


The Role of D-Serine and D-Aspartate in the Pathogenesis and Therapy of Treatment-Resistant Schizophrenia
 ,
,  ,
, 
Abstract
:1. Introduction
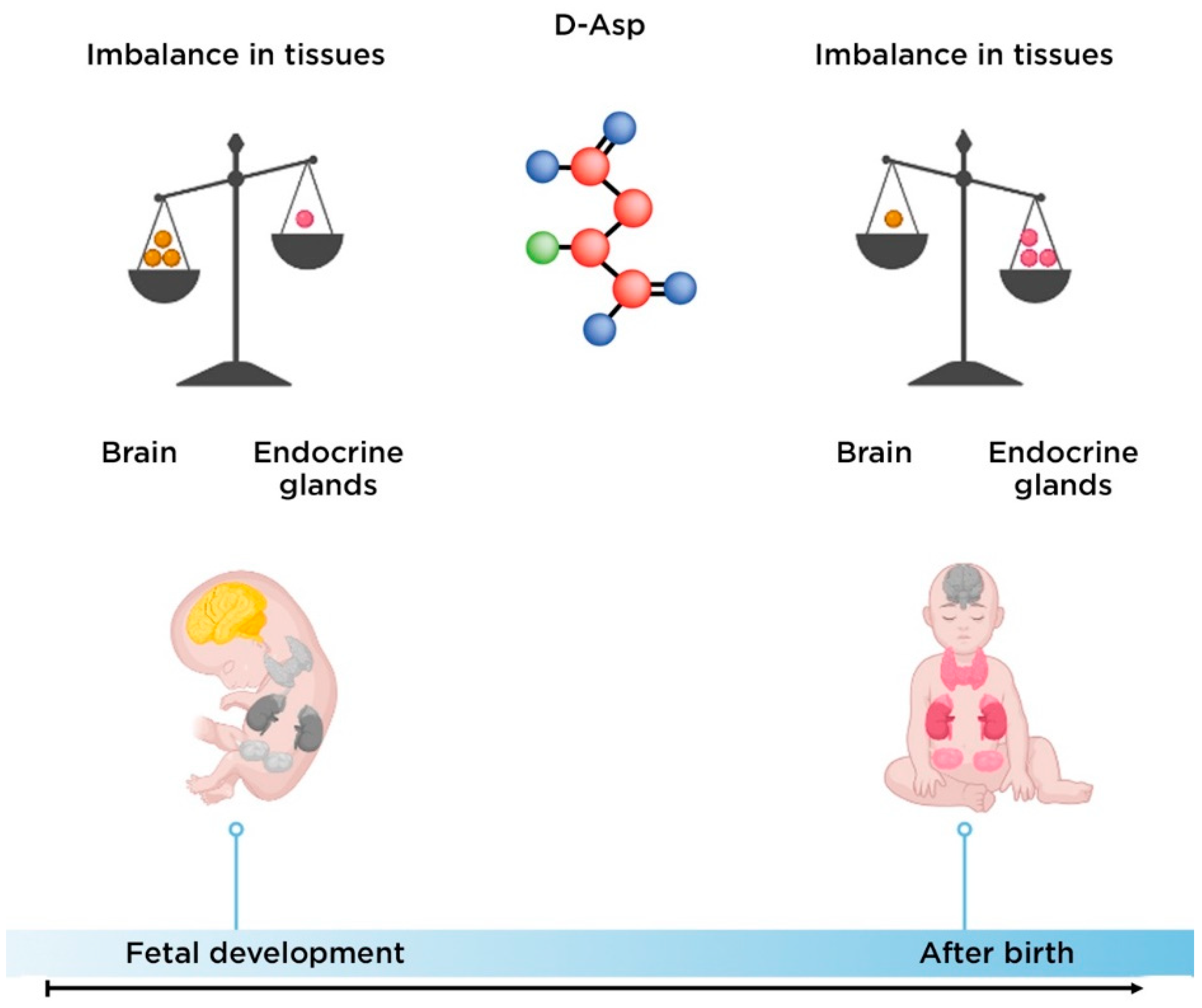
2. D-Aspartate
2.1. The Biological Role of D-Aspartate
2.2. The Role of D-Aspartate in the Pathogenesis of Schizophrenia
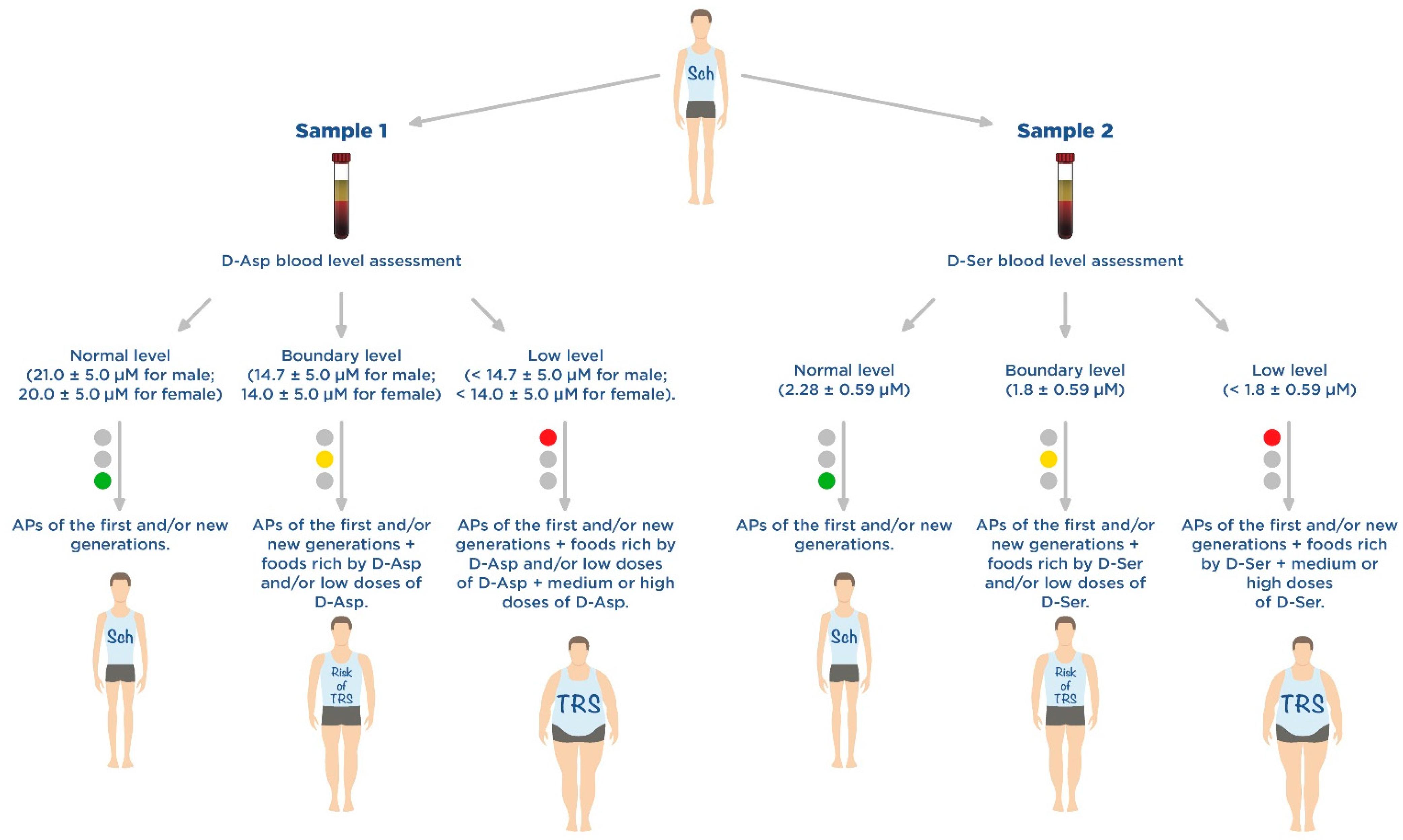
2.3. D-Aspartate as a Promising Component of Disease-Modifying Therapy for Treatment-Resistant Schizophrenia
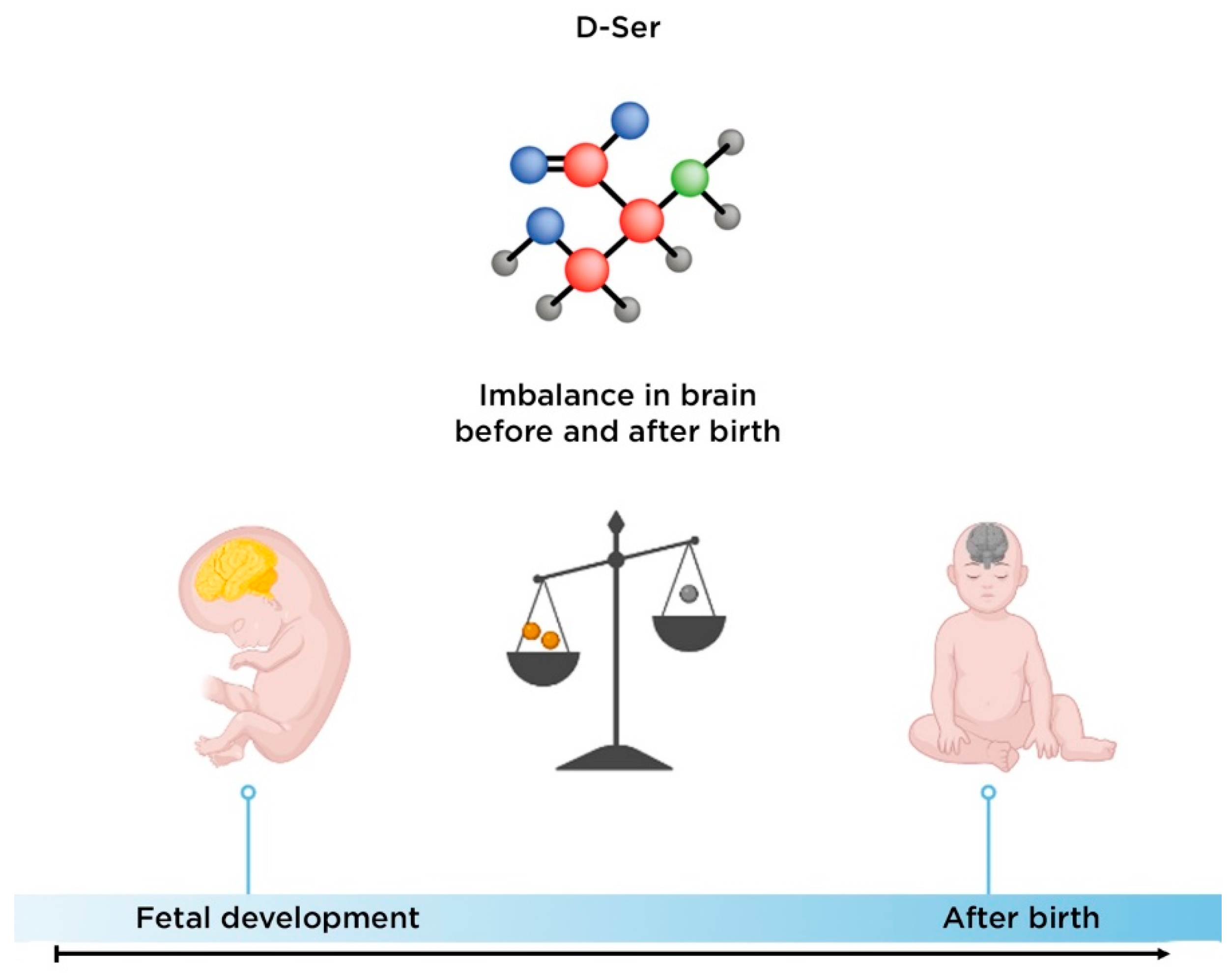
3. D-Serine
3.1. Biological Role of D-Serine
3.2. The Role of D-Serine in the Development of Schizophrenia
3.3. D-Serine as a Promising Component of Disease-Modifying Therapy for Treatment-Resistant Schizophrenia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.F.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.J.; Whiteford, H.A. Global epidemiology and burden of schizophrenia: Findings from the global burden of disease study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Vita, A.; Minelli, A.; Barlati, S.; Deste, G.; Giacopuzzi, E.; Valsecchi, P.; Turrina, C.; Gennarelli, M. Treatment-resistant schizophrenia: Genetic and neuroimaging correlates. Front. Pharmacol. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Shatalina, E. Integrating the neurodevelopmental and dopamine hypotheses of schizophrenia and the role of cortical excitation-inhibition balance. Biol. Psychiatry 2022, 92, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Plitman, E.; Iwata, Y.; Caravaggio, F.; Nakajima, S.; Chung, J.K.; Gerretsen, P.; Kim, J.; Takeuchi, H.; Chakravarty, M.M.; Remington, G.; et al. Kynurenic acid in schizophrenia: A systematic review and meta-analysis. Schizophr. Bull. 2017, 43, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Uno, Y.; Coyle, J.T. Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Adell, A. Brain NMDA receptors in schizophrenia and depression. Biomolecules 2020, 10, 947. [Google Scholar] [CrossRef]
- Seeman, P.; Kapur, S. Schizophrenia: More dopamine, more D2 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 7673–7675. [Google Scholar] [CrossRef] [Green Version]
- Nasyrova, R.F.; Neznanov, N.G. Clinical Psychopharmacogenetics, 1st ed.; DEAN Publishing House: Saint Petersburg, Russia, 2019; pp. 93–174. (In Russian) [Google Scholar]
- Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Al-Zamil, M.; Otmakhov, A.P.; Neznanov, N.G.; Shipulin, G.A.; Petrova, M.M.; Garganeeva, N.P.; Nasyrova, R.F. Cytokine imbalance as a biomarker of treatment-resistant schizophrenia. Int. J. Mol. Sci. 2022, 23, 11324. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Errico, F.; Aceto, G.; Tomasetti, C.; Usiello, A.; Iasevoli, F. D-aspartate dysregulation in Ddo (−/−) mice modulates phencyclidine-induced gene expression changes of postsynaptic density molecules in cortex and striatum. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 62, 35–43. [Google Scholar] [CrossRef]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 20 September 2022).
