
Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome?


1
Departamento de Fisiología y Farmacología, Facultad de Medicina, Universidad de Cantabria, 39011 Santander, Spain
2
Department of Biomedical and Neuromotor Sciences, University of Bologna, 40126 Bologna, Italy
*
Author to whom correspondence should be addressed.
Nutrients 2022, 14(14), 2880; https://doi.org/10.3390/nu14142880
Submission received: 1 June 2022
/
Revised: 11 July 2022
/
Accepted: 12 July 2022
/
Published: 13 July 2022
(This article belongs to the Special Issue Update on Fatty Acids and the Brain)
Abstract
:The triplication of chromosome 21 causes Down syndrome (DS), a genetic disorder that is characterized by intellectual disability (ID). The causes of ID start in utero, leading to impairments in neurogenesis, and continue into infancy, leading to impairments in dendritogenesis, spinogenesis, and connectivity. These defects are associated with alterations in mitochondrial and metabolic functions and precocious aging, leading to the early development of Alzheimer’s disease. Intense efforts are currently underway, taking advantage of DS mouse models to discover pharmacotherapies for the neurodevelopmental and cognitive deficits of DS. Many treatments that proved effective in mouse models may raise safety concerns over human use, especially at early life stages. Accumulating evidence shows that fatty acids, which are nutrients present in normal diets, exert numerous positive effects on the brain. Here, we review (i) the knowledge obtained from animal models regarding the effects of fatty acids on the brain, by focusing on alterations that are particularly prominent in DS, and (ii) the progress recently made in a DS mouse model, suggesting that fatty acids may indeed represent a useful treatment for DS. This scenario should prompt the scientific community to further explore the potential benefit of fatty acids for people with DS.
1. Trisomy 21: Overview
Down syndrome (DS) is a relatively high-incidence chromosomal disorder (about 1 in 800–1000 live births) (see [1,2]) due to the triplication of chromosome 21. For this reason, it is also called trisomy 21 (T21). This gene burden causes a series of clinical manifestations, such as heart defects, hematologic, intestinal, and stomatognathic disturbances, and vision and hearing defects, which may not be present in all individuals with DS [3]. Intellectual disability (ID) present from infancy, however, is a constant hallmark of DS [4,5,6]. Although it exhibits a wide spectrum of severity, from very severe (IQ of 20–35) to mild (IQ of 50–70) [3], it prevents a completely autonomous life. Moreover, people with DS are particularly prone to developing Alzheimer’s disease (AD) starting from 35–40 years of age, which worsens an already compromised brain functioning because it leads to dementia [7]. ID is undoubtedly a major concern of the parents and guardians of children with DS and, additionally, it represents a social burden in view of its increase in during the last 10–20 years due to advances in medical care. Therefore, a pressing question is whether it is possible to ameliorate the damage to the cognitive abilities of people with DS, in addition to their general health, and to provide them with a greater degree of autonomy. In order to answer this question, it is necessary to know the causes of ID in DS.
In individuals with DS, the brain is smaller in comparison with the brain in the general population. This typical feature, which is particularly prominent in the cerebellum and hippocampus, is detectable in fetal life stages (see [8]). Early hypotrophy in the brain is not caused by a loss of neurons due to neurodegeneration but by impairment of the process of neuron generation (neurogenesis), which causes a −20 to −30% reduction in the number of neurons populating the brain (see [8]). Neurogenesis is reduced due to (i) a reduction in the proliferation rate of neural stem cells and progenitor cells (collectively, NPCs), and (ii) preferential differentiation of the newborn cells into astrocytes/oligodendrocytes rather than into neurons, i.e., a reduction in neurogenesis sensu stricto (see [9]). It must be noted that, although gliogenesis is not compromised in DS, astrocytes and oligodendrocytes exhibit various functional abnormalities that affect neuronal function [10,11,12]. This reduction in the number of neurons that populate the DS brain is worsened by the fact that their maturation is also compromised, which causes the generation of dendritic arbors scarcely ramified and endowed with dendritic spines that are reduced in number and have an abnormal shape [13,14,15,16,17,18]. Moreover, various neurotransmitter and receptor systems are altered in DS (see [19,20]), which, by building upon these ”gross” abnormalities, further impair brain-circuit functioning. At the cellular level, in addition to alterations to numerous pathways, mitochondrial, morphological, and functional alterations impair cell metabolism and cause oxidative stress [21,22]. These alterations may compromise neurogenesis due to an overall impairment in cell functioning [23,24] and may promote the precocious aging that characterizes individuals with DS [25]. Cell metabolic dysfunction, in turn, causes neuroinflammation, which further impairs neuronal functions [26]. Although the paucity of neurons and their aberrant maturation are most likely crucial determinants of ID in DS, all of the above-mentioned alterations promote ID. The term ID generically signifies an alteration in normal brain functioning. In the case of DS, ID includes various cognitive domains, among which the impairment of visual, spatial, and verbal memory, language, and executive behaviors stand out as particularly severe [27,28,29,30,31,32,33]. ID may be accompanied by psychiatric comorbidities, such as attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), mood disorders, psychosis, and regression [34,35,36,37,38].
2. The Search for Therapies for DS and the Hypothesis: Can Fatty Acids Become a Therapeutic Strategy for DS?
The fact that an entire chromosome is triplicated makes it very difficult to establish which of the triplicated genes or sets of genes is responsible for the multifaceted alterations of the DS brain. However, accumulating evidence suggests that among the triplicated genes, DYRK1A, APP, RCAN1, OLIG2, and DSCAM are certainly major determinants of the neurodevelopmental alterations in DS (see [9]). The mechanisms regarding psychiatric comorbidities are less well understood, although alterations in GABA signaling and monoaminergic signaling are likely to play a prominent role (see [39]). Regarding AD development in DS, APP is thought to be the main candidate gene, although the mechanisms whereby increased APP gene dose causes AD in DS have only been partially clarified [40,41,42].
There are no treatments for ID in DS, no specific treatment for psychiatric comorbidities, and no treatment to prevent AD. During the last 20 years, great efforts have been undertaken to establish whether it is possible to improve ID in DS by taking advantage of murine models. Various mouse models of DS are now available (see [43,44]), among which the Ts65Dn mouse model has been extensively exploited because it exhibits numerous phenotypes that are typical of DS. These studies have shown that a variety of agents either targeting or not targeting triplicated genes can benefit cognition and may counteract age-related neurodegeneration, which encourages the search for treatments for DS (see [9,45,46,47,48,49,50]). Most of the treatments used in DS mouse models may raise safety concerns for human use, especially at early life stages. Natural substances have been traditionally used by humans to combat a myriad of illnesses and disturbances. Many natural substances are currently attracting attention as possible tools to ameliorate developmental brain disorders because they are able to change the expression of genes and the activity of pathways involved in brain development (see [51]). Accumulating evidence shows that fatty acids, which are nutrients present in a normal diet, exert many positive effects on the body and brain, affecting cardiovascular disease, diabetes, cancer, Alzheimer’s disease, dementia, depression, and visual and neurological development [52,53,54].
The advantage of fatty acids is their generally safe profile, which is why they are useful during gestation and infancy. This is relevant for DS because the fetal period is the only time window during which cortical neurogenesis can be rescued. In this study, we review the knowledge obtained so far in various animal models regarding the impact of fatty acids on brain alterations that are particularly prominent in DS (Figure 1). This knowledge is fundamental for the design of future studies specifically devoted to establishing whether treatment with fatty acids may represent a useful tool for different DS-related phenotypes. Very few studies have explored the hypothesis that fatty acids may be a potential strategy for improvements in the neurodevelopmental and cognitive defects in DS. We also review recent achievements in relation to mouse models and individuals with DS, suggesting that fatty acids may indeed become a useful treatment for DS. These preliminary achievements should prompt researchers to further explore the potential benefits of fatty acids for people with DS.
3. Overview of Fatty Acids: Chemistry and Nomenclature
Below, we provide a short description of fatty acids, mainly based on the reviews by Petrovic and Arsic [55] and Hussain et al. [56]. “Fatty acids” is an umbrella term including various chemical compounds, such as triglycerides, phospholipids, and cholesterol. Triglycerides make up 90% of dietary lipids and provide 30–40% of daily energy intake in Western societies. Triglycerides are the esters of glycerol and free fatty acids. Most fatty acids in the body take the form of glycerol esters. Fatty acids (FAs) are long hydrocarbon chain molecules (C4 to C28 atoms). Most FAs have an unbranched chain with a carboxyl group (–COOH) at one end and a methyl group (–CH3) at the other end. The naming of FAs is based on three main criteria. The first is the length of the hydrocarbon chain, which divides FAs into short-, medium-, long-, and very-long-chain FAs (Table 1). The second is the degree of unsaturation, i.e., the number of double bonds. Based on the degree of unsaturation, FAs are classified as either saturated if they have no C-C double bond (saturated fatty acids; SFAs) or unsaturated if they have at least one C-C double bond. Unsaturated FAs, in turn, are classified as monounsaturated FAs (MUFAs) if they have one double bond and polyunsaturated FAs (PUFAs) if they have two or more double bonds. The third is the position of the double bonds. According to one classification, this position refers to the distance from the carboxyl terminus, C1 (C-x). Alternatively, numbering refers to the distance of the double bond from the C atom of the methyl group, which is designated as n or ω (omega). In n minus x (also ω-x), a double bond of the fatty acid is located on the xth carbon–carbon bond counting from the terminal methyl carbon (designated as n or ω) toward the carbonyl carbon (Figure 2; see Table 2 for examples of FAs).
Unlike unsaturated FAs, SFAs are solid at room temperature. Thus, humans have FA desaturases that introduce a double bond. In addition, elongases increase the lengths of FAs. The most important PUFAs for humans are derived from two parent acids, linoleic acid (LA, 18:2 n-6) and α-linolenic acid (ALA, 18:3 n-3). Humans can synthesize SFAs and MUFAs, but they are not able to synthesize LA and ALA [57]. Both must be consumed with the diet and, therefore, they are termed “essential” fatty acids. Through the actions of desaturases and elongases, LA produces principally arachidonic acid (ARA, 20:4 n-6), whereas ALA produces mainly eicosapentaenoic acid (EPA, 20:5 n-3) and, subsequently, docosahexaenoic acid (DHA, 22:6 n-3). Lipid mediators derived from n-6 and n-3 PUFA are metabolically distinct and often have opposing physiological and pathological functions. While EPA, DPA, and DHA produce anti-inflammatory metabolites (e.g., resolvins), ARA produces pro-inflammatory metabolites (Figure 3).
