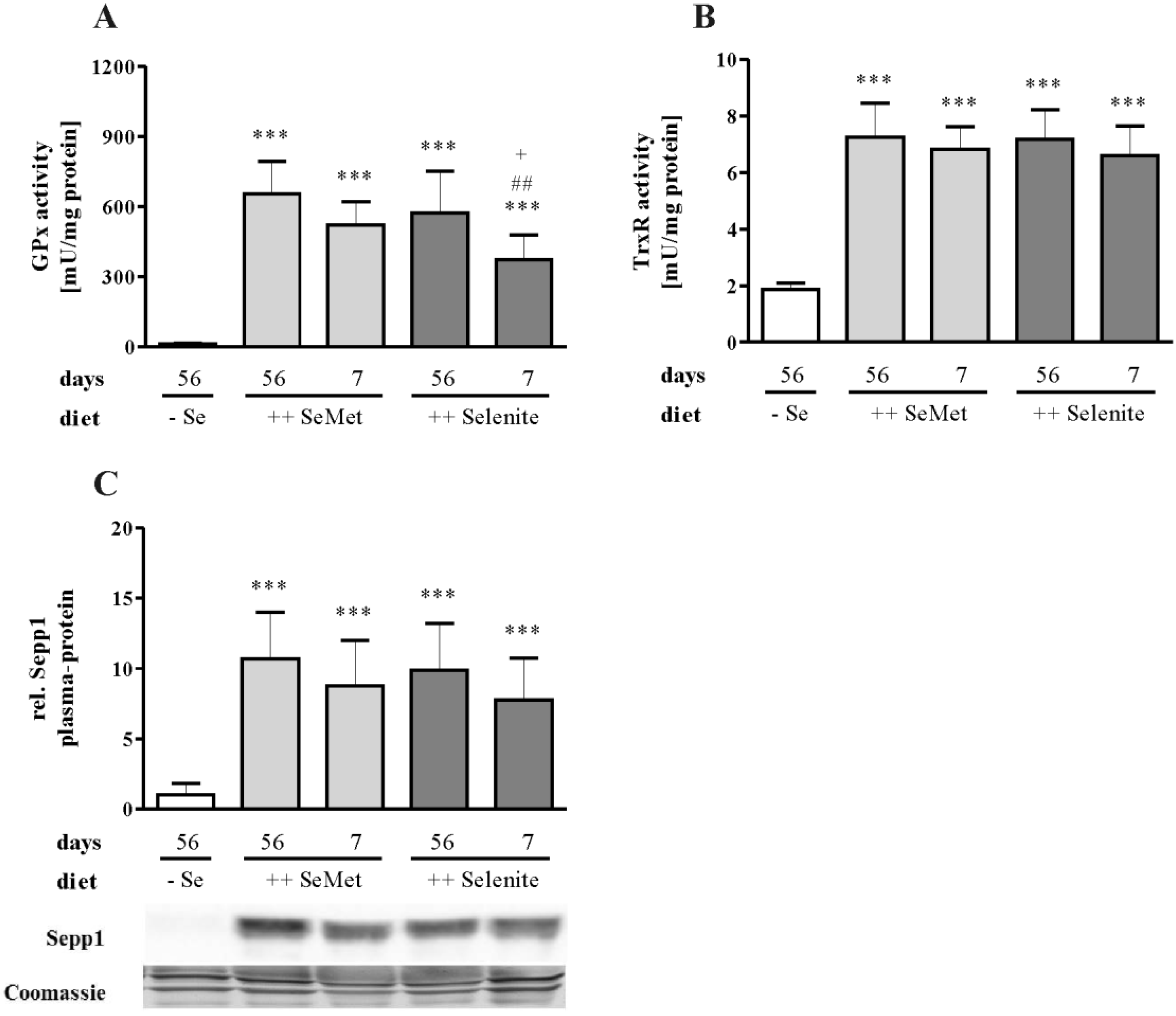
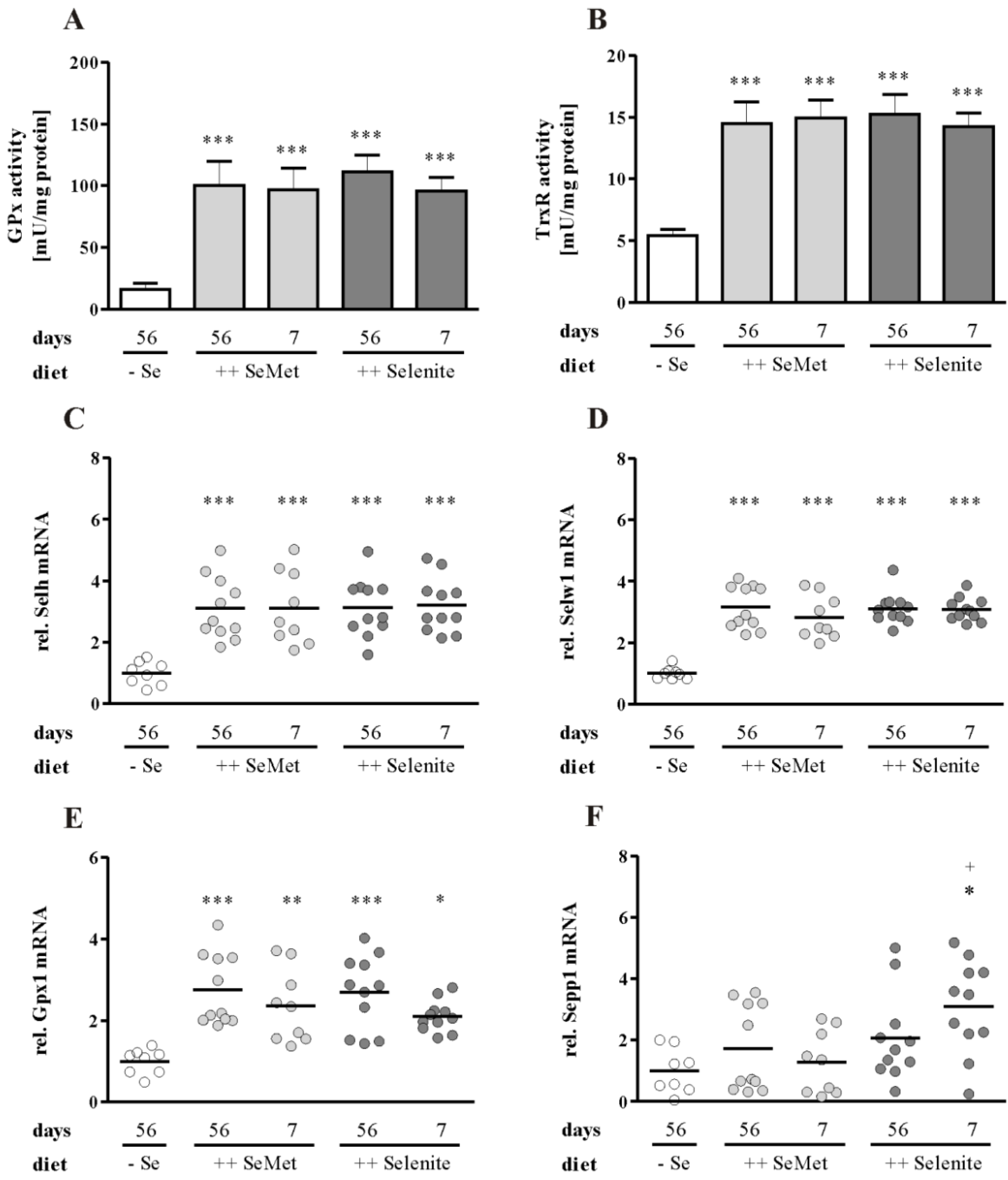
3.3. Enhancement of Colitis by Short-Term Selenite Supplementation Did Not Correlate with Changes in Selenoprotein Expression



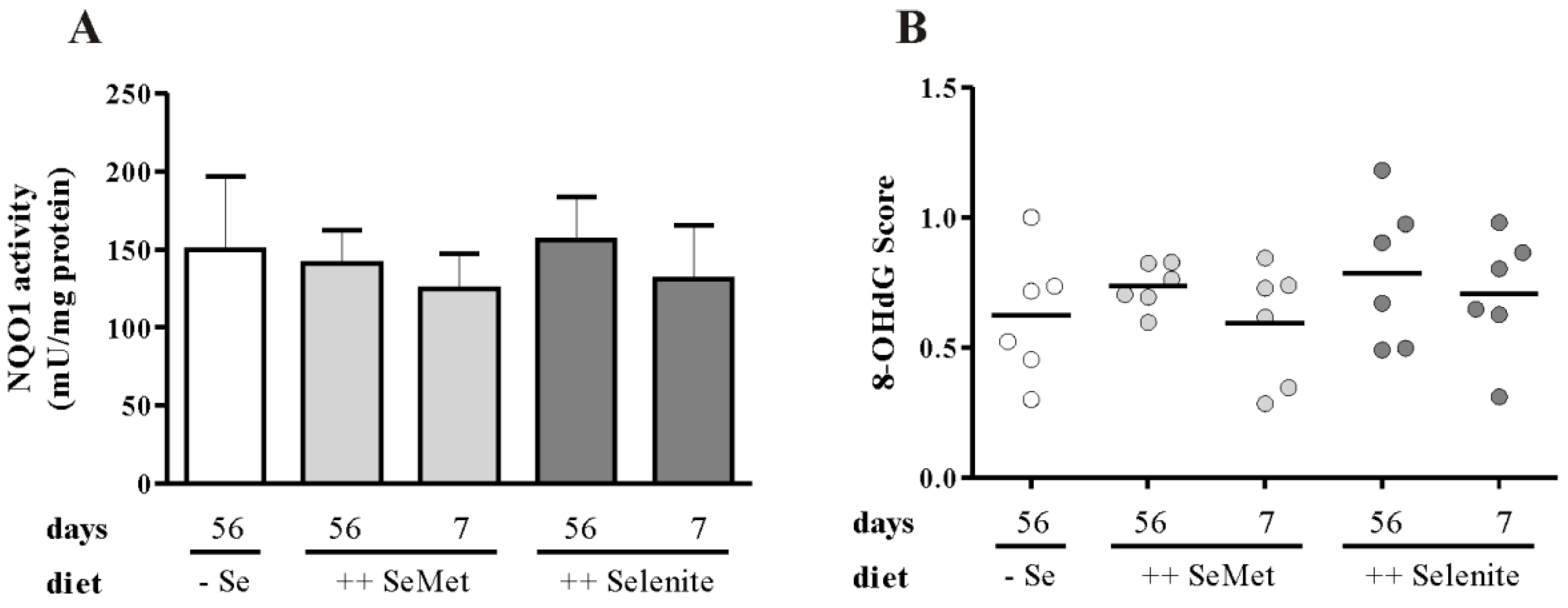
3.4. The Cellular Redox Status Is Unaffected after Seven Days of Selenite Supplementation


4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Hartman, C.; Eliakim, R.; Shamir, R. Nutritional status and nutritional therapy in inflammatory bowel diseases. World J. Gastroenterol. 2009, 15, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Hirashima, M.; Maeda, H.; Hata, K.; Inatomi, O.; Tsujikawa, T.; Sasaki, M.; Takahashi, K.; Fujiyama, Y. Serum selenoprotein-P levels in patients with inflammatory bowel disease. Nutrition 2005, 21, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Zinoni, F.; Birkmann, A.; Leinfelder, W.; Böck, A. Cotranslational insertion of selenocysteine into formate dehydrogenase from Escherichia coli directed by a UGA codon. Proc. Natl. Acad. Sci. USA 1987, 84, 3156–3160. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; Höge, S.C.; Li, P.A.; Hoffmann, F.W.; Hashimoto, A.C.; Berry, M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007, 35, 3963–3973. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The role of selenium in inflammation and immunity: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.S.; Cohen, H.J. Altered oxidative metabolism in selenium-deficient rat granulocytes. J. Immunol. 1983, 130, 2856–2860. [Google Scholar] [PubMed]