- Wagner, E.; Kane, J.M.; Correll, C.U.; Howes, O.; Siskind, D.; Honer, W.G.; Lee, J.; Falkai, P.; Schneider-Axmann, T.; Hasan, A.; et al. Clozapine combination and augmentation strategies in patients with schizophrenia -recommendations from an international expert survey among the treatment response and resistance in psychosis (TRRIP) working group. Schizophr. Bull. 2020, 46, 1459–1470. [Google Scholar] [CrossRef]
- Polese, D.; Fornaro, M.; Palermo, M.; De Luca, V.; de Bartolomeis, A. Treatment-resistant to antipsychotics: A resistance to everything? psychotherapy in treatment-resistant schizophrenia and nonaffective psychosis: A 25-year systematic review and exploratory meta-analysis. Front. Psychiatry 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry 1988, 45, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Ajnakina, O.; Horsdal, H.T.; Lally, J.; MacCabe, J.H.; Murray, R.M.; Gasse, C.; Wimberley, T. Validation of an algorithm-based definition of treatment resistance in patients with schizophrenia. Schizophr. Res. 2018, 197, 294–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkis, H.; Buckley, P.F. Treatment-Resistant Schizophrenia. Psychiatr. Clin. N. Am. 2016, 39, 239–265. [Google Scholar] [CrossRef]
- Remington, G.; Addington, D.; Honer, W.; Ismail, Z.; Raedler, T.; Teehan, M. Guidelines for the pharmacotherapy of schizophrenia in adults. Can. J. Psychiatry 2017, 62, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, S.M.; Bellack, A.S. A scientific agenda for the concept of recovery as it applies to schizophrenia. Clin. Psychol. Rev. 2008, 28, 1108–1124. [Google Scholar] [CrossRef]
- Kennedy, J.L.; Altar, C.A.; Taylor, D.L.; Degtiar, I.; Hornberger, J.C. The social and economic burden of treatment-resistant schizophrenia: A systematic literature review. Int. Clin. Psychopharmacol. 2014, 29, 63–76. [Google Scholar] [CrossRef]
- Lally, J.; Ajnakina, O.; Di Forti, M.; Trotta, A.; Demjaha, A.; Kolliakou, A.; Mondelli, V.; Reis Marques, T.; Pariante, C.; Dazzan, P.; et al. Two distinct patterns of treatment resistance: Clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychol. Med. 2016, 46, 3231–3240. [Google Scholar] [CrossRef] [Green Version]
- Sheitman, B.B.; Lieberman, J.A. The natural history and pathophysiology of treatment resistant schizophrenia. J. Psychiatr. Res. 1998, 32, 143–150. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current concepts and treatments of schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Bendikov, I.; Nadri, C.; Amar, S.; Panizzutti, R.; De Miranda, J.; Wolosker, H.; Agam, G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr. Res. 2007, 90, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Teo, C.; Zai, C.; Borlido, C.; Tomasetti, C.; Strauss, J.; Shinkai, T.; Le Foll, B.; Wong, A.; Kennedy, J.L.; De Luca, V. Analysis of treatment-resistant schizophrenia and 384 markers from candidate genes. Pharm. Genom. 2012, 22, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Lv, X.; Huang, H.; Li, M.; Huai, C.; Wu, X.; Wu, H.; Ma, J.; Chen, L.; Wang, T.; et al. Genome-wide analysis of DNA methylation in 106 schizophrenia family trios in Han Chinese. eBioMedicine 2021, 72, 103609. [Google Scholar] [CrossRef] [PubMed]
- Wagh, V.V.; Vyas, P.; Agrawal, S.; Pachpor, T.A.; Paralikar, V.; Khare, S.P. Peripheral blood-based gene expression studies in schizophrenia: A systematic review. Front. Genet. 2021, 12, 736483. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 2015, 593, 83–95. [Google Scholar] [CrossRef]
- Vieira, M.; Yong, X.; Roche, K.W.; Anggono, V. Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef] [Green Version]
- Radulovic, J.; Ren, L.Y.; Gao, C. N-Methyl D-aspartate receptor subunit signaling in fear extinction. Psychopharmacology 2019, 236, 239–250. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Krashia, P.; Ledonne, A.; Nobili, A.; Cordella, A.; Errico, F.; Usiello, A.; D′Amelio, M.; Mercuri, N.B.; Guatteo, E.; Carunchio, I. Persistent elevation of D-Aspartate enhances NMDA receptor-mediated responses in mouse substantia nigra pars compacta dopamine neurons. Neuropharmacology 2016, 103, 69–78. [Google Scholar] [CrossRef]
- Cristino, L.; Luongo, L.; Squillace, M.; Paolone, G.; Mango, D.; Piccinin, S.; Zianni, E.; Imperatore, R.; Iannotta, M.; Longo, F.; et al. d-Aspartate oxidase influences glutamatergic system homeostasis in mammalian brain. Neurobiol. Aging 2015, 36, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Mothet, J.P.; Usiello, A. D-aspartate: An endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J. Pharm. Biomed. Anal. 2015, 116, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Nisticò, R.; Di Giorgio, A.; Squillace, M.; Vitucci, D.; Galbusera, A.; Piccinin, S.; Mango, D.; Fazio, L.; Middei, S.; et al. Free D-aspartate regulates neuronal dendritic morphology, synaptic plasticity, gray matter volume and brain activity in mammals. Transl. Psychiatry 2014, 4, e417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulbagi, M.; Wang, L.; Siddig, O.; Di, B.; Li, B. D-amino acids and d-amino acid-containing peptides: Potential disease biomarkers and therapeutic targets? Biomolecules 2021, 11, 1716. [Google Scholar] [CrossRef]
- Biomolecules, Science: Amino Acids: Building Blocks of Proteins. Available online: https://conductscience.com/amino-acids-building-blocks-of-proteins/ (accessed on 20 September 2022).
- de Bartolomeis, A.; Vellucci, L.; Austin, M.C.; De Simone, G.; Barone, A. Rational and translational implications of d-amino acids for treatment-resistant schizophrenia: From neurobiology to the clinics. Biomolecules 2022, 12, 909. [Google Scholar] [CrossRef]
- Billard, J.M. D-Amino acids in brain neurotransmission and synaptic plasticity. Amino Acids 2012, 43, 1851–1860. [Google Scholar] [CrossRef]
- Balu, D.T. The NMDA receptor and schizophrenia: From pathophysiology to treatment. Adv. Pharmacol. 2016, 76, 351–382. [Google Scholar]
- Jiménez-Sánchez, L.; Campa, L.; Auberson, Y.P.; Adell, A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacology 2014, 39, 2673–2680. [Google Scholar] [CrossRef] [Green Version]
- Metabocard for D-Aspartic Acid (HMDB0006483). Human Metabolome Database. Available online: https://hmdb.ca/metabolites/HMDB0006483 (accessed on 20 September 2022).
- Kiriyama, Y.; Nochi, H. D-amino acids in the nervous and endocrine systems. Scientifica 2016, 2016, 6494621. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Robb, F.T.; Onstott, T.C. Aspartic acid racemization and repair in the survival and recovery of hyperthermophiles after prolonged starvation at high temperature. FEMS Microbiol. Ecol. 2021, 97, fiab112. [Google Scholar] [CrossRef]
- Bastings, J.; van Eijk, H.M.; Olde Damink, S.W.; Rensen, S.S. D-amino acids in health and disease: A focus on cancer. Nutrients 2019, 11, 2205. [Google Scholar] [CrossRef] [PubMed]
- Usiello, A.; Di Fiore, M.M.; De Rosa, A.; Falvo, S.; Errico, F.; Santillo, A.; Nuzzo, T.; Chieffi Baccari, G. New evidence on the role of d-aspartate metabolism in regulating brain and endocrine system physiology: From preclinical observations to clinical applications. Int. J. Mol. Sci. 2020, 21, 8718. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Napolitano, F.; Nisticò, R.; Usiello, A. New insights on the role of free d-aspartate in the mammalian brain. Amino Acids 2012, 43, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, M.M.; Santillo, A.; Chieffi Baccari, G. Current knowledge of d-aspartate in glandular tissues. Amino Acids 2014, 46, 1805–1818. [Google Scholar] [CrossRef]
- Li, Y.; Han, H.; Yin, J.; Li, T.; Yin, Y. Role of D-aspartate on biosynthesis, racemization, and potential functions: A mini-review. Anim. Nutr. 2018, 4, 311–315. [Google Scholar] [CrossRef] [PubMed]
- D′Aniello, A.; Guiditta, A. Identification of d-aspartic acid in the brain of octopus vulgaris lam. J. Neurochem. 1977, 29, 1053–1057. [Google Scholar] [CrossRef]