Thus, while n-3 PUFA-derived lipid mediators largely inhibit inflammation, n-6 PUFA-derived eicosanoids tend to promote inflammation (see [58,60]). However, it must be noted that not all metabolites of ARA are inflammatory. In particular, lipoxin A4, which derives from the metabolism of ARA, powerfully inhibits the synthesis of pro-inflammatory cytokines while increasing anti-inflammatory cytokines (see [59]). The overall effects of PUFAs are dependent on the n-3 and n-6 ratio. A ratio of 1:4 is recommended for health benefits to be obtained, although the ratio is about 1:10 in modern diets. Since the metabolites derived from n-6 and n-3 PUFAs have opposing physiological and pathological activities, the deleterious consequences associated with the consumption of n-6 PUFA-rich diets are thought to reflect the excessive production of n-6 PUFA-derived eicosanoids, while the beneficial effects associated with the consumption of n-3 PUFA-rich diets reflect the large production and activities of n-3 PUFA-derived mediators.
Gut microbiota comprise trillions of microorganisms, including bacteria, archaea, and eukaryotes [61]. Gut microbiota can alter n-3 PUFA metabolism to generate increased levels of long-chain PUFA metabolites, such as EPA and DHA; furthermore, they can produce conjugated fatty-acid derivatives of PUFAs, such as conjugated linoleic acid (CLA) and conjugated α-linolenic acid, and they can increase the production of short-chain FAs (see [62,63]). Short-chain FAs are produced by microbiota through fermentation of nondigestible carbohydrates, such as dietary fibers. Among short-chain FAs, butyrate has multiple beneficial effects because it reduces mucosal inflammation, enhances FA oxidation, lowers total cholesterol, and prevents metabolic diseases [64,65].
In addition to inducing local changes in the gut epithelium, dietary metabolites derived from gut microbiota can affect multiple metabolic and physiological processes through epigenetic changes of gene expression (see [62]). Importantly, gut metabolites can influence the epigenetic machinery of brain cells, leading to changes in neuronal expressions that may cause changes in host behavior (microbiota–gut–brain axis). Consequently, any alteration in microbiota composition can lead to various disorders, such as inflammatory diseases, psychiatric disorders, and cognitive alterations [66]. Different factors affect the microbiota composition [67] and, thus, the epigenetic mechanisms that regulate gene expression. One of the most important factors affecting microbiota composition and microbiota-dependent epigenetic effects is the diet and the amount and type of dietary fat, with a high-fat diet being implicated in reduced gut microbiota richness [68].
4. Overview of Fatty Acids in the Brain
4.1. Major FAs in the Brain
Dietary fatty acids are currently of great interest because they act as signaling molecules responsible for a range of effects in the developing and adult brain [52,53,54]. Western fetuses accumulate FAs in the order of LA > ARA > DHA at all stages of pregnancy, and the highest accretion rates are reached in the last 5 weeks of gestation [59]. During pregnancy, most of the infant LA, ARA, and DHA are located in adipose tissue (68, 44, and 50%, respectively). Substantial amounts of LA (17%) and ALA (40%) are also located in skeletal muscle, and substantial amounts of ALA (11%) and DHA (23%) are also located in the brain [59]. Maternal omega-3 DHA and omega-6 ARA accumulate rapidly within the cerebral cortex during the last trimester of pregnancy, and the balance between DHA and ARA appears to play a fundamental role during brain development [60,61], as well as throughout life [62].
Approximately 50–60% of the adult dry brain weight consists of lipids, mostly structural lipids in the form of phospholipids (see [63]). These include large amounts of long-chain polyunsaturated fatty acids (LCPUFAs), mainly ARA and DHA, and very small amounts of LA and ALA. [57]. The lipids in the brain are both structural components and regulators of cell physiology. As components of plasma membranes, lipids provide fluidity and a framework for receptors, enzymes, transporters, and ion channels, and they function as reservoirs of lipid mediators. The LCPUFAs in the brain are important building blocks of neuronal membranes. The lipid bilayer of neuronal membranes consists of phospholipids, with DHA, ARA, and EPA as their main components. In addition to having a structural role, LCPUFAs support and improve brain structure and functioning. As detailed below, LCPUFAs (mainly in a healthy balance of n-6/n-3) play an important role during brain development by modulating neurogenesis, the outgrowth of axons and dendrites, and cognition, as well as normal aging by decreasing neuroinflammation, thereby preventing cognitive deterioration.
4.2. Sources of Transport of FAs in Neuronal Cells
Intracellular FAs may derive from (i) the lipolysis of complex lipids, (ii) de novo lipogenesis, or (iii) extracellular sources (see [69]). In the process of lipolysis, triglycerides are hydrolyzed by various lipases, producing FAs. In the process of de novo lipogenesis, fatty acid synthase (FASN) elicits the formation of palmitic acid (PA) from acetyl-CoA and, subsequently, malonyl-CoA. PA can then be expanded by elongases to produce stearic acid and/or desaturated to produce MUFAs, such as oleic acid (OA) (see [56,69]). Extracellular FAs are bound to fatty-acid-binding proteins (FABP), albumin, or in triglyceride form within apolipoproteins (Apo). Extracellular fatty acids can enter the cell by passive diffusion (short-chain FAs) or via transporters, such as FAT/CD36 or fatty-acid-transport protein (FATP) (see [69]).
Since they have low solubility, FAs diffuse through the cytosol bound to proteins. Fatty-acid-binding proteins (FABPs) are a family of proteins that reversibly bind FAs and shuttle them to different intracellular compartments, such as mitochondria, peroxisome, endoplasmic reticulum, and the nucleus (see [70,71]). FABPs are expressed in NPCs in vivo and in vitro [72]. Fabp7 is expressed in embryonic and hippocampal NPCs and is necessary for the maintenance of NPCs [73]. Fabp7 is also required for the differentiation of neuronal and glial cells [72].
4.3. Functions of FAs in Neuronal Cells
4.3.1. Components of Cell Membranes
FAs can serve as components of neuronal membranes, into which they are integrated in the form of phospholipids or glycolipids. The types of FA (MUFAs or PUFAs) in the membranes contribute to their physicochemical properties, such as fluidity, which in turn may affect the functions of proteins embedded in the membranes, such as receptors or transporters.
4.3.2. Production of Energy
FAs are stored as triglycerides in lipid droplets within cells. Following lipolysis, they enter the mitochondria for ATP production through β-oxidation.
4.3.3. Signaling
FA signaling can be mediated by membrane and nuclear receptors. The membranes of peripheral and neuronal cells contain G-protein-coupled free-fatty-acid receptors (FFARs) (see [74]). Among the FFARs, there are two forms with prominent levels of expression in neural tissues: FFAR1 (also known as GPR40) and FFAR4 (GPR120 or O3FAR) [71]. Although the signaling pathways of FFAR1 and FFRAE4 in neural cells are not well understood, the effects of FFAR1 might be mediated by the phosphorylation of CREB (see [71]). In addition, FAs derived from membrane lipids can play a role as second messengers. For instance, diacylglycerol, which is formed by glycerol plus two FAs and is derived from the hydrolysis of membrane phosphatidylinositols operated by phospholipase C, can activate protein kinase C. The effects of FAs in the nucleus are mediated by the peroxisome-proliferator-activated receptors (PPARs). PPARs are ligand-activated nuclear-receptor proteins with a transcriptional function and represent the best-recognized nuclear sensor system for fatty acids [75,76]. There are three classes of PPAR: PPARα, PPARβ/δ, and PPARγ [76]. All three PPARs are able to bind FAs, with a general preference for LCPUFAs [75]. PPARs form heterodimers with their obligate partners, the members of another subfamily of nuclear receptors, the retinoid X receptors (RXRs). Upon ligand binding, PPAR/RXR heterodimers recognize and bind to specific DNA sequences, called PPAR response elements (PPRE). The chain length and degree of unsaturation of FAs affect their affinity to PPARs [77], which helps explain differences in the actions exerted by different FAs in the brain.
5. Fatty Acids and Neurogenesis
The intrinsic genetic programs that regulate prenatal and postnatal neurogenesis can be modulated by many factors, including hormones, neurotransmitters, growth factors, cytokines, drugs, physical activity, and the environment (see [78,79]). Importantly, recent evidence indicates that FAs are not merely key elements of cell-membrane molecules or sources of energy, but rather appear to play a significant role in brain development by regulating NPC proliferation, differentiation, and maturation (see [56,69,71,80]).
5.1. Short Outline of Neurogenesis in Humans and Rodents
The brain is formed by neurons and glia (astrocytes and oligodendrocytes). The term neurogenesis includes two major processes: (i) the active proliferation of neural stem cells; and (ii) the differentiation of their daughter cells into either neurons or glia. The production of neurons and glia takes place during gestation. Neurons are produced first, followed by the production of astrocytes and, subsequently, of oligodendrocytes (for references and details regarding neurogenesis and gliogenesis, see [81,82,83,84,85]). Neurons and glia are derived from NPCs that are located in the germinative epithelium lining the wall of the neural tube. This epithelium is formed, initially, by the ventricular zone (VZ) and, subsequently, by the VZ plus the subventricular zone (SVZ). Before neurogenesis begins, NPCs divide symmetrically, forming two equal daughter cells, thereby expanding the pool of NPCs. At a certain time point, NPCs produce radial glial cells (RGC) that are NPCs themselves and divide asymmetrically, producing one RGC that remains in the VZ/SVZ and one neuron that migrates radially to the primordium of the future cortex. RGCs can also produce a population of cells called intermediate precursor cells (IPCs) or transit-amplifying cells (TAP), which, after a certain number of divisions, create neurons that migrate to the respective cortex. The formation of the cortex follows an inside-out pattern, according to which neurons migrating from the VZ/SVZ settle in increasingly superficial layers of the developing cortex. Cortical neurogenesis is completed by GW24–25 [84]. By contrast, cerebellar neurogenesis continues to the fifth postnatal month [86], while neurogenesis in the subgranular zone (SGZ) of the hippocampal dentate gyrus extends to the first postnatal year [82,87], and continues at a very slow rate throughout life [88,89,90,91]. Although cortical neurogenesis is completed before birth, the NPCs in the VZ/SVZ do not lose their proliferative capacity, but the neurons they produce are specifically destined for the olfactory bulb. In mice, cortical neurogenesis occurs between E11 and E17, hippocampal neurogenesis occurs between E10 and E18, and cerebellar granule cell neurogenesis starts at E13 and is completed at postnatal day 14 (see [46]). In rodents, the dentate gyrus produces most of its neurons (~80%) in the first two neonatal weeks and continues, slowly, to produce neurons throughout life. Similarly to humans, the postnatal VZ/SVZ of mice produce granule neurons destined for the olfactory bulb.