- Hattori, H.; Imai, H.; Furuhama, K.; Sato, O.; Nakagawa, Y. Induction of phospholipid hydroperoxide glutathione peroxidase in human polymorphonuclear neutrophils and HL60 cells stimulated with TNF-alpha. Biochem. Biophys. Res. Commun. 2005, 337, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Won, H.Y.; Sohn, J.H.; Min, H.J.; Lee, K.; Woo, H.A.; Ho, Y.S.; Park, J.W.; Rhee, S.G.; Hwang, E.S. Glutathione peroxidase 1 deficiency attenuates allergen-induced airway inflammation by suppressing Th2 and Th17 cell development. Antioxid. Redox Signal. 2010, 13, 575–587. [Google Scholar] [PubMed]
- Verma, S.; Hoffmann, F.W.; Kumar, M.; Huang, Z.; Roe, K.; Nguyen-Wu, E.; Hashimoto, A.S.; Hoffmann, P.R. Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J. Immunol. 2011, 186, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.E.; Jowett, J.B.; Elliott, K.S.; Gao, Y.; Gluschenko, K.; Wang, J.; Abel Azim, D.M.; Cai, G.; Mahaney, M.C.; Comuzzie, A.G.; et al. Genetic variation in selenoprotein S influences inflammatory response. Nat. Genet. 2005, 37, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Kipp, A.P.; Banning, A.; van Schothorst, E.M.; Méplan, C.; Coort, S.L.; Evelo, C.T.; Keijer, J.; Hesketh, J.; Brigelius-Flohé, R. Marginal selenium deficiency down-regulates inflammation-related genes in splenic leukocytes of the mouse. J. Nutr. Biochem. 2012, 23, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Yoo, M.H.; Sano, Y.; Sengupta, A.; Kim, J.Y.; Irons, R.; Gladyshev, V.N.; Hatfield, D.L.; Park, J.M. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Irons, R.D.; Carlson, B.A.; Sano, Y.; Gladyshev, V.N.; Park, J.M.; Hatfield, D.L. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J. Biol. Chem. 2008, 283, 20181–20185. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Hasegawa, G.; Ido, R.; Okuno, T.; Nakamuro, K. Effects of selenium status and supplementary seleno-chemical sources on mouse T-cell mitogenesis. J. Trace Elem. Med. Biol. 2008, 22, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, C.W.; Singh, K.; Motley, A.K.; Lintel, M.K.; Matafonova, E.; Bradley, A.M.; Ning, W.; Poindexter, S.V.; Parang, B.; Reddy, V.K.; et al. Dietary selenium deficiency exacerbates DSS-induced epithelial injury and AOM/DSS-induced tumorigenesis. PloS ONE 2013, 8, e67845. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, N.; Kudva, A.K.; Patterson, A.D.; Chiaro, C.; Kennett, M.J.; Desai, D.; Amin, S.; Carlson, B.A.; Cantorna, M.T.; Prabhu, K.S. Crucial role of macrophage selenoproteins in experimental colitis. J. Immunol. 2014, 193, 3683–3692. [Google Scholar] [CrossRef] [PubMed]
- McConnell, K.P.; Cho, G.J. Transmucosal movement of selenium. Am. J. Physiol. 1965, 208, 1191–1195. [Google Scholar] [PubMed]
- Anundi, I.; Högberg, J.; Ståhl, A. Absorption of selenite in the rat small intestine: Interactions with glutathione. Acta Pharmacol. Toxicol. (Copenh.) 1984, 54, 273–277. [Google Scholar] [CrossRef]