- D′Aniello, G.; Ronsini, S.; Guida, F.; Spinelli, P.; D′Aniello, A. Occurrence of D-aspartic acid in human seminal plasma and spermatozoa: Possible role in reproduction. Fertil. Steril. 2005, 84, 1444–1449. [Google Scholar] [CrossRef]
- D′Aniello, G.; Grieco, N.; Di Filippo, M.A.; Cappiello, F.; Topo, E.; D′Aniello, E.; Ronsini, S. Reproductive implication of D-aspartic acid in human pre-ovulatory follicular fluid. Hum. Reprod. 2007, 22, 3178–3183. [Google Scholar] [CrossRef] [Green Version]
- Ota, N.; Shi, T.; Sweedler, J.V. D-Aspartate acts as a signaling molecule in nervous and neuroendocrine systems. Amino Acids 2012, 43, 1873–1886. [Google Scholar] [CrossRef] [Green Version]
- Wolosker, H.; D′Aniello, A.; Snyder, S.H. D-aspartate disposition in neuronal and endocrine tissues: Ontogeny, biosynthesis and release. Neuroscience 2000, 100, 183–189. [Google Scholar] [CrossRef]
- Hons, J.; Zirko, R.; Ulrychova, M.; Cermakova, E.; Doubek, P.; Libiger, J. Glycine serum level in schizophrenia: Relation to negative symptoms. Psychiatry Res. 2010, 176, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Singh, V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 2011, 25, 859–885. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, T.; Anil, A.E.; Jin, D.; Jayathilake, K.; Lee, M.; Meltzer, H.Y. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: Relation to negative symptoms. Int. J. Neuropsychopharmacol. 2004, 7, 1–8. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, G.E.; Yang, P.; Chung, L.C.; Tsai, I.C.; Tsai, C.W.; Coyle, J.T. D-serine added to clozapine for the treatment of schizophrenia. Am. J. Psychiatry 1999, 156, 1822–1825. [Google Scholar] [CrossRef]
- Tsai, G.E.; Lin, P.Y. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr. Pharm. Des. 2010, 16, 522–537. [Google Scholar] [CrossRef]
- Verrall, L.; Walker, M.; Rawlings, N.; Benzel, I.; Kew, J.N.; Harrison, P.J.; Burnet, P.W. D-amino acid oxidase and serine racemase in human brain: Normal distribution and altered expression in schizophrenia. Eur. J. Neurosci. 2007, 26, 1657–1669. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.Q.; Frandsen, A.; Lu, W.Y.; Wan, Y.; Zabek, R.L.; Pickering, D.S.; Bai, D. D-aspartate and NMDA, but not L-aspartate, block AMPA receptors in rat hippocampal neurons. Br. J. Pharm. 2005, 145, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Errico, F.; Nuzzo, T.; Carella, M.; Bertolino, A.; Usiello, A. The emerging role of altered d-aspartate metabolism in schizophrenia: New insights from preclinical models and human studies. Front. Psychiatry 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Weiser, M.; Heresco-Levy, U.; Davidson, M.; Javitt, D.C.; Werbeloff, N.; Gershon, A.A.; Abramovich, Y.; Amital, D.; Doron, A.; Konas, S.; et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J. Clin. Psychiatry 2012, 73, e728–e734. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Malhotra, A.K.; Cornblatt, B.; Silipo, G.; Balla, A.; Suckow, R.F.; D′Souza, C.; Saksa, J.; Woods, S.W.; Javitt, D.C. High dose D-serine in the treatment of schizophrenia. Schizophr. Res. 2010, 121, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Wolosker, H.; Sheth, K.N.; Takahashi, M.; Mothet, J.P.; Brady, R.O.; Ferris, C.D., Jr.; Snyder, S.H. Purification of serine racemase: Biosynthesis of the neuromodulator D-serine. Proc. Natl. Acad. Sci. USA 1999, 96, 721–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Ohnishi, T.; Hashimoto, K.; Ohba, H.; Iwayama-Shigeno, Y.; Toyoshima, M.; Okuno, A.; Takao, H.; Toyota, T.; Minabe, Y.; et al. Identification of multiple serine racemase (SRR) mRNA isoforms and genetic analyses of SRR and DAO in schizophrenia and D-serine levels. Biol. Psychiatry 2005, 57, 1493–1503. [Google Scholar] [CrossRef]
- Yamamori, H.; Hashimoto, R.; Fujita, Y.; Numata, S.; Yasuda, Y.; Fujimoto, M.; Ohi, K.; Umeda-Yano, S.; Ito, A.; Ohmori, T.; et al. Changes in plasma D-serine, L-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci. Lett. 2014, 582, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Assisi, L.; Botte, V.; D′Aniello, A.; Di Fiore, M.M. Enhancement of aromatase activity by D-aspartic acid in the ovary of the lizard Podarcis s. sicula. Reproduction 2001, 121, 803–808. [Google Scholar] [CrossRef]
- Bi, C.; Zheng, X.; Azaria, S.; Beeram, S.; Li, Z.; Hage, D.S. Chromatographic studies of protein-based chiral separations. Separations 2016, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Kantrowitz, J.T.; Epstein, M.L.; Lee, M.; Lehrfeld, N.; Nolan, K.A.; Shope, C.; Petkova, E.; Silipo, G.; Javitt, D.C. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: Correlation with symptoms. Schizophr. Res. 2018, 191, 70–79. [Google Scholar] [CrossRef]
- Dunlop, D.S.; Neidle, A.; McHale, D.; Dunlop, D.M.; Lajtha, A. The presence of free D-aspartic acid in rodents and man. Biochem. Biophys. Res. Commun. 1986, 141, 27–32. [Google Scholar] [CrossRef]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.H.; Wykes, T.; Kurtz, M.M. Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: Meta-analytical investigation of efficacy. Br. J. Psychiatry 2013, 203, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Olney, J.W.; Farber, N.B. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, J.; Wu, R. Drugs Based on NMDAR Hypofunction Hypothesis in Schizophrenia. Front. Neurosci. 2021, 15, 641047. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, A. D-Aspartic acid: An endogenous amino acid with an important neuroendocrine role. Brain Res. Rev. 2007, 53, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Rossi, S.; Napolitano, F.; Catuogno, V.; Topo, E.; Fisone, G.; D′Aniello, A.; Centonze, D.; Usiello, A. D-aspartate prevents corticostriatal long-term depression and attenuates schizophrenia-like symptoms induced by amphetamine and MK-801. J. Neurosci. 2008, 28, 10404–10414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errico, F.; D′Argenio, V.; Sforazzini, F.; Iasevoli, F.; Squillace, M.; Guerri, G.; Napolitano, F.; Angrisano, T.; Di Maio, A.; Keller, S.; et al. A role for D-aspartate oxidase in schizophrenia and in schizophrenia-related symptoms induced by phencyclidine in mice. Transl. Psychiatry 2015, 5, e512. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shu, N.; Liu, Y.; Song, M.; Hao, Y.; Liu, H.; Yu, C.; Liu, Z.; Jiang, T. Altered resting-state functional connectivity and anatomical connectivity of hippocampus in schizophrenia. Schizophr. Res. 2008, 100, 120–132. [Google Scholar] [CrossRef]
- Kitamura, A.; Hojo, Y.; Ikeda, M.; Karakawa, S.; Kuwahara, T.; Kim, J.; Soma, M.; Kawato, S.; Tsurugizawa, T. Ingested d-aspartate facilitates the functional connectivity and modifies dendritic spine morphology in rat hippocampus. Cereb. Cortex 2019, 29, 2499–2508. [Google Scholar] [CrossRef]
- Errico, F.; Napolitano, F.; Squillace, M.; Vitucci, D.; Blasi, G.; de Bartolomeis, A.; Bertolino, A.; D′Aniello, A.; Usiello, A. Decreased levels of D-aspartate and NMDA in the prefrontal cortex and striatum of patients with schizophrenia. J. Psychiatr. Res. 2013, 47, 1432–1437. [Google Scholar] [CrossRef]
- Nuzzo, T.; Sacchi, S.; Errico, F.; Keller, S.; Palumbo, O.; Florio, E.; Punzo, D.; Napolitano, F.; Copetti, M.; Carella, M.; et al. Decreased free d-aspartate levels are linked to enhanced d-aspartate oxidase activity in the dorsolateral prefrontal cortex of schizophrenia patients. NPJ Schizophr. 2017, 3, 16. [Google Scholar] [CrossRef]
- Sacchi, S.; Novellis, V.; Paolone, G.; Nuzzo, T.; Iannotta, M.; Belardo, C.; Squillace, M.; Bolognesi, P.; Rosini, E.; Motta, Z.; et al. Olanzapine, but not clozapine, increases glutamate release in the prefrontal cortex of freely moving mice by inhibiting D-aspartate oxidase activity. Sci. Rep. 2017, 7, 46288. [Google Scholar] [CrossRef] [Green Version]
- Metabocard for D-Serine (HMDB0003406). Human Metabolome Database. Available online: https://hmdb.ca/metabolites/HMDB0003406 (accessed on 20 September 2022).