The proliferation and differentiation of NPCs is controlled by genes that encode basic helix–loop–helix (bHLH) transcription factors [92,93]. Neuronal differentiation is promoted by activator-type bHLH factors, such as Mash1 and NeuroD, whereas repressor-type bHLH factors, such as Notch1, Hes1, Hes3, and Hes5, promote the proliferation of NPCs. In the absence of repressor-type bHLH factors, NPCs do not proliferate sufficiently, but prematurely differentiate into neurons and become depleted. Hes genes antagonize the activator-type bHLH genes that promote neuronal fate determination. The balance of activity among these factors is thought to determine cell fate [92,93].
5.2. Effects of Fatty Acids on Neurogenesis
5.2.1. Overview
Accumulating evidence shows that FAs play an important role in controlling neurogenesis in the two major forebrain neurogenic niches, the VZ/SVZ of the lateral ventricle and the SGZ of the hippocampal dentate gyrus [94].
Under normal conditions, the NPCs of the VZ/SVZ depend on fatty-acid oxidation for proliferation [95]. The NPCs of the SVZ do not require glucose to sustain aerobic respiration, unlike mature neurons and astrocytes. The NPCs within the adult SVZ oxidize fatty acids to produce energy, and this process is fundamental to supporting neurogenic activity. Indeed, the inhibition of fatty-acid oxidation decreases proliferation in cultured NPCs both in vitro and in the SVZ [95]. By contrast, the inhibition of fatty-acid oxidation does not decrease proliferation in the SGZ of the dentate gyrus. In this region, however, baseline and exercise-induced hippocampal neurogenesis require fatty-acid synthesis [96,97].
The relevance of FAs to the process of neurogenesis is highlighted by the fact that a doubling of membrane phospholipids is required in the S phase of mitosis for the creation of daughter cells [98]. Although the mechanisms whereby FAs affect proliferation are not well understood, emerging evidence (described below) shows that FAs are able to affect cell-cycle regulatory proteins and to change the expression of factors promoting NPC proliferation, e.g., Notch1 and Hes1, and differentiation, e.g., Mash1 and NeuroD [99,100,101,102].
Since the chemical structures of FAs may affect their function, and the cellular responses to FAs are characterized by distinct gene-expression profiles [103], we describe separately the effects of the different classes of FAs (SFAs, MUFAs, and PUFAs) on NPC neurogenesis.
5.2.2. Effects of Saturated Fatty Acids
PA is the most common SFA, accounting for 20–30% of the total fatty acids in the human body. It has been considered detrimental to the health because it causes lipotoxicity [104], although its negative effects may be related to an imbalance of dietary PA/PUFAs [105]. PA appears to inhibit the proliferation of mouse-derived NPCs and favor astrogliogenesis, an effect associated with the activation of Stat3 (a transcription factor that promotes astrogliogenesis) [106]. In cultures of human embryonic stem cells, PA does not affect pluripotency or differentiation into neural progenitor cells, but reduces NPC proliferation [107]. Unlike in mouse-derived NPCs, however, PA increases the expression of genes needed for neuronal differentiation (e.g., TBR2, TBR1, and NEUROD1) and neurogenesis [107]. Adult mice that engage in voluntary exercise exhibit upregulation of hippocampal FA synthase, increased levels of PA (and stearic acid) in the hippocampus (but not the cortex or cerebellum), an increase in NPC proliferation in the SGZ of the dentate gyrus, and cognitive enhancement [96]. Taken together, experiments in vitro suggest that PA inhibits NPC proliferation [106,107] and favors gliogenesis in mouse-derived NPCs [106], but favors neurogenesis in human-derived NPCs [107]. It remains to be established whether these differences in cell fate are due to differences in the experimental protocols used in these studies or reflect differences between humans and rodents. Interestingly, the available in vivo evidence suggests that de novo synthesis of FAs, including PA, may be involved in the exercise-induced stimulation of neurogenesis in the adult dentate gyrus [96].
5.2.3. Effects of Unsaturated Fatty Acids: MUFAs
Recent evidence shows that OA is the most abundant MUFA in human neural stem/progenitor cells and is necessary for their survival [108]. TLX (also known as NR2E1) is an orphan nuclear receptor that regulates the self-renewal and proliferation of NPCs [109]. OA was found to bind to TLX and to convert it from a transcriptional repressor to a transcriptional activator of cell-cycle and neurogenesis genes. This effect led to an increased proliferation rate of NPCs and their differentiation into neurons [108]. Nitro fatty acids (NO2-FAs) are naturally occurring compounds generated through the actions of reactive nitrogen species on cellular lipids, mainly PUFAs [110]. A recent study shows that nitro-oleic acid (NO2-OA) inhibits the expression of pluripotency markers (NANOG, SOX2, and OCT4) in murine embryonic stem cells and upregulates the expression of neuronal progenitor markers, such as Nestin [111], suggesting that OA, when nitrated, inhibits the proliferative potency of NPCs and enhances neurogenesis.
These studies show that OA increases the proliferation of NPCs and drives the progeny toward the neuronal lineage [108]. Although OA nitration inhibits stemness maintenance, nitrated OA retains the ability to favor the acquisition of a neuronal phenotype [111]. It must be observed that diets high in MUFAs decrease the number of proliferating NPCs in the hippocampus of adult male rats [112], suggesting that excessive doses of MUFAs may be deleterious for neurogenesis.
5.2.4. Effects of Unsaturated Fatty Acids: PUFAs
LA was shown to favor the proliferation of mouse-derived NPCs [113], an effect that was accompanied by an increase in cell-cycle regulatory proteins (such as cyclin D1, cyclin E) and mediated via Ca2+, PLC/PKC, PI3K/Akt, and p44/42 MAPKs signaling pathways. In line with this study, LA was found to promote the maintenance of NPCs derived from rat embryos [114]. Treatment with safflower-seed oil, which contains a high concentration of LA (74%) (as well as 15% OA), was found to enhance the proliferation rate of mouse-derived cultured NPCs and, additionally, to favor neuronal differentiation [115]. LA alone, however, had no effects on proliferation or differentiation, suggesting the necessity of a synergic action of LA and OA (and/or other components of safflower seed oil). In line with this latter evidence, Hejr et al. found no effect of LA on either the proliferation or differentiation of mouse-derived embryonic NPCs [102]. The discrepancies between all these studies regarding the effect of LA on NPC proliferation may be explained by differences in the tissues used to derive NPCs and/or embryonic age of animals. Cis-9, trans-11-conjugated linoleic acid (CLA), is the primary isomer of LA naturally present in milk. In rat-derived embryonic NPCs, treatment with CLA was found to reduce the proliferation rate and to promote neuronal differentiation [101]. This indicates that LA and its major isomer may have opposite effects on proliferation, but that both may favor neurogenesis.
Hejr et al. found that ALA increased the proliferation rate of mouse-derived embryonic NPCs [102]. Treatment with LA had no effect, but various ratios of LA/ALA (1:3, 2:2, or 3:1) increased cell proliferation more than ALA alone, with the largest effect was at the ratio of 2:2 (LA 50: ALA 50), suggesting the relevance of a suitable ratio between these two PUFAs. Under differentiation conditions, the ALA increased the acquisition of an astrocytic and oligodendrocytic phenotype, but did not increase acquisition of a neuronal phenotype. ALA is the major component (90%) of Alyssum-homolocarpum-seed oil (AHSO). Mouse-derived embryonic NPCs treated with either AHSO or ALA were found to undergo an increase in proliferation rate and, under differentiation conditions, to preferentially acquire astrocyte and oligodendrocyte phenotypes [116]. Both these studies suggest that ALA has a pro-proliferative effect on NPCs and, under differentiation conditions, increases the acquisition of an astrocytic and oligodendrocytic phenotype.
Kawakita et al. examined the effect of DHA on rat-derived embryonic NPCs [99,117]. They found that DHA reduced the number of proliferating cells, an effect that was associated with decreased levels of Hes1 (a transcription factor that maintains NSCs in an undifferentiated state), and increased the number of cells differentiated into neurons, an effect that was associated with increased levels of NeuroD, a neurogenic differentiation factor. The pro-neurogenic role of DHA was confirmed by another study [118] showing that in embryonic NPCs induced to differentiate, exposure to DHA increases the number of cells exhibiting a neuronal phenotype. The pro-neurogenic effect of DHA has also been found to be associated with the increased activation of WNT and CREB signaling [119]. Docosahexaenoylethanolamine (DEA, also called synaptamide) is a metabolite of DHA. Treatment with DEA was found to induce neuronal but not astrocytic differentiation of cultured rat NPCs, suggesting that endogenously produced synaptamide may promote neurogenesis [120].
Rats fed from postnatal day 2 until 31 days of age with a diet enriched with ARA, but not with DHA or ARA + DHA, were found to undergo an increase in the number of NPCs in the dentate gyrus, indicating that ARA, unlike DHA, exerts pro-proliferative effects [121]. Accordingly, in rats fed with ARA, but not DHA, there was an amelioration in the age-related decline in the number of hippocampal NPCs [122]. Under differentiation conditions, ARA was also found to promote the glial differentiation of NPCs [123]. Taken together, the studies on DHA and ARA suggest that DHA does not foster the maintenance of an NPC pool because it enhances neurogenesis. By contrast, ARA contributes to maintain the pool of NPC and does not favor neurogenesis, but favors gliogenesis.