- Krehl, S.; Loewinger, M.; Florian, S.; Kipp, A.P.; Banning, A.; Wessjohann, L.A.; Brauer, M.N.; Iori, R.; Esworthy, R.S.; Chu, F.F.; et al. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis 2012, 33, 620–628. [Google Scholar] [CrossRef] [PubMed]
- McConnell, K.P.; Hoffman, J.L. Methionine-selenomethionine parallels in rat liver polypeptide chain synthesis. FEBS Lett. 1972, 24, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Esaki, N.; Tanaka, H.; Uemura, S.; Suzuki, T.; Soda, K. Catalytic action of L-methionine gamma-lyase on selenomethionine and selenols. Biochemistry 1979, 18, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Esaki, N.; Nakamura, T.; Tanaka, H.; Suzuki, T.; Morino, Y.; Soda, K. Enzymatic synthesis of selenocysteine in rat liver. Biochemistry 1981, 20, 4492–4496. [Google Scholar] [CrossRef] [PubMed]
- Wallenberg, M.; Olm, E.; Hebert, C.; Björnstedt, M.; Fernandes, A.P. Selenium compounds are substrates for glutaredoxins: A novel pathway for selenium metabolism and a potential mechanism for selenium-mediated cytotoxicity. Biochem. J. 2010, 429, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Björnstedt, M.; Holmgren, A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur. J. Biochem. 1992, 207, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ganther, H.E. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry 1968, 7, 2898–2905. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Nutrient Requirements of Laboratory Animals, 4th ed.The National Academies Press: Washington, DC, USA, 1995; p. 192.