- Ito, T.; Hamauchi, N.; Hagi, T.; Morohashi, N.; Hemmi, H.; Sato, Y.G.; Saito, T.; Yoshimura, T. D-serine metabolism and its importance in development of dictyostelium discoideum. Front. Microbiol. 2018, 9, 784. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Kumashiro, S.; Nishikawa, T.; Oka, T.; Takahashi, K.; Mito, T.; Takashima, S.; Doi, N.; Mizutani, Y.; Yamazaki, T. Embryonic development and postnatal changes in free D-aspartate and D-serine in the human prefrontal cortex. J. Neurochem. 1993, 61, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.M.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: A pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412. [Google Scholar] [CrossRef]
- Yang, S.; Qiao, H.; Wen, L.; Zhou, W.; Zhang, Y. D-serine enhances impaired long-term potentiation in CA1 subfield of hippocampal slices from aged senescence-accelerated mouse prone/8. Neurosci. Lett. 2005, 379, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ge, W.; Chen, Y.; Zhang, Z.; Shen, W.; Wu, C.; Poo, M.; Duan, S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. USA 2003, 100, 15194–15199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimazaki, T.; Kaku, A.; Chaki, S. D-Serine and a glycine transporter-1 inhibitor enhance social memory in rats. Psychopharmacology 2010, 209, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Durrant, A.R.; Heresco-Levy, U. D-Serine in neuropsychiatric disorders: New advances. Adv. Psychiatry 2014, 2014, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S., Jr. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Fujita, Y.; Ishima, T.; Hashimoto, K. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci. Rep. 2016, 6, 37261. [Google Scholar] [CrossRef]
- Andersen, J.; Pouzet, B. Spatial Memory Deficits Induced by Perinatal Treatment of Rats with PCP and Reversal Effect of D-Serine. Neuropsychopharmacology 2004, 29, 1080–1090. [Google Scholar] [CrossRef] [Green Version]
- MacKay, M.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-serine: Potential therapeutic agent and/or biomarker in schizophrenia and depression? Front. Psychiatry 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sawamura, H.; Ikeda, Y.; Tsuji, A.; Kitagishi, Y.; Matsuda, S. D-amino acids as a biomarker in schizophrenia. Diseases 2022, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Hons, J.; Zirko, R.; Vasatova, M.; Doubek, P.; Klimova, B.; Masopust, J.; Valis, M.; Kuca, K. Impairment of executive functions associated with lower d-serine serum levels in patients with schizophrenia. Front. Psychiatry 2021, 12, 514579. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: Evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch. Gen. Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balu, D.T.; Li, Y.; Puhl, M.D.; Benneyworth, M.A.; Basu, A.C.; Takagi, S.; Bolshakov, V.Y.; Coyle, J.T. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. USA 2013, 110, E2400–E2409. [Google Scholar] [CrossRef] [Green Version]
- Habl, G.; Zink, M.; Petroianu, G.; Bauer, M.; Schneider-Axmann, T.; von Wilmsdorff, M.; Falkai, P.; Henn, F.A.; Schmitt, A. Increased D-amino acid oxidase expression in the bilateral hippocampal CA4 of schizophrenic patients: A post-mortem study. J. Neural Transm. 2009, 116, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Burnet, P.W.; Eastwood, S.L.; Bristow, G.C.; Godlewska, B.R.; Sikka, P.; Walker, M.; Harrison, P.J. D-amino acid oxidase activity and expression are increased in schizophrenia. Mol. Psychiatry 2008, 13, 658–660. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.R.; Hewedi, D.H.; Eissa, A.M.; Moustafa, A.A. The cerebellum and psychiatric disorders. Front. Public Health 2015, 3, 66. [Google Scholar] [CrossRef] [Green Version]
- Labrie, V.; Fukumura, R.; Rastogi, A.; Fick, L.J.; Wang, W.; Boutros, P.C.; Kennedy, J.L.; Semeralul, M.O.; Lee, F.H.; Baker, G.B.; et al. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum. Mol. Genet. 2009, 18, 3227–3243. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.C.; Luo, D.Z.; Gau, S.S.; Chang, C.Y.; Lai, W.S. Directly and Indirectly Targeting the Glycine Modulatory Site to Modulate NMDA Receptor Function to Address Unmet Medical Needs of Patients with Schizophrenia. Front. Psychiatry 2021, 12, 742058. [Google Scholar] [CrossRef]