ALA and DHA are omega-3 FAs, and LA and ARA are omega-6 FAs. The effects of different ratios of omega-3 and omega-6 FA may induce effects that are more complex than those elicited by ALA, DHA, LA, and ARA alone. In rats, a maternal diet deficient in ALA causes a reduction in brain phospholipid levels of DHA (which is derived from ALA) and an increase in the levels of ARA (which is derived from LA) in comparison with controls, with no change in proliferation in the SVZ and SGZ, but a reduction in neurogenesis [124]. An increase in dietary omega-3 relative to omega-6 fatty acids (2:1) favors neurogenesis and reduces apoptosis in the lobster brain [125]. In mice, a higher intake of omega-3 PUFAs with a lower ratio of n-6/n-3 PUFAs (between approx. 6:1 and 1:1) supplied to mothers during pregnancy favors NPC proliferation and counteracts apoptosis in the progeny [126]. Conversely, an increase in the ratio of n-6/n-3 due to a maternal diet deficient in omega-3 reduces neurogenesis in the offspring of rats [124,127].
5.2.5. Conclusions
The data reported above show that different types of FA differentially affect the proliferation of NPCs and the differentiation of the progeny of NPCs into neurons or glia. The main effects of different FAs are summarized in Figure 4. These studies provide convincing evidence that fatty acids are useful for fundamental processes, such as energy metabolism and lipid membrane formation, but they could also serve as signaling molecules in neurogenic niches, where they regulate neurogenesis. NPCs appear to be sensitive to specific fatty acids. Suitable amounts and types of fatty acids may have a positive impact on NPC proliferation and neurogenesis, but excessive levels, improper ratios of different types of fatty acids, and disease-linked alterations to their metabolism may induce adverse effects.
6. Fatty Acids and Neuron Maturation
6.1. Overview of Neuron Maturation in Humans and Rodents
Once neurons are generated, they develop dendrites and an axon in order to establish appropriate synaptic connections. In humans, the development of the dendrites of cortical neurons starts at gestational week (GW) 13.5–GW15. An apical and basal dendritic tree is recognizable at GW17–GW25. Dendrites exhibit a growth spurt at GW26–GW32 [128,129]. At GW27, fibers from the thalamus invade the cortical plate [130]. Dendrites attain their maximum size between 4 months and 2 years of age [131]. Dendritic spines appear at GW25/26, and the process is completed by the end of postnatal months 5–6 [18,128,129,132].
In mice, an apical and basal dendritic arbor is already detectable in neocortical pyramidal neurons at postnatal day (PD) 2, but the apical arbor is less well developed than the basal arbor [133]. Similarly, in rats, the basal and apical dendritic arbor of CA1 pyramidal neurons has a different time course; the number of basal dendrites is already established at the age of 5 days, while the lateral and terminal branches of the apical arbor emerge later [134]. In rats, the apical dendritic branches of cortical pyramidal neurons increase in number up to PD15 [135]. Dendritic spines appear at approximately PD8 in both the hippocampus and the neocortex [133,135,136,137,138,139].
6.2. Effects of FA on Neuron Maturation
6.2.1. Axon
Numerous studies show that OA synthesized by astrocytes fosters axon development in rat cultured neurons [140,141,142,143] and rat pups [144] and exerts this effect through the PPARα receptor [142]. Importantly, an oleate-rich diet restores the severe peripheral neuropathy induced in mice by a palmitate-rich diet [145]. Treatment with DHA fosters axon development in rat cultured neurons [146,147], and this effect is mediated by the activation of the Akt-mTOR-S6K pathway [147].
6.2.2. Dendrites
In rat cultured neurons, OA increases neurite elongation, and this effect is prevented by inhibiting protein kinase C [148]. Ample evidence shows that DHA enhances neurite development in rat [117,146,149,150] and mouse [118] cultured neurons. Evidence in rat cultured neurons [151] and human iPSC-derived NPCs [119] shows that this effect is not mediated by nuclear retinoid X receptors (RXR) [151], but is associated with increased WNT and CREB signaling. [119]. Combined treatment with DHA and ARA also increases neurite development in cultures of human mesenchymal stem cells induced to differentiate into neurons [152]. ARA, EPA, and DHA exert robust effects on neurite development in cultures of primary sensory neurons from rat dorsal root ganglia, isolated at different ages; these effects are present from an early postnatal stage and persist into adulthood [153]. There is evidence that the neurite-outgrowth-promoting effect of NGF on PC12 cells is potentiated by DHA but reduced by ARA [154]. Another study, however, shows that DHA, EPA, and ARA (but not OA) enhance NGF-induced neurite growth in PC12 cells [155].
6.2.3. Dendritic Spines and Synaptic Proteins
Growing evidence suggests that protein complexes that regulate spine plasticity depend on specific interactions with membrane lipids for proper function and accurate intracellular signaling (see [156]). In rat neuronal cultures, the local synthesis of OA increases the expression of synaptotagmin and postsynaptic density protein 95 (PDS-95), which are markers of the pre- and postsynaptic compartments, respectively [157], indicating that OA fosters connectivity. Ample evidence shows that treatment with DHA administered orally to rat dams during gestation and nursing [158], to adult gerbils [159], or supplemented to mice in neuronal cultures [118] increases the density of dendritic spines and the expression of pre- and postsynaptic proteins (Syntaxin-3, PSD-95, GluR-1, and Synapsin-1) and membrane phosphatides in the adult hippocampus [150,159,160]. By contrast, ARA has no effect on either dendritic spine density or membrane phosphatides [159,160], suggesting that the effect of DHA on spine formation may be related to enhancements in the synthesis of membrane phospholipids.
6.2.4. Conclusions
Taken together, these studies suggest that OA favors both axon development and connectivity. DHA favors axon development, neuritic development, spinogenesis, and connectivity. No evidence is available for ARA regarding axon development. ARA may favor dendritic development, although results are not consistent across studies, and does not foster spinogenesis. The effects of different FAs are summarized in Figure 5.
7. Fatty Acids and Mitochondrial Function
In humans, the highest concentration of LCPUFAs is found in the central nervous system. There is increasing evidence that, in addition to playing a structural role in membranes, LCPUFAs are crucial for energy production. LCPUFAs promote mitochondrial biogenesis [161,162] and modulate the expression of genes associated with ATP production and energy metabolism [163]. MUFAs may also have a positive effect on mitochondrial function because oleate prevents palmitate-induced mitochondrial dysfunction, insulin resistance, and inflammatory signaling in rat cortical neurons [164].
Brain regions with high energy consumption and high rates of oxidative metabolism have a high concentration of DHA and high levels of DHA in mitochondria [165,166]. In mice, DHA obtained from the diet is integrated into mitochondrial membranes [167], where it plays a crucial role in the mitochondrial oxidative phosphorylation system (OXPHOS), which is the biochemical pathway responsible for the production of energy in the form of ATP [165]. In addition, the mitochondrial respiratory chain, which is dependent on oxygen, generates reactive oxygen species (ROS). In fact, mitochondria are the main sources of ROS, since 90% of ROS is produced by the OXPHOS system as a byproduct of electron-transport-chain activity [166]. Impaired mitochondrial function is very commonly due to excessive ROS, affecting proteins of the OXPHOS system as well as the PUFAs of the mitochondrial membrane [168,169]. Moreover, alterations to the OXPHOS system produce carnitine deficiency, which impairs the mitochondrial pathway necessary for the synthesis of DHA-containing phospholipids [170].
During aging, the amount of mitochondrial cholesterol and phospholipid content decreases, membrane fluidity is reduced, and oxidative damage is enhanced [166,171]. In addition, mitochondrial respiratory capacity is decreased, and mitochondrial DNA (mtDNA) accumulates mutations due to enhanced ROS production [172,173]. Unfortunately, attempts to reduce mitochondrial decay by using antioxidants proved to have very limited efficacy [174]. Thus, it has been proposed that different compounds targeting other mechanisms underlying mitochondrial dysfunction could be useful. Among them, the supplementation of FAs to overcome their decay during aging could be a promising strategy. Indeed, FAs have been shown to reduce age-related mitochondrial alterations. Ochoa et al. compared the effects of the administration of MUFAs from virgin olive oil with those of the administration of omega-6 PUFAs from sunflower oil in aged mice [175]. While the MUFA diet led to oxidative-stress reduction and mtDNA deletions, the omega-6 PUFA diet increased lipid peroxidation in the mitochondria and caused an attenuation of mitochondrial membrane potential [175]. The administration of fish oil, which is rich in PUFAs, including the omega-3 DHA and EPA, attenuated mitochondrial dysfunction in aged mice [166]. Finally, supplementation with DHA had a protective effect in the mitochondria of a murine model of accelerated aging, the SAMP8 mouse [176], and protected against Aβ-mediated mitochondrial deficits in an AD animal model [177].
Oxidative-stress-related mitochondrial dysfunction may contribute to brain damage and neurodegeneration [178]. Several fatty acids have been demonstrated to induce neuroprotection. The oral administration of EPA inhibited irradiation-induced ROS production [179]. This neuroprotective effect seems to be mediated by the inhibition of JNK activation through the PPAR-α receptor, which suppresses the JNK-c-Jun pathways [180]. DHA protects from prenatal stress-induced cognitive dysfunction caused by alterations in mitochondrial function [181]. The beneficial effects of DHA are mediated by nuclear receptors, such as PPAR-α, HNF-4α, RXR, and LXRα, which induce changes in the expression of genes that encode transcription factors, fatty-acid-binding proteins, and inflammatory proteins [77,166,182].
Taken together, these data suggest that FAs, particularly LCPUFAs, play a physiological role in mitochondrial functioning and may improve mitochondrial dysfunction associated with aging and brain damage.