- Hrdina, J.; Banning, A.; Kipp, A.; Loh, G.; Blaut, M.; Brigelius-Flohé, R. The gastrointestinal microbiota affects the selenium status and selenoprotein expression in mice. J. Nutr. Biochem. 2009, 20, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, D.; Lehmann, C.; Florian, S.; Barknowitz, G.; Haack, M.; Mewis, I.; Wiesner, M.; Schreiner, M.; Glatt, H.; Brigelius-Flohé, R.; et al. Glucosinolates from pak choi and broccoli induce enzymes and inhibit inflammation and colon cancer differently. Food Funct. 2014, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Wingler, K.; Schmehl, K.; Jacobasch, G.; Kreuzer, O.J.; Meyerhof, W.; Brigelius-Flohé, R. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic. Res. 2001, 35, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Florian, S.; Pommer, S.; Osterhoff, M.; Esworthy, R.S.; Chu, F.F.; Brigelius-Flohé, R.; Kipp, A.P. Deletion of glutathione peroxidase-2 inhibits azoxymethane-induced colon cancer development. PloS ONE 2013, 8, e72055. [Google Scholar] [CrossRef] [PubMed]
- Florian, S.; Krehl, S.; Loewinger, M.; Kipp, A.; Banning, A.; Esworthy, S.; Chu, F.F.; Brigelius-Flohé, R. Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med. 2010, 49, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Banning, A.; Brigelius-Flohé, R.; Kipp, A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010, 5, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Böl, G.F.; Jurrmann, N.; Brigelius-Flohé, R. Recruitment of the interleukin-1 receptor (IL-1RI)-associated kinase IRAK to the IL-1RI is redox regulated. Biol. Chem. 2003, 384, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Kipp, A.P.; Frombach, J.; Deubel, S.; Brigelius-Flohé, R. Selenoprotein W as biomarker for the efficacy of selenium compounds to act as source for selenoprotein biosynthesis. Methods Enzym. 2013, 527, 87–112. [Google Scholar]
- Hill, K.E.; Wu, S.; Motley, A.K.; Stevenson, T.D.; Winfrey, V.P.; Capecchi, M.R.; Atkins, J.F.; Burk, R.F. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 2012, 287, 40414–40424. [Google Scholar] [CrossRef] [PubMed]
- Persson-Moschos, M.; Huang, W.; Srikumar, T.S.; Åkesson, B.; Lindeberg, S. Selenoprotein P in serum as a biochemical marker of selenium status. Analyst 1995, 120, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Spallholz, J.E. On the nature of selenium toxicity and carcinostatic activity. Free Radic. Biol. Med. 1994, 17, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.S.; Spallholz, J.E.; Neldner, K.H.; Pence, B.C. Selenium compounds have disparate abilities to impose oxidative stress and induce apoptosis. Free Radic. Biol. Med. 1999, 26, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Wycherly, B.J.; Moak, M.A.; Christensen, M.J. High dietary intake of sodium selenite induces oxidative DNA damage in rat liver. Nutr. Cancer 2004, 48, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional prevention of cancer study group. Jama 1996, 276, 1957–1963. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and Vitamin E on risk of prostate cancer and other cancers: The selenium and Vitamin E cancer prevention trial (SELECT). Jama 2009, 301, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Food and Nutrition Board. Dietary Reference Intakes for Vitamin C, Vitamin E, selenium and carotenoid; The National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Fairweather-Tait, S.J.; Collings, R.; Hurst, R. Selenium bioavailability: Current knowledge and future research requirements. Am. J. Clin. Nutr. 2010, 91, 1484S–1491S. [Google Scholar] [CrossRef] [PubMed]