- Ermilov, M.; Gelfin, E.; Levin, R.; Lichtenberg, P.; Hashimoto, K.; Javitt, D.C.; Heresco-Levy, U. A pilot double-blind comparison of d-serine and high-dose olanzapine in treatment-resistant patients with schizophrenia. Schizophr. Res. 2013, 150, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol. Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Nakajima, S.; Suzuki, T.; Keefe, R.S.; Plitman, E.; Chung, J.K.; Caravaggio, F.; Mimura, M.; Graff-Guerrero, A.; Uchida, H. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: A systematic review and meta-analysis of double-blind randomized controlled trials. Mol. Psychiatry 2015, 20, 1151–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, E.A.; MacKenzie, E.M.; Rossolatos, D.; Perez-Parada, J.; Baker, G.B.; Dursun, S.M. D-serine and schizophrenia: An update. Expert Rev. Neurother. 2012, 12, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kondori, N.R.; Paul, P.; Robbins, J.P.; Liu, K.; Hildyard, J.C.W.; Wells, D.J.; de Belleroche, J.S. Focus on the role of d-serine and d-amino acid oxidase in amyotrophic lateral sclerosis/motor neuron disease (ALS). Front. Mol. Biosci. 2018, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Our World in Data: Schizophrenia Prevalence. Available online: https://ourworldindata.org/grapher/share-of-population-with-schizophrenia (accessed on 20 September 2022).
- Onaolapo, O.J.; Onaolapo, A.Y. Nutrition, nutritional deficiencies, and schizophrenia: An association worthy of constant reassessment. World J. Clin. Cases 2021, 9, 8295–8311. [Google Scholar] [CrossRef] [PubMed]
- Dipasquale, S.; Pariante, C.M.; Dazzan, P.; Aguglia, E.; McGuire, P.; Mondelli, V. The dietary pattern of patients with schizophrenia: A systematic review. J. Psychiatr. Res. 2013, 47, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Joy, C.B.; Mumby-Croft, R.; Joy, L.A. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst. Rev. 2006, 2006, CD001257. [Google Scholar]
- Aucoin, M.; LaChance, L.; Cooley, K.; Kidd, S. Diet and psychosis: A scoping review. Neuropsychobiology 2020, 79, 20–42. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Vieira, K.F. Nutritional therapies for mental disorders. Nutr. J. 2008, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Szeligowski, T.; Yun, A.L.; Lennox, B.R.; Burnet, P. The gut microbiome and schizophrenia: The current state of the field and clinical applications. Front. Psychiatry 2020, 11, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrono, E.; Svoboda, J.; Stuchlík, A. Schizophrenia, the gut microbiota, and new opportunities from optogenetic manipulations of the gut-brain axis. Behav. Brain Funct. 2021, 17, 7. [Google Scholar] [CrossRef] [PubMed]








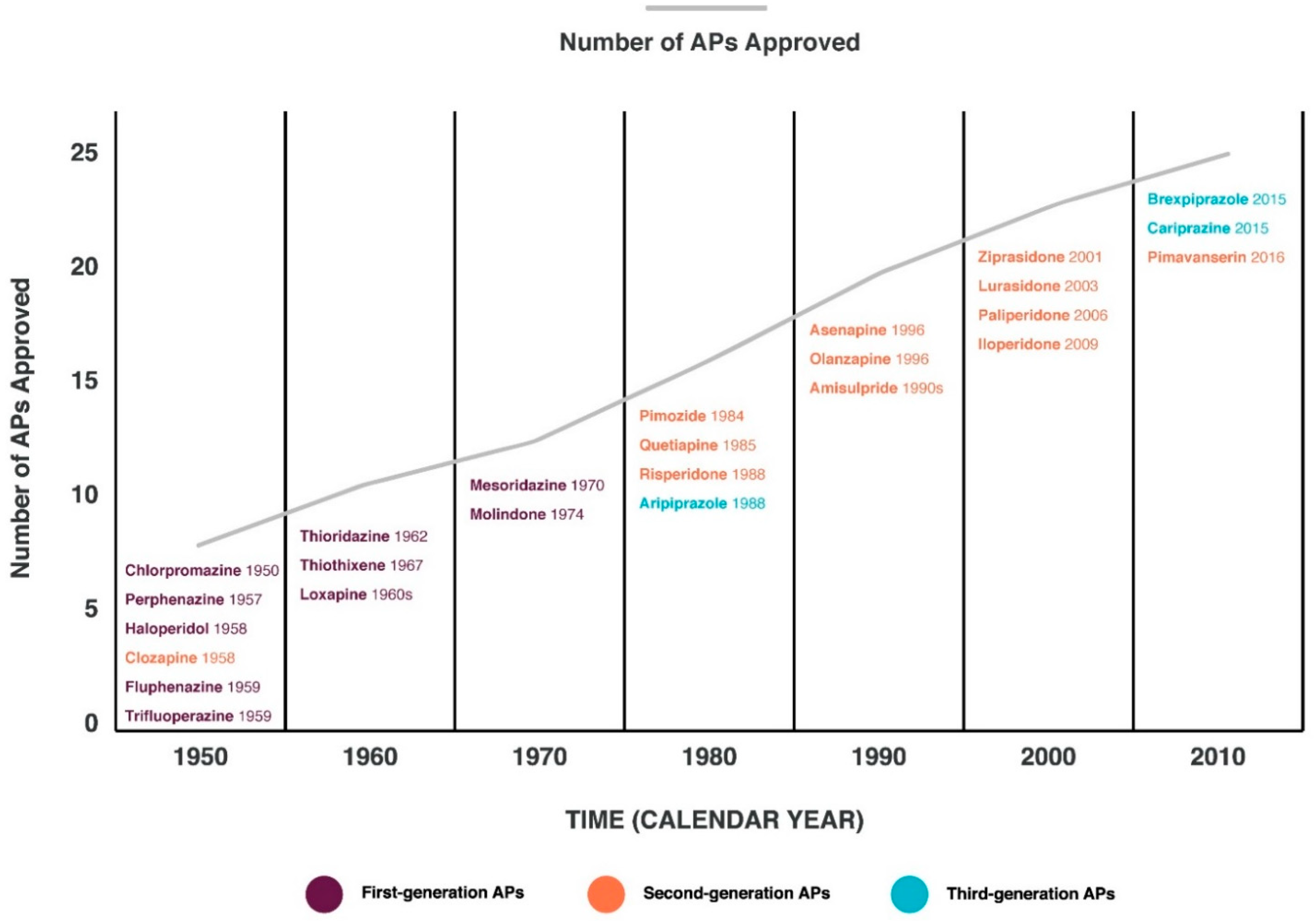{kind=link}
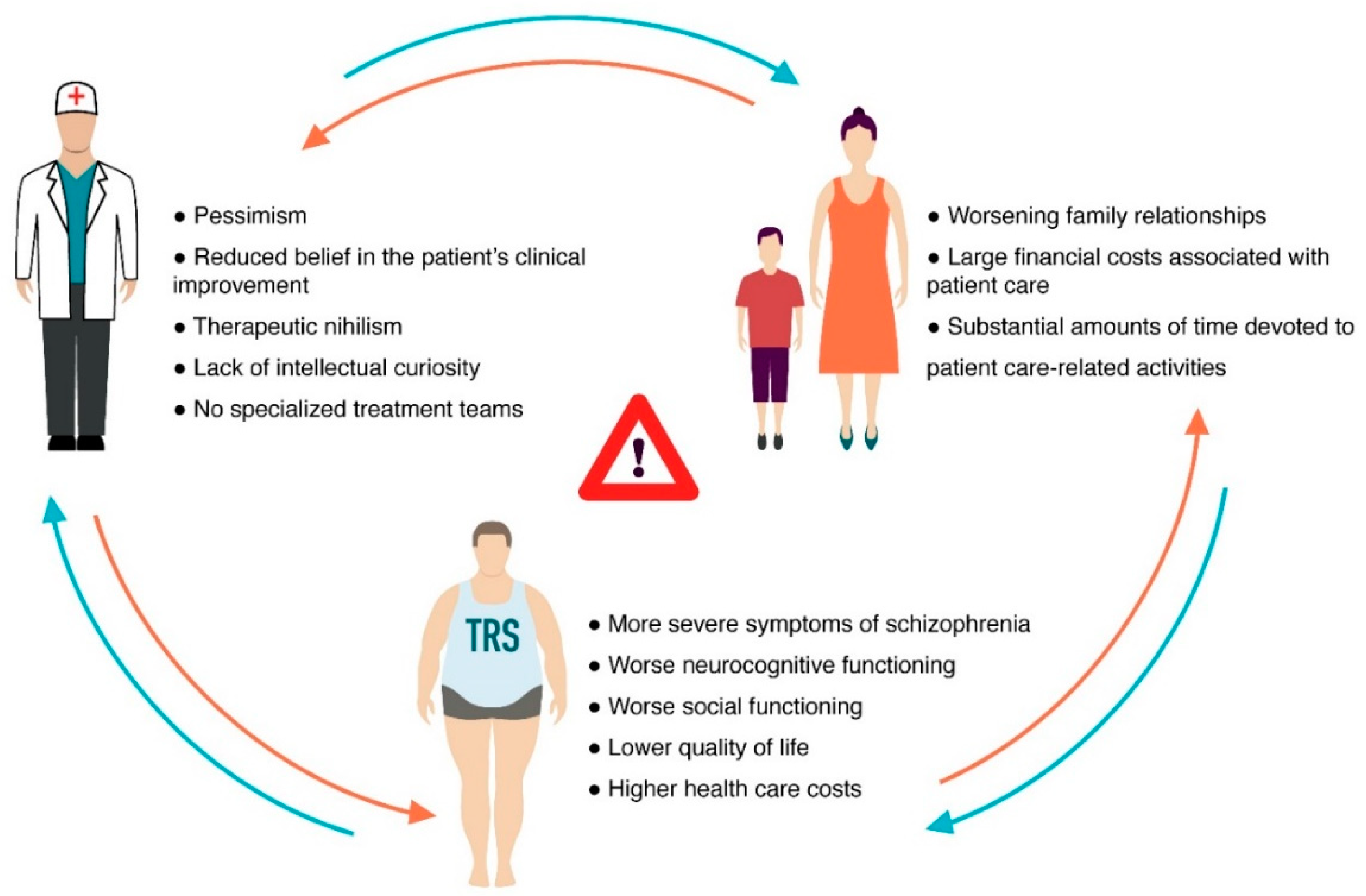{kind=link}
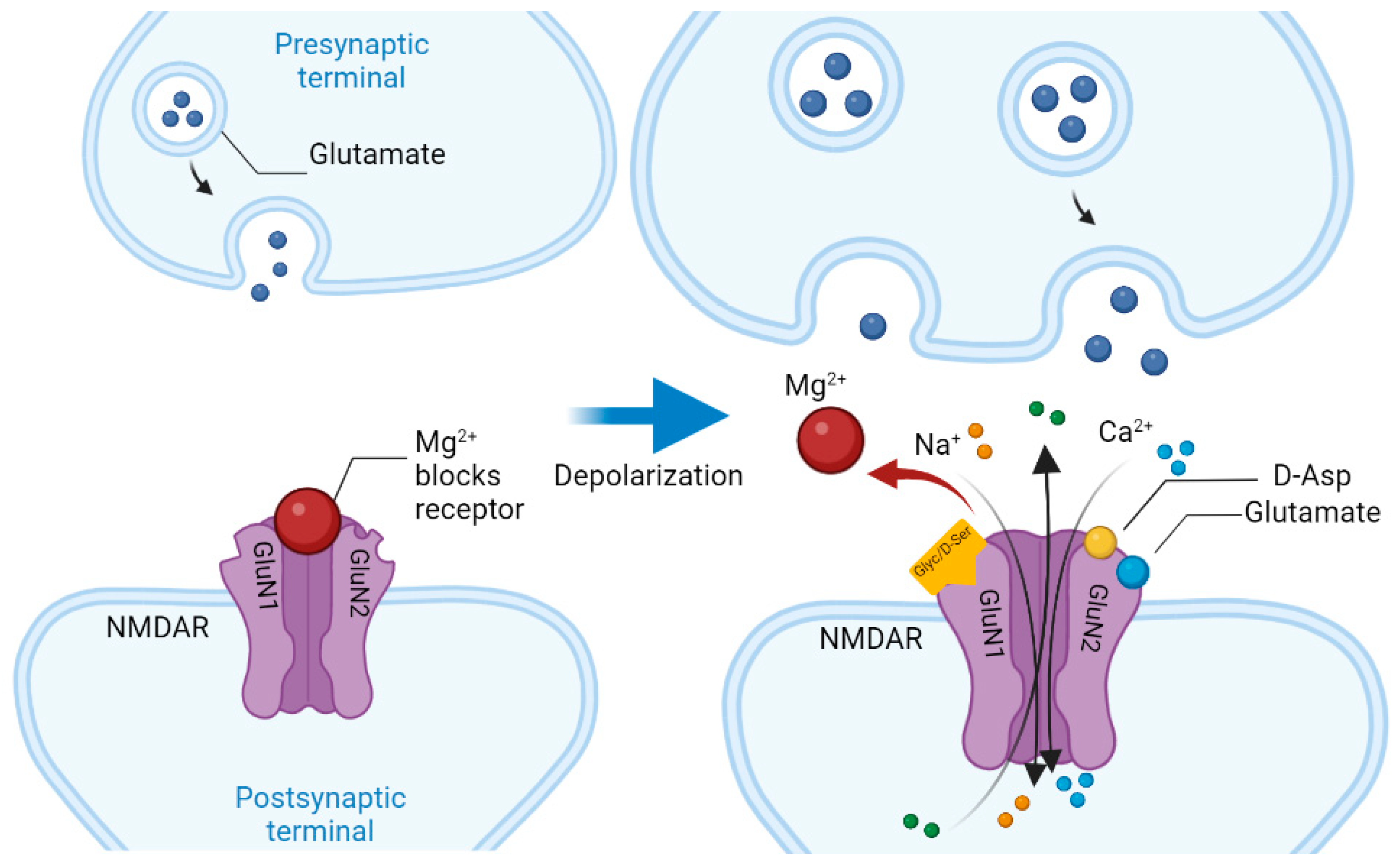{kind=link}
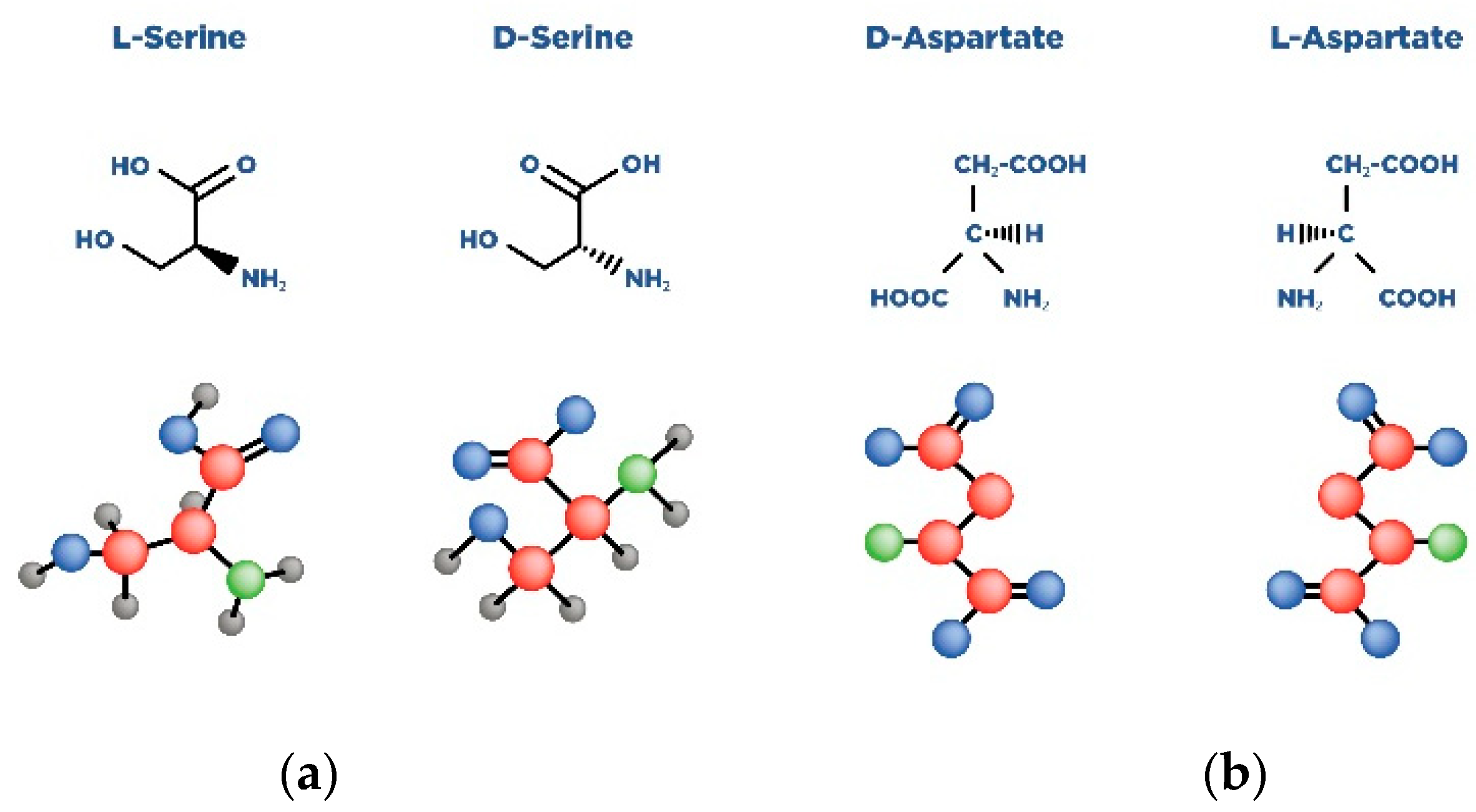{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Receptor | Function |
|---|---|---|
| D-aspartate | Synaptic NMDAR | Binds to the L-Glut region of ionotropic NMDARs. |
| D-serine | Synaptic NMDAR | A full agonist of the NMDAR Glyc modulatory site. Physiological ligand of the co-agonist site. Reduces NMDAR-mediated transmission in vitro via non-vesicular release via ASC-1 transporters. |
| Authors, Year | Country | Study Design | Results |
|---|---|---|---|
| Errico et al. [78] | Italy | Prospective study. Mice with a knockout of the DDO gene. The dose of D-Asp is 20 mM per os. | Increase in NMDARs activity. Decrease in pre-pulse inhibition deficit. Prevention of long-term cortical-striate depression. Enhancement of NMDAR-dependent memory. |