8. Fatty Acids and Cognition
A wide body of evidence relates FA to cognition. As mentioned in previous sections, LA and ALA are obtained from the diet. Maternal LCPUFAs are delivered to the fetal brain during the third trimester of pregnancy via the placenta and postnatally in breast milk. Deficits in maternal omega-3 FA, especially DHA, affect the metabolic and epigenetic factors of the fetus that deteriorate neurodevelopment and cognition [183,184]. When omega-3 deficiency is maintained across consecutive generations in rats, cognitive deficits are evident in the younger offspring [185]. There is a direct association between the percentage of fatty acids in maternal plasma and the development of cognitive function in neonates [186]. Moreover, maternal supplementation with DHA during pregnancy is associated with higher scores on tests of cognition in infants and preschool children, and a relationship between in utero DHA deprivation and several neurological birth defects has been proposed [187,188,189,190]. Deficits in DHA also produce cognitive deficits in humans during childhood and adulthood [183,191]. Among the cognitive domains affected by low levels of DHA are attention and working-memory deficits, reading difficulties, dyslexia, and dyspraxia. Children with attention deficit hyperactivity disorder (ADHD) and/or learning difficulties very commonly show low levels of omega-3 PUFAs in their blood [191]. Montgomery et al. evaluated the relationship between omega-3 PUFAs in blood and cognitive performance in children. They found that low omega-3 levels were strongly correlated with cognitive performance and proposed that supplementation with DHA or DHA-rich fish oil could reduce these deficits.
Animal studies have demonstrated the beneficial effects of supplementation with omega-3 PUFAs on mice or rats with cognitive impairments due to different causes. Supplementation with ALA, or its derivative, DHA, in the diet of pregnant female rodents enhances the cognition of their offspring [58,120,150,192,193,194,195,196,197]. The pro-cognitive effects of these diets were associated with the enhancement of neurogenesis, dendritogenesis, and synaptogenesis, suggesting that they were mediated by an improvement in brain development. Other processes, such as the restoration of the lipid composition of the membrane and reduced neuroinflammation, are also likely to be implicated in these cognitive improvements.
As mentioned in previous sections, the relative content of the different fatty acids is a critical factor determining the outcome of FA supplementation. Rats fed with oils that contain LA and ALA presented an improvement in spatial cognition in a Morris water maze, and the relative content of ALA appeared to be the critical variable [198]. High LA and low ALA supplementation for pregnant and lactating primates, mice, and rats leads to biochemical, neural, and cognitive impairments in their offspring [199], indicating that higher amounts of ALA in comparison with LA are necessary to induce cognitive improvements.
Given the positive effects of FA supplementation on animals’ cognitive abilities, it is expected that similar effects will be elicited in humans. Accordingly, many older adults consume omega-3 supplements to prevent cognitive decline. However, a systematic review and meta-analysis of randomized trials performed on adults, assessing the effect of higher vs. lower omega-3, omega-6, or total PUFA on cognition, did not confirm this presumption [200]. This meta-analysis suggested very low or no effect of omega-3 on cognitive impairment or global cognition. In addition, the effects of increasing the relative concentration of ALA, LA, or total PUFA were not clear. Thus, omega-3 PUFAs supplements do not seem to protect against cognitive decline.
9. Fatty Acids and Alzheimer’s Disease-Related Dementia
9.1. Altered Brain- Lipid Profile in AD
The most common type of dementia is caused by Alzheimer’s disease (AD). The etiopathology of this disease is multifactorial and includes enhanced oxidative stress and altered mitochondrial function, altered energy metabolism, neuroinflammation, reduced neurogenesis, loss of synapses, neurodegeneration, plaques composed of β-amyloid peptides, and neurofibrillary tangles of hyperphosphorylated tau protein. AD is also characterized by dysregulated FA composition and metabolism [69]. Studies have shown that long- and short-chain FAs play an important role in AD pathology, exerting both protective and pathogenic effects [201]. Because reduced levels of FA induce many of these pathological processes, it has been proposed that supplementation with medium-chain fatty acids (MCFAs) and omega-3 PUFAs may palliate the neuropathology and dementia found in mild cognitive impairment (MCI) and AD [202]. In fact, epidemiological and experimental research indicates that MCFAs and omega-3 PUFA supplementation can be beneficial in these diseases [56].
Elderly people consuming lower amounts of omega-3 FA have an elevated risk of AD, and several studies have implicated lower brain DHA in the pathogenesis of AD (see [190]). Moreover, post-mortem AD brains and plasma from patients with AD present lower levels of EPA and DHA [203,204,205]. In addition, an increased omega-3/omega-6 ratio negatively correlates with cognitive decline and the incidence of AD [206]. Furthermore, the level of PUFA oxidation products and the consequent oxidative damage is increased in AD brain tissue [207,208,209]. Interestingly, the main genetic risk factor for sporadic AD is a polymorphism of the ApoE gene [210], which encodes a lipid-carrying apolipoprotein in the brain. Alterations in lipid metabolism have also been found in animal models of AD, such as the 3 x Tg mouse [69]. These mice, as with humans with AD, present lipid droplets, which are intracellular stores of neutral lipids, such as triglycerides and cholesterol.
9.2. Supplementation with FA to Prevent or Reduce AD-Related Dementia
Several clinical trials have demonstrated that diets providing high levels of EPA and DHA are correlated with a reduced risk of developing AD [211,212,213]. However, controversial results have been obtained on the effectiveness of supplementation with FA at reducing dementia in MCI or AD. Some studies report that dietary supplementation with EPA and DHA improves cognitive performance in patients diagnosed with MCI or AD [214,215,216]. However, other studies failed to establish any benefit of omega-3 PUFA supplementation or cognitive improvements [217,218,219].
Despite the discrepancies, a wide body of literature suggests that FA supplementation might be beneficial in patients with MCI or in initial phases of AD in which dementia is relatively mild. These beneficial effects on the cognitive performances of patients have been attributed to: (i) an improvement in cerebral energy metabolism; (ii) a reduction in β-amyloid deposition, through a shift in the proportion of amyloidogenic and non-amyloidogenic APP processing; (iii) an increase in the clearance of Aβ42; (iv) a decrease in the aggregation of these peptides into amyloid plaques; (v) a reduction in oxidative stress and the enhancement of mitochondrial function and cerebral glucose metabolism; and (vi) a reduction in neuroinflammation (see [202]). The mechanistic effects of omega-3 PUFAs, especially DHA, on most of these mechanisms were reviewed in previous sections.
In conclusion, AD is characterized by altered homeostasis of brain lipids and changes in FA composition. Although studies conducted on animal models provided evidence that supplementation with omega 3-PUFAs could prevent or reduce cognitive decline in murine models of AD, the evidence in human trials is controversial (see [220] for a review). Nonetheless, most of the studies provide evidence for a potential benefit of these FAs in patients with MCI or in the early stages of AD.
10. Fatty Acids and Down Syndrome
In view of the positive effect of FAs on a range of brain functions in healthy and diseased brains, it is surprising that scarce attention has been devoted to FAs in the context of DS. Below, we report what is known regarding FAs in the DS brain and discuss the results of pioneering studies that have explored the potential benefits of FAs for ID in DS.
10.1. Brain-Lipid-Profile Alterations in DS
Individuals with DS are characterized by impaired lipid metabolism [221] and alterations in the brain’s lipidomic profile, which are associated with a decrease in plasma-membrane fluidity, an important biological parameter affecting membrane biochemical and physiological properties [222]. The most frequent changes in the dyslipidemia of DS are low levels of high-density lipoprotein (HDL) and elevated levels of triglycerides (see [223]). The levels of carnitine, which transports long-chain FAs to mitochondria, are reduced in DS children [224]. This defect, in conjunction with impaired mitochondrial oxidative phosphorylation, may impair FA catabolism and, consequently, cause triglyceride imbalances. Individuals with DS exhibit reduced levels of some FABPs in the brain [225]. Since FABPs bind to PUFAs, stimulate cellular FA uptake, and enhance the intracellular diffusion of FAs, these reduced levels may also contribute to FA imbalance in DS. Whether alterations in the lipidic profile in DS are due to specific overexpressed genes on HSA21 remains to be established. ABCG1, a gene that encodes for proteins involved in the efflux of cholesterol from peripheral cells onto HDL [226], localizes to the long arm of chromosome 21 [227], suggesting that excess ABCG1 may underlie some of the differences in lipid metabolism in DS. Individuals with DS exhibit a higher prevalence of diabetes, which is associated with dyslipidemia (increase in triglycerides and low HDL). Some HSA21 genes, such as BACE2, RCAN1, DYRK1A, and S100B, are thought to be related to diabetes phenotypes in DS (see [228,229]), suggesting that these HSA21 genes may also indirectly alter lipid metabolism.
The few available studies reviewed below show alterations in the plasma lipid profiles of women carrying fetuses with DS and in the brains of fetuses and adults with DS.
10.1.1. Maternal Blood
The maternal blood of women carrying DS fetuses presents lower circulating high-density lipoprotein (HDL) in the first trimester of gestation and higher levels of low-density lipoproteins (LDL) during the second trimester of gestation [230]. In addition, during the first trimester, plasma-lipid extracts from DS mothers show decreased levels of 18:2 FA; during the second trimester, they present lower levels, of 20:4 and 22:6 FA [230].
10.1.2. Fetuses with DS
Regarding MUFAs, reduced levels of OA have been detected in serine phosphoglycerides in the cortexes of fetuses with DS [231]. Albumin, which induces the synthesis and release of OA in astrocytes [232], is present at high levels in the brain during development [144,233,234]. DS individuals, however, present lower serum albumin concentrations [235,236], which might cause a lower synthesis of OA in this population. In cells from the T16 mouse model of DS, the uptake of OA is higher than in euploid cells; in the plasma membranes of euploid cells, an increase in phosphatidylcholine concentrations occurs in the presence of OA; and in trisomic cells, OA fails to increase phosphatidylcholine incorporation into the plasma membrane [234]. Regarding PUFAs, a study in DS and age-matched fetuses (during the fifth gestational month) quantified the PUFAs of ethanolamine and serine phosphoglycerides (EPG and SPG) [231]. The total percentage of PUFAs was similar in both groups. However, the ratio of omega-3 to omega-6 FAs was much higher in the DS fetuses. The levels of docosahexaenoyl, 22:6 (n-3) were enhanced, whereas the proportion of ARA 20:4 (n-6) and of its docosatetraenoic and docosapentaenoic homologues, 22:4 (n-6) and 22:5 (n-6), respectively, was significantly lower in the DS fetuses than in the controls. As DHA is more susceptible to peroxidation than ARA, an increased proportion of DHA in membranes could partially explain the excessive oxidative stress found in DS fetuses [231].