- Kipp, A.; Banning, A.; van Schothorst, E.M.; Méplan, C.; Schomburg, L.; Evelo, C.; Coort, S.; Gaj, S.; Keijer, J.; Hesketh, J.; et al. Four selenoproteins, protein biosynthesis, and Wnt signalling are particularly sensitive to limited selenium intake in mouse colon. Mol. Nutr. Food Res. 2009, 53, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Yoo, M.H.; Shrimali, R.K.; Irons, R.; Gladyshev, V.N.; Hatfield, D.L.; Park, J.M. Role of selenium-containing proteins in T-cell and macrophage function. Proc. Nutr. Soc. 2010, 69, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Speckmann, B.; Pinto, A.; Winter, M.; Förster, I.; Sies, H.; Steinbrenner, H. Proinflammatory cytokines down-regulate intestinal selenoprotein P biosynthesis via NOS2 induction. Free Radic. Biol. Med. 2010, 49, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Renko, K.; Hofmann, P.J.; Stoedter, M.; Hollenbach, B.; Behrends, T.; Köhrle, J.; Schweizer, U.; Schomburg, L. Down-regulation of the hepatic selenoprotein biosynthesis machinery impairs selenium metabolism during the acute phase response in mice. FASEB J. 2009, 23, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Hiller, F.; Besselt, K.; Deubel, S.; Brigelius-Flohé, R.; Kipp, A.P. GPx2 induction is mediated via STAT transcription factors during acute colitis. Inflamm. Bowel Dis. 2015. accepted manuscript. [Google Scholar]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. Toxicol. 2014, 114, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:Quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Tobe, R.; Yoo, M.H.; Fradejas, N.; Carlson, B.A.; Calvo, S.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency enhances selenite toxicity in cancer cells via a thioredoxin-independent mechanism. Biochem. J. 2012, 445, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Last, K.; Maharaj, L.; Perry, J.; Strauss, S.; Fitzgibbon, J.; Lister, T.A.; Joel, S. The activity of methylated and non-methylated selenium species in lymphoma cell lines and primary tumours. Ann. Oncol. 2006, 17, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Carrier, J.; Aghdassi, E.; Cullen, J.; Allard, J.P. Iron supplementation increases disease activity and Vitamin E ameliorates the effect in rats with dextran sulfate sodium-induced colitis. J. Nutr. 2002, 132, 3146–3150. [Google Scholar] [PubMed]
- Carrier, J.; Aghdassi, E.; Platt, I.; Cullen, J.; Allard, J.P. Effect of oral iron supplementation on oxidative stress and colonic inflammation in rats with induced colitis. Aliment. Pharmacol. Ther. 2001, 15, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Chandel, N.S. Rethinking antioxidants in the intensive care unit. Am. J. Respir. Crit. Care Med. 2013, 188, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiller, F.; Oldorff, L.; Besselt, K.; Kipp, A.P. Differential Acute Effects of Selenomethionine and Sodium Selenite on the Severity of Colitis. Nutrients 2015, 7, 2687-2706. https://doi.org/10.3390/nu7042687
Hiller F, Oldorff L, Besselt K, Kipp AP. Differential Acute Effects of Selenomethionine and Sodium Selenite on the Severity of Colitis. Nutrients. 2015; 7(4):2687-2706. https://doi.org/10.3390/nu7042687
Chicago/Turabian StyleHiller, Franziska, Lisa Oldorff, Karolin Besselt, and Anna Patricia Kipp. 2015. "Differential Acute Effects of Selenomethionine and Sodium Selenite on the Severity of Colitis" Nutrients 7, no. 4: 2687-2706. https://doi.org/10.3390/nu7042687