| Sacchi et al. [84] | Italy | Prospective study. Mice with a knockout of the DDO gene. The dose of D-Asp is 20 mM D-Asp per os. The dose of olanzapine is 5 mg/kg intraperitoneal. | Modulation of the therapeutic response to APs via D-Asp. Inhibition of the enzymatic activity of DDO. Increase in D-Asp and L-Glut extracellular level in the prefrontal cortex via olanzapine. |
| Authors, Year | Country | Study Design | Results |
|---|---|---|---|
| MacKay et al. [96] | Canada | Prospective study. Mice with a mutation in the SR gene. Biochemical markers, mRNA expression, behavioral phenotypes and pharmacological responses were examined. | Appearance of Sch symptoms: stereotypy, cognitive impairment, impaired prepulse inhibition, lack of social interaction. |
| Balu et al. [100] | USA | Prospective study. Mice with a knockout of the SR gene. The dose of D-Ser is 300 mg/kg day 1, 150 mg/kg days 2–21 subcutaneously. | Restoration of neuroplasticity. Increase in BDNF protein expression. Reversing the freezing deficit. |
| Authors, Year | Country | Study Design | Results |
|---|---|---|---|
| Kantrowitz et al. [65] | USA | Open prospective study. 42 adults with SSDS. The dose of D-Ser: 30, 60 or 120 mg/kg/day in addition to APs (the first and new generations). | Positive, negative and general PANSS symptoms improvement, reduction in neurocognitive dysfunction. |
| Pei et al. [105] | Taiwan | Open placebo-controlled prospective studies; 37 adults with SSDS. The dose of D-Ser: 1500–4000 mg/day in addition to APs (the first and new generations). | MMN and cortical plasticity improvement. Weak decrease in the severity of negative and positive Sch symptoms. Improvement of negative symptoms at doses above 3600 mg/day of D-Ser. |
| Kantrowitz et al. [88] | USA | Pilot, double-blind, placebo-controlled randomized parallel-group study. 20 patients aged between 13 and 35 years with clinical high risk of Sch. The dose of D-Ser: 60 mg/kg. | Improvement of negative symptoms. |
| Weiser et al. [64] | Israel | Multicenter, double-blind, randomized, placebo-controlled study; 97 adults with SSDS. The dose of D-Ser: 2000 mg/day in addition to APs (the first and new generations). | No significant difference between drug and placebo. |
| Tsai et al. [60] | USA and Taiwan | Meta-analysis; 102 adults with SSDS. The dose of D-Ser: 2000 mg/day in addition to APs (the first and new generations). | Reduction in Sch symptoms, improvement of cognitive functions and reduction in affective symptoms. |
| Kantrowitz et al. [71] | USA | Double-blind, placebo-controlled study; 21 adults with SSDS. The dose of D-Ser: 60 mg/kg/day. | MMN improvement. Reduction in PANSS overall scores. |
| Ermilov et al. [106] | Israel | Pilot, double-blind, placebo-controlled study; 10 adults with SSDS. The dose of D-Ser: 3000 mg/day. | Significant reduction in PANSS positive symptom scores and overall PANSS score. |
| MacKay et al. [96] | Canada | Double-blind, placebo-controlled studies, open-label studies. Different number of adults with SSDS. The dose of D-Ser: 30–120 mg/kg/day alone or in addition to APs (non-clozapine APs). | Reduction in negative and positive symptoms of Sch. Improvement of cognitive functions. |
| Heresco-Levy et al. [107] | USA | Double-blind, placebo-controlled study; 39 adults with SSDS. The dose of D-Ser: 30 mg/kg/day in addition to APs (olanzapine, risperidone). | Improvement of cognitive functions, reduction in positive and negative symptoms of Sch and reduction in comorbid depressive disorders. |
| Singh et al. [56] | UK | Meta-analysis; 99 adults with SSDS. The dose of D-Ser: 2000 mg/day in addition to APs (the first and new generations). | Reduction in Sch symptoms, improvement of cognitive functions and reduction in affective symptoms. |
| Iwata et al. [108] | Canada | Meta-analysis; 175 adults with SSDS. The dose of D-Ser—30 mg/kg/day in addition to APs (the first and new generations). | No significant difference between drug and placebo. |
| Tsai et al. [59] | Taiwan | Double-blind study; 10 adults with SSDS. The dose of D-Ser: 30 mg/kg/day in addition to APs (clozapine). | No increase in therapeutic efficacy of clozapine. |
| D-aspartate | D-serine |
|---|---|
| Berries | |
| Green plum | Black raisin |