10.1.3. Adults with DS
In adults with DS, the phospholipids in myelin and synaptosomes contain a reduced amount of MUFA and PUFA [237]. The total phospholipid content is reduced by approximately 20% in the cortex and cerebellum in adults with DS [238]. Small but significant differences were found in the proportion of several FAs between DS and age-matched controls (mean: 55.7 years) in the phospholipids of the frontal cortex [239]. In particular, choline phosphoglycerides in the DS frontal cortex exhibited an increase in OA (16:0) and 18:1 (n-7), a decrease in 18:0 and 18:1 (n-9), and small decreases in omega-6 PUFAs. Altogether, the ratio of total omega-3 to omega-6 PUFAs was increased in the DS cortices in comparison with the controls [239]. Adults with DS and AD undergo an increase in OA levels in the brain [239]; however, this is an adverse event that may promote AD development [240].
10.2. Effects of Fatty Acids in a DS Model
10.2.1. Fetal Treatment
The evidence obtained in various cellular and animal models reviewed above (Section 5 and Section 6) shows that FAs exert beneficial effects on neurogenesis, neuron maturation, and synaptogenesis. As these processes are altered in DS, from the early stages of brain development, it can be hypothesized that the administration of FAs during critical developmental time windows (the prenatal and early postnatal periods) may improve these DS-linked alterations and, possibly, enhance cognition.
In a recent study, prenatal administration of OA or ALA to a murine model of DS, the Ts65Dn (TS) mouse, from embryonic day 10 (ED10) to postnatal day 2 (PD2), was found to rescue several trisomy-related neuromorphological alterations [241]. In particular, both treatments increased the brain weight, the volume of the granule cell layer (GCL), and the number of proliferating and mature granule cells in the hippocampus of PD2 TS mice, suggesting that treatment with either OA or ALA during embryonic brain development can foster prenatal brain neurogenesis.
In the same study, another cohort of mice was submitted to a washout drug-free period of 6 weeks after the discontinuation of the treatment. At this time, OA- and ALA-treated TS mice showed enhanced reference, working, and spatial memory in a hippocampal-dependent task (assessed with a Morris water maze) when compared to vehicle-treated TS mice. These results agree with the numerous reports of the pro-cognitive effects of the prenatal supplementation of OA, ALA, and its derivative, DHA, in different models of altered cognition (see Section 8). By contrast, adult TS animals did not differ from euploid mice in their GCL volume or in the number of proliferating cells. In the prenatal stages, brain development is regulated by the α-fetoprotein (AFP)/albumin ratio, which modulates the neurotrophic effects of oleic acid [242]. Thus, the balance between these two signals is crucial to induce neurogenesis during neurodevelopment. DS individuals present a reduction in the serum concentrations of AFP and albumin [236,243] that correlates with a lower concentration of OA in the brain [244]. In a study by Garcia-Cerro et al. [241], bovine serum albumin (BSA) was co-administered with OA (and ALA) to all animals. Thus, it is possible that during the administration of OA in the prenatal stages, adequate concentrations of this FA were reached, thereby correcting the impairment of hippocampal neurogenesis in the TS mice. However, after the discontinuation of treatment, the concentrations of both the OA and the ALA are likely to have returned to their previous deficit levels. This might explain why the beneficial effects of FA on neurogenesis were no longer present in TS mice after the discontinuation of treatment and suggests that the continuous administration of OA or ALA may be necessary to induce permanent effects on neurogenesis.
Since the effects of OA and ALA on neurogenesis were not retained in the adult TS mice, the beneficial effects of prenatal treatment with OA and ALA on cognition cannot be attributed to permanent changes in neurogenesis. However, the TS mice did show an increase in the postsynaptic marker, PSD-95, in the hippocampus when compared to the vehicle-treated animals. PSD-95 is predominantly present in excitatory synapses [245,246]. OA promotes the expression of PSD-95 [157], which is implicated in long-term potentiation (LTP) [247]. TS mice are characterized by excessive over-inhibition due to an imbalance between GABAergic and glutamatergic transmission, which affects LTP and cognition [248]. Thus, an enhancement in the expression of PSD-95 induced by OA and ALA treatment could be implicated in improving the functioning of the brain circuits of TS mice prenatally treated with these FAs.
Given that both OA and ALA are natural substances present in the human diet, and that they have no side effects, they could be safely administered prenatally, thereby fostering brain development and, possibly, improving cognitive abilities in DS infants.
10.2.2. Early Postnatal Treatment
As previously mentioned, hippocampal neurogenesis continues after birth and, in rodents, it is very prominent in the early postnatal stages. Dendritic and synaptic development largely take place neonatally. Thus, the early postnatal period may provide a useful opportunity to enhance hippocampal neurogenesis and dendritic development in DS through treatment with FA.
Based on this rationale, in a recent study, we analyzed the effect of the early postnatal administration (from PD3 to PD15) of OA or ALA on the hippocampal development and cognitive abilities of a TS mouse [249]. We found that neonatal treatment with OA or ALA did not exert any effects on the hippocampal NPC proliferation or the total size of the granule cell layer in the TS mice. These results are not in agreement with studies demonstrating that OA plays an essential role in postnatal neurogenesis from PD5 onwards [140,144]. However, consistent with the fact that OA also participates in neural differentiation, we found that early postnatal OA administration enhanced the percentage of the NPC that differentiated into neurons (BrdU+/NeuN+ cells) and the number of mature granule cells in TS mice. Thus, early postnatal OA treatment was sufficient to enhance differentiation but not proliferation in TS mice. Regarding ALA, neonatal treatment did not exert any effects on proliferation, differentiation, or survival, although it showed some benefit when prenatally administered [241]. Regarding cognition, while the early postnatal administration of OA improved reference, working, and spatial memory abilities when TS mice reached adulthood, treatment with ALA improved spatial memory only in the probe trial of the Morris water maze [249]. In view of the scarce (or absent) effects of treatment with OA or ALA on neurogenesis, the improvement in cognitive abilities found in treated TS mice’s cognitive abilities might be partially due to changes in synaptogenesis. Indeed, both OA and ALA increased the expression of PSD-95, which is implicated in LTP [247] and, thus, in learning processes. Moreover, OA and ALA might ameliorate other processes that are altered in the TS brain, such as oxidative stress, neuroinflammation, and altered energy metabolism, which might also contribute to the pro-cognitive effects of treatment.
10.2.3. Adult Treatment
As described in previous sections, brain development is affected not only by the total concentration of FA, but also by the ratio and relationships between them. Corn oil, which is extracted from the germs of corn, contains a high percentage of both LA and OA. LA is a precursor to the PUFAs of the omega-6 series, which produces ARA. Giacomini et al. [250] evaluated the ability of corn-oil administration for one month to young-adult (4-month-old) TS mice to improve neuromorphological and cognitive alterations. They demonstrated that adult treatment with corn oil restored hippocampal neurogenesis in TS mice. Moreover, the treatment enhanced dendritic length by increasing the number of branches of intermediate order and causing the de novo appearance of high-order branches. Importantly, the treatment enhanced hippocampal-dependent learning and memory (assessed with the Morris water maze and fear-conditioning tests), an effect that was very likely related to the restoration of neurogenesis and dendritogenesis. An evaluation of the separate effects of LA and OA in cultures of trisomic NPCs showed that both LA and OA increased their proliferation rate, although the effects of the LA were greater than those of the OA. The effects of the LA were mediated by the PPARβ/δ and PPARγ receptors, suggesting that these receptors might be involved in the effects of corn oil observed in vivo in TS mice.
10.3. Effects of Fatty Acids on Individuals with DS
To the best of our knowledge, very few studies have been conducted to evaluate the effects of PUFAs on the DS population, and most of these studies evaluated the efficacy of PUFAs in association with other supplements, which makes it difficult to determine the relative contribution of PUFAs to the overall effects.
In one study, the biochemical responses of DS children after a nutritional regimen supplemented with amino acids, vitamins, and PUFAs were evaluated [251]. This nutritional supplementation regimen lasted 12 months and consisted of the oral administration of essential amino acids (acetylcarntine, citrulline, glutathione, histidine, alpha-ketoglutaric acid, methionine, ornithine, proline, serine, tryptophan, tyrosine, taurine, and hydrochloride betaine), vitamin B6, vitamin B12, vitamin C, vitamin E, and omega-3 and omega-6 PUFAs. This study was performed on 86 DS children and 80 healthy children divided into two groups, one ranging between 0 and 6 years of age and the other between 6 and 12 years of age. The study evaluated the plasma levels of amino acids, antioxidant enzyme activities, and ROS before and after supplementation. The authors reported that the supplementation they used had some benefits for the DS individuals, such as a recovery of the ratio of methionine to cysteine, the normalization of the levels of homocysteine (which was thought to have been produced by vitamin B6 and B12 supplementation), and a reduction in malondialdehyde, due to the antioxidant action of vitamins C and E. However, given the high amount of nutrients administered, this preliminary study provides little information regarding the contribution of PUFAs to the benefits found in this group of DS children.
Two other studies evaluated the safety and the effect of the green tea polyphenol epigallocatechin-3-gallate (EGCG) combined with fish oil omega-3 [252,253] on the mitochondrial dysfunctions that characterize the DS population. EGCG is a nutraceutical with antioxidant properties that also inhibits the activity of the protein product of the T21 gene DYRK1A. Although there are several reports regarding the efficacy of EGCG in animal models, and clinical trials have shown that administration of EGCG is safe and provides cognitive benefits in the DS population [254], treatment with EGCG has raised concerns because other groups have reported deleterious effects on bone or a lack of pro-cognitive effect of EGCG on the Ts65Dn model [255,256]. There is evidence that EGCG decreases saturated fatty acids (C17:0 and C22:0), increases unsaturated fatty acids (C18:1 and C18:2) in the serum, and decreases the liver-protein expression of SCD1 and FADS2 [257]. Thus, a lack of data on the individual effects of fish oil and EGCG prevents the determination of the synergistic/antagonistic effects of treatment with EGCG plus fish oil. Vacca et al. reported that the administration of EGCG and fish oil containing omega-3 FA EPA and DHA rescues mitochondrial dysfunction and is safe in a 10-year-old DS child [252]. As mentioned in previous sections, omega-3 FAs reverse mitochondrial dysfunction. In addition, in vitro studies have demonstrated the ability of EGCG to counteract oxidative stress, prevent OXPHOS deficits, and promote mitochondrial biogenesis [258]. Vacca et al. report that, after 3 months of supplementation, EPA and DHA enhanced EGCG bioavailability and its beneficial effects on mitochondrial function in a DS child without altering hepatic function and lipid profile. They also observed an improvement in auditory attention and verbal strategic tests [252]. In a pilot clinical trial, 14 DS children (1–8 years of age) received EGCG suspended in omega-3 FAs for 6 months, and 10 DS children and 6 healthy children who did not receive any substance were used as controls [253]. The authors reported that EGCG (>90%) plus omega-3 is safe in DS children and effective at reverting deficits in mitochondrial complex I and V activities. However, this treatment did not improve the developmental performance of DS children [253].
The scarcity of research and the fact that different agents were co-administered makes it difficult to draw conclusions regarding the effects of FAs in the DS brain. However, it seems reasonable to conclude that FAs might be involved in the improvement of mitochondrial function observed in subjects with DS and that treatment with FAs is free of adverse effects.
10.4. Early Therapies with FA in DS: A Promising Strategy
The results from a model of DS summarized above [241,249,250] and in Figure 6 provide the first demonstration that treatment with FAs may be a useful and safe strategy for the improvement of brain alterations and cognitive performance in DS.
Taken together, the results of prenatal and neonatal treatment with OA and ALA [241,249] suggest that both exert positive effects on brain development, and some of these effects are retained in adulthood and translate into improved cognitive abilities. Prenatal treatment is much more effective than neonatal treatment, and treatment with OA is much more powerful than treatment with ALA. The superior effects of OA are possibly explained by a rescue of the intrinsic defects in OA levels/metabolism that characterize DS [144,231,232,234,235,236]. The data from adult Ts65Dn mice [250] suggest that a diet based on a combination of FAs may benefit the cognitive performance of DS individuals, even if administered during adult life stages. Given that these essential FAs are taken in the diet and that they are devoid of adverse effects, the provision of supplementation with these acids to the DS population could be a safe and promising strategy for improving DS cognitive alterations.
It remains to be established whether other types of FA may be more beneficial than those tested so far in the Ts65Dn model of DS. In relation to this, it is worthwhile to mention the results of a recent study showing that supplementation with fish oil reduces the levels of RCAN1 in normal mice pups [259]. RCAN1 is one of the triplicated genes that is thought to be strongly involved in many DS brain phenotypes (see [9]). In line with the data on mice, the bioinformatic mining of human fish-oil studies also revealed reduced RCAN1 mRNA expression [259]. This study shows that FAs may target triplicated genes and supports the hypothesis that dietary supplements with FAs may represent a potential treatment for improvements in brain development in individuals with DS.
11. Concluding Remarks
The large body of evidence in animal models reviewed above shows that FAs are able to restore many of the brain defects that are found in DS, including neurogenesis, dendritic development, connectivity, mitochondrial function, neurodegeneration, and cognitive impairment. Thus, FAs may be rationally conceived as a potential tool for improvements in brain development and, possibly, the prevention/amelioration of AD in DS.
Although the paucity of studies available for DS individuals prevents the determination of the correctness of this idea, the data obtained so far in a mouse model of DS clearly show that FAs restore or improve neurogenesis, dendritogenesis, connectivity, and cognitive performance, and that some of the effects of treatment leave a long-lasting trace on the brain. Therefore, we believe that these promising achievements should stimulate new research aimed at better delineating the effects of FAs. In view of the multifaceted effects of the large family of FAs on the brain, studies should examine the effects of different SFAs, MUFAs, PUFAs, and/or a combination of FAs, on the DS brain, in order to discover the optimal treatment.
New medical treatments should take into account two key prerequisites: efficacy and safety. In the case of DS, it must be remembered that neurogenesis failure is a key determinant of ID in DS and that cortical neurogenesis is completed before birth. Thus, treatment aimed at correcting neurogenesis failure should be administered prenatally to expectant mothers. This makes the issue of safety even more important, because the safety of both mothers and children must be taken into account. In this regard, natural substances may really become an option of choice because, at proper doses, they offer safety. We hope that this review may stimulate the scientific community to explore in depth the potential of FAs for DS, thereby providing a rational basis for new treatments.
Author Contributions
C.M.-C. and R.B. conceptualized the review; C.M.-C. and R.B. wrote the review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant to R.B. from the “Fondazione Generali e Assicurazione Generali”, Italy, and by a grant to C.M.C. by the Spanish “Agencia Estatal de Investicación, MICIN” (grant number: PID2020-117601RB-I00).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the review; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish it.
References
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef]
- Hughes-McCormack, L.A.; McGowan, R.; Pell, J.P.; Mackay, D.; Henderson, A.; O’Leary, L.; Cooper, S.A. Birth incidence, deaths and hospitalisations of children and young people with Down syndrome, 1990–2015: Birth cohort study. BMJ Open 2020, 10, e033770. [Google Scholar] [CrossRef] [Green Version]
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef]
- Zigman, W.B. Atypical aging in Down syndrome. Dev. Disabil. Res. Rev. 2013, 18, 51–67. [Google Scholar] [CrossRef]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in Down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Zigman, W.B.; Lott, I.T. Alzheimer’s disease in Down syndrome: Neurobiology and risk. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 237–246. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Bartesaghi, R. Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic. Biol. Med. 2018, 114, 15–32. [Google Scholar] [CrossRef]
- Stagni, F.; Bartesaghi, R. The Challenging Pathway of Treatment for Neurogenesis Impairment in Down Syndrome: Achievements and Perspectives. Front. Cell. Neurosci. 2022, 16, 903729. [Google Scholar] [CrossRef]
- Ponroy Bally, B.; Murai, K.K. Astrocytes in Down Syndrome Across the Lifespan. Front. Cell. Neurosci. 2021, 15, 334. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Kang, H.J.; Tyler, W.A.; Silbereis, J.C.; Cheng, F.; Zhu, Y.; Pletikos, M.; Jankovic-Rapan, L.; Cramer, N.P.; Galdzicki, Z.; et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 2016, 89, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Abraham, H.; Vincze, A.; Veszpremi, B.; Kravjak, A.; Gomori, E.; Kovacs, G.G.; Seress, L. Impaired myelination of the human hippocampal formation in Down syndrome. Int. J. Dev. Neurosci. 2012, 30, 147–158. [Google Scholar] [CrossRef]
- Takashima, S.; Becker, L.E.; Armstrong, D.L.; Chan, F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down’s syndrome. A quantitative and qualitative Golgi study. Brain Res. 1981, 225, 1–21. [Google Scholar] [CrossRef]
- Takashima, S.; Iida, K.; Mito, T.; Arima, M. Dendritic and histochemical development and ageing in patients with Down’s syndrome. J. Intellect. Disabil. Res. 1994, 38, 265–273. [Google Scholar] [CrossRef]
- Becker, L.E.; Armstrong, D.L.; Chan, F. Dendritic atrophy in children with Down’s syndrome. Ann. Neurol. 1986, 20, 520–526. [Google Scholar] [CrossRef]
- Prinz, M.; Prinz, B.; Schulz, E. The growth of non-pyramidal neurons in the primary motor cortex of man: A Golgi study. Histol. Histopathol. 1997, 12, 895–900. [Google Scholar]
- Marin-Padilla, M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. J. Comp. Neurol. 1976, 167, 63–81. [Google Scholar] [CrossRef]
- Purpura, D.P. Normal and aberrant neuronal development in the cerebral cortex of human fetus and young infant. UCLA Forum. Med. Sci. 1975, 18, 141–169. [Google Scholar] [CrossRef]
- Gotti, S.; Caricati, E.; Panzica, G. Alterations of brain circuits in Down syndrome murine models. J. Chem. Neuroanat. 2011, 42, 317–326. [Google Scholar] [CrossRef]
- Bartesaghi, R.; Guidi, S.; Ciani, E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011, 22, 419–455. [Google Scholar] [CrossRef]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [Green Version]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Esbensen, A.J. Chapter Four-Health Conditions Associated with Aging and End of Life of Adults with Down Syndrome. In International Review of Research in Mental Retardation; Urbano, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 39, pp. 107–126. [Google Scholar]
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down syndrome and Alzheimer’s disease. Free Radic Biol. Med. 2018, 114, 110–121. [Google Scholar] [CrossRef]
- Godfrey, M.; Lee, N.R. A comprehensive examination of the memory profile of youth with Down syndrome in comparison to typically developing peers. Child Neuropsychol. 2020, 26, 721–738. [Google Scholar] [CrossRef]
- Pennington, B.F.; Moon, J.; Edgin, J.; Stedron, J.; Nadel, L. The neuropsychology of Down syndrome: Evidence for hippocampal dysfunction. Child Dev. 2003, 74, 75–93. [Google Scholar] [CrossRef]
- Byrne, A.; MacDonald, J.; Buckley, S. Reading, language and memory skills: A comparative longitudinal study of children with Down syndrome and their mainstream peers. Br. J. Educ. Psychol. 2002, 72, 513–529. [Google Scholar] [CrossRef]
- Jarrold, C.; Baddeley, A.D.; Phillips, C. Long-term memory for verbal and visual information in Down syndrome and Williams syndrome: Performance on the Doors and People test. Cortex 2007, 43, 233–247. [Google Scholar] [CrossRef]
- Vicari, S. Implicit versus explicit memory function in children with Down and Williams syndrome. Downs Syndr. Res. Pract. 2001, 7, 35–40. [Google Scholar] [CrossRef]
- Grieco, J.; Pulsifer, M.; Seligsohn, K.; Skotko, B.; Schwartz, A. Down syndrome: Cognitive and behavioral functioning across the lifespan. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Tungate, A.S.; Conners, F.A. Executive function in Down syndrome: A meta-analysis. Res. Dev. Disabil. 2021, 108, 103802. [Google Scholar] [CrossRef]
- Del Hoyo Soriano, L.; Rosser, T.; Hamilton, D.; Wood, T.; Abbeduto, L.; Sherman, S. Gestational age is related to symptoms of attention-deficit/hyperactivity disorder in late-preterm to full-term children and adolescents with down syndrome. Sci. Rep. 2020, 10, 20345. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.C.; Dosen, A.; Buitelaar, J.K.; Janzing, J.G. Depression in Down syndrome: A review of the literature. Res. Dev. Disabil. 2011, 32, 1432–1440. [Google Scholar] [CrossRef]