| Goji | Chinese plum |
| Blackcurrant | Green plum |
| Redcurrant | Wampee |
| Gooseberry | Goji |
| Vaccinium (Blueberry, Cranberry, Huckleberry) | Monk fruit |
| Bilberry | Hawthorn |
| Lingonberry | Lantern fruit |
| Elderberry | Cape gooseberry |
| Grape | Mulberry |
| Loganberry | Black mulberry |
| Strawberry guava | Black chokeberry |
| Strawberry | Blackcurrant |
| Redcurrant | |
| Gooseberry | |
| Cloudberry | |
| Red raspberry | |
| Black raspberry | |
| Black elderberry | |
| Rowanberry | |
| Vaccinium (Blueberry, Cranberry, Huckleberry) | |
| Lowbush blueberry | |
| Sparkleberry | |
| Highbush blueberry | |
| American cranberry | |
| Bilberry | |
| Lingonberry | |
| Muscadine grape | |
| Common grape | |
| Beans | |
| Lima bean | Cannellini bean |
| Common bean | Scarlet bean |
| Adzuki bean | Lima bean |
| Mung bean | Common bean |
| Hyacinth bean | Broad bean |
| Winged bean | Adzuki bean |
| Gram bean | |
| Mung bean | |
| Climbing bean | |
| Hyacinth bean | |
| Moth bean | |
| Winged bean | |
| Bean | |
| Yellow wax bean | |
| Green bean | |
| Fruits | |
| Lichee | Pitaya |
| Mango | Lichee |
| Passion fruit | Mango |
| Pineapple | Passion fruit |
| Guava | Pineapple |
| Pomegranate | Custard apple |
| Tamarind | Guava |
| Banana | Pomegranate |
| Longan | Tamarind |
| Star fruit | Banana |
| Cherimoya | Longan |
| Coconut | Rambutan |
| Jackfruit | Star fruit |
| Kumquat | Abiyuch |
| Papaya | Acerola |
| Common persimmon | Breadfruit |
| French plantain | Natal plum |
| Prickly pear | Cherimoya |
| Sapodilla | Coconut |
| Mamey sapote | Durian |
| Kiwi | Jackfruit |
| Feijoa | Java plum |
| Persimmon | Kumquat |
| Apple | Mammee apple |
| Pear | Purple mangosteen |
| Papaya | |
| Common persimmon | |
| Pitanga | |
| Plains prickly pear | |
| French plantain | |
| Prickly pear | |
| Malabar plum | |
| Sapodilla | |
| Mamey sapote | |
| Soursop | |
| Sugar apple | |
| Kiwi | |
| Feijoa | |
| Persimmon | |
| Mushrooms | |
| Common mushroom | Jew’s ear |
| Shiitake | Common mushroom |
| Oyster mushroom | Shiitake |
| Maitake | Enokitake |
| Chanterelle | Oyster mushroom |
| Cloud ear fungus | |
| Maitake | |
| Chanterelle | |
| Morchella (Morel) | |
| Vegetables | |
| Spinach | Iceberg lettuce |
| Swiss chard | Pea shoots |
| Common beet | Water spinach |
| Jute | Spinach |
| Endive | Chicory leaves |
| Lettuce | Common beet |
| Garden cress | Corn salad |
| Fruit vegetables | Jute |
| Olive | Malabar spinach |
| Avocado | Rocket salad |
| Garden tomato | Swamp cabbage |
| Eggplant | Garden cress |
| Groundcherry | Radish |
| Pepper | Burdock |
| Parsnip | Celeriac |
| Radish | Carrot |
| Burdock | Potato |
| Celeriac | Asparagus |
| Carrot | Onion |
| Rape | |
| Carrot | |
| Garden rhubarb | |
| Celery stalks | |
| Aquatic foods | |
| Fishes | Fishes |
| Seaweed | Seaweed |
| Crustaceans | Mollusks |
| Mollusks | |
| Roe | |
| Meat | |
| Cattle (Beef, Veal) | Cattle (Beef, Veal) |
| Chicken | Chicken |
| Pork | Pork |
| Coffee | |
| Eggs | |
| Milk products | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasyrova, R.F.; Khasanova, A.K.; Altynbekov, K.S.; Asadullin, A.R.; Markina, E.A.; Gayduk, A.J.; Shipulin, G.A.; Petrova, M.M.; Shnayder, N.A. The Role of D-Serine and D-Aspartate in the Pathogenesis and Therapy of Treatment-Resistant Schizophrenia. Nutrients 2022, 14, 5142. https://doi.org/10.3390/nu14235142
Nasyrova RF, Khasanova AK, Altynbekov KS, Asadullin AR, Markina EA, Gayduk AJ, Shipulin GA, Petrova MM, Shnayder NA. The Role of D-Serine and D-Aspartate in the Pathogenesis and Therapy of Treatment-Resistant Schizophrenia. Nutrients. 2022; 14(23):5142. https://doi.org/10.3390/nu14235142
Chicago/Turabian StyleNasyrova, Regina F., Aiperi K. Khasanova, Kuanysh S. Altynbekov, Azat R. Asadullin, Ekaterina A. Markina, Arseny J. Gayduk, German A. Shipulin, Marina M. Petrova, and Natalia A. Shnayder. 2022. "The Role of D-Serine and D-Aspartate in the Pathogenesis and Therapy of Treatment-Resistant Schizophrenia" Nutrients 14, no. 23: 5142. https://doi.org/10.3390/nu14235142