- Marino, M.; Scala, I.; Scicolone, O.; Strisciuglio, P.; Bravaccio, C. Distribution and age of onset of psychopathological risk in a cohort of children with Down syndrome in developmental age. Ital. J. Pediatr. 2019, 45, 92. [Google Scholar] [CrossRef] [Green Version]
- Dykens, E.M.; Shah, B.; Davis, B.; Baker, C.; Fife, T.; Fitzpatrick, J. Psychiatric disorders in adolescents and young adults with Down syndrome and other intellectual disabilities. J. Neurodev. Disord. 2015, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, M.L.; McDougle, C.J. Pharmacotherapy of Down syndrome. Expert Opin. Pharm. 2018, 19, 1875–1889. [Google Scholar] [CrossRef]
- Bartesaghi, R.; Vicari, S.; Mobley, W.C. Prenatal and Postnatal Pharmacotherapy in Down Syndrome: The Search to Prevent or Ameliorate Neurodevelopmental and Neurodegenerative Disorders. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 211–233. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Herault, Y.; Delabar, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Yu, E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Model. Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñiz Moreno, M.D.M.; Brault, V.; Birling, M.C.; Pavlovic, G.; Herault, Y. Modeling Down syndrome in animals from the early stage to the 4.0 models and next. Prog. Brain Res. 2020, 251, 91–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.C.; Scott-McKean, J.J. Prospects for improving brain function in individuals with down syndrome. CNS Drugs 2013, 27, 679–702. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesaghi, R. Timing of therapies for Down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, K.J. Pharmacological approaches to improving cognitive function in Down syndrome: Current status and considerations. Drug Des. Devel. Ther. 2015, 9, 103–125. [Google Scholar] [CrossRef] [Green Version]
- Rueda, N.; Florez, J.; Dierssen, M.; Martinez-Cue, C. Translational validity and implications of pharmacotherapies in preclinical models of Down syndrome. Prog. Brain Res. 2020, 251, 245–268. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- Hart, S.J.; Visootsak, J.; Tamburri, P.; Phuong, P.; Baumer, N.; Hernandez, M.C.; Skotko, B.G.; Ochoa-Lubinoff, C.; Liogier D’Ardhuy, X.; Kishnani, P.S.; et al. Pharmacological interventions to improve cognition and adaptive functioning in Down syndrome: Strides to date. Am. J. Med. Genet. A 2017, 173, 3029–3041. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Tian, D.; Guo, Y.; Yan, Y.; Yang, H. Regulation of Neurogenesis and Neuronal Differentiation by Natural Compounds. Curr. Stem Cell Res. Ther. 2021, 34493197. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef] [PubMed]
- Djuricic, I.; Calder, P.C. Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty Acids on Human Health: An Update for 2021. Nutrients 2021, 13, 2421. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance-A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, S.; Arsic, A. Fatty Acids. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 623–631. [Google Scholar] [CrossRef]
- Hussain, G.; Schmitt, F.; Loeffler, J.P.; Gonzalez de Aguilar, J.L. Fatting the brain: A brief of recent research. Front. Cell Neurosci. 2013, 7, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, C.I.; Kiliaan, A.J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: The influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef]
- Kang, J.X.; Wan, J.B.; He, C. Concise review: Regulation of stem cell proliferation and differentiation by essential fatty acids and their metabolites. Stem. Cells 2014, 32, 1092–1098. [Google Scholar] [CrossRef]
- Tułowiecka, N.; Kotlęga, D.; Bohatyrewicz, A.; Szczuko, M. Could Lipoxins Represent a New Standard in Ischemic Stroke Treatment? Int. J. Mol. Sci. 2021, 22, 4207. [Google Scholar] [CrossRef]
- Ghasemi Fard, S.; Cameron-Smith, D.; Sinclair, A.J. n-3 Docosapentaenoic acid: The iceberg n-3 fatty acid. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 134–138. [Google Scholar] [CrossRef]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef]
- Patterson, E.; Cryan, J.F.; Fitzgerald, G.F.; Ross, R.P.; Dinan, T.G.; Stanton, C. Gut microbiota, the pharmabiotics they produce and host health. Proc. Nutr. Soc. 2014, 73, 477–489. [Google Scholar] [CrossRef]
- Bhat, M.I.; Kapila, R. Dietary metabolites derived from gut microbiota: Critical modulators of epigenetic changes in mammals. Nutr. Rev. 2017, 75, 374–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. MMBR 2017, 81, e00036-17. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The Healthy Microbiome-What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Cândido, F.G.; Valente, F.X.; Grześkowiak, Ł.M.; Moreira, A.P.B.; Rocha, D.; Alfenas, R.C.G. Impact of dietary fat on gut microbiota and low-grade systemic inflammation: Mechanisms and clinical implications on obesity. Int. J. Food Sci. Nutr. 2018, 69, 125–143. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Fernandes, K.J.L. Neural stem cells and adult brain fatty acid metabolism: Lessons from the 3xTg model of Alzheimer’s disease. Biol. Cell 2018, 110, 6–25. [Google Scholar] [CrossRef]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Reviews. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Giménez, E.; Toscani, A.M. Fatty Acid Signaling Mechanisms in Neural Cells: Fatty Acid Receptors. Front. Cell Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef]
- Feng, L.; Hatten, M.E.; Heintz, N. Brain lipid-binding protein (BLBP): A novel signaling system in the developing mammalian CNS. Neuron 1994, 12, 895–908. [Google Scholar] [CrossRef]
- Arai, Y.; Funatsu, N.; Numayama-Tsuruta, K.; Nomura, T.; Nakamura, S.; Osumi, N. Role of Fabp7, a downstream gene of Pax6, in the maintenance of neuroepithelial cells during early embryonic development of the rat cortex. J. Neurosci. 2005, 25, 9752–9761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Kersten, S. Mechanisms of gene regulation by fatty acids. Adv. Nutr. 2012, 3, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidaleo, M.; Fanelli, F.; Ceru, M.P.; Moreno, S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARalpha) and its lipid ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 253–264. [Google Scholar] [CrossRef]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors that influence adult neurogenesis as potential therapy. Transl. Neurodegener. 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Guillemot, F. Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [Green Version]
- Basak, S.; Mallick, R.; Banerjee, A.; Pathak, S.; Duttaroy, A.K. Maternal Supply of Both Arachidonic and Docosahexaenoic Acids Is Required for Optimal Neurodevelopment. Nutrients 2021, 13, 2061. [Google Scholar] [CrossRef]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Rice, D.; Barone, S. Critical periods of vulnerabiliy for the developing nervpus system: Evidence from humans and animal models. Environ. Health Perspect. 2010, 108, 511–533. [Google Scholar]
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Kostović, I.; Sedmak, G.; Judaš, M. Neural histology and neurogenesis of the human fetal and infant brain. Neuroimage 2019, 188, 743–773. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Seki, T.; Imayoshi, I.; Tamamaki, N.; Hayashi, Y.; Tatebayashi, Y.; Hitoshi, S. Neural stem cells and neuro/gliogenesis in the central nervous system: Understanding the structural and functional plasticity of the developing, mature, and diseased brain. J. Physiol. Sci. 2016, 66, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, H.; Tornoczky, T.; Kosztolanyi, G.; Seress, L. Cell formation in the cortical layers of the developing human cerebellum. Int. J. Dev. Neurosci. 2001, 19, 53–62. [Google Scholar] [CrossRef]
- Seress, L.; Abraham, H.; Tornoczky, T.; Kosztolanyi, G. Cell formation in the human hippocampal formation from mid-gestation to the late postnatal period. Neuroscience 2001, 105, 831–843. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; John Mann, J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef] [Green Version]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Bostrom, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Jiménez, E.P.; Terreros-Roncal, J.; Flor-García, M.; Rábano, A.; Llorens-Martín, M. Evidences for Adult Hippocampal Neurogenesis in Humans. J. Neurosci. 2021, 41, 2541–2553. [Google Scholar] [CrossRef]
- Wilkinson, G.; Dennis, D.; Schuurmans, C. Proneural genes in neocortical development. Neuroscience 2013, 253, 256–273. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Jessberger, S. Metabolism and neurogenesis. Curr. Opin. Neurobiol. 2017, 42, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, E.A.; Makin, R.; Sweet, I.R.; Trevelyan, A.J.; Miwa, S.; Horner, P.J.; Turnbull, D.M. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem. Cells 2015, 33, 2306–2319. [Google Scholar] [CrossRef]
- Chorna, N.E.; Santos-Soto, I.J.; Carballeira, N.M.; Morales, J.L.; de la Nuez, J.; Cátala-Valentin, A.; Chornyy, A.P.; Vázquez-Montes, A.; De Ortiz, S.P. Fatty Acid Synthase as a Factor Required for Exercise-Induced Cognitive Enhancement and Dentate Gyrus Cellular Proliferation. PLoS ONE 2013, 8, e77845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knobloch, M.; Braun, S.M.G.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, Ö.; Suter, U.; Machado, R.A.C.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Jackowski, S. Cell cycle regulation of membrane phospholipid metabolism. J. Biol. Chem. 1996, 271, 20219–20222. [Google Scholar] [CrossRef] [Green Version]
- Katakura, M.; Hashimoto, M.; Shahdat, H.M.; Gamoh, S.; Okui, T.; Matsuzaki, K.; Shido, O. Docosahexaenoic acid promotes neuronal differentiation by regulating basic helix-loop-helix transcription factors and cell cycle in neural stem cells. Neuroscience 2009, 160, 651–660. [Google Scholar] [CrossRef]
- Insua, M.F.; Garelli, A.; Rotstein, N.P.; German, O.L.; Arias, A.; Politi, L.E. Cell cycle regulation in retinal progenitors by glia-derived neurotrophic factor and docosahexaenoic acid. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2235–2244. [Google Scholar] [CrossRef]
- Okui, T.; Hashimoto, M.; Katakura, M.; Shido, O. Cis-9,trans-11-conjugated linoleic acid promotes neuronal differentiation through regulation of Hes6 mRNA and cell cycle in cultured neural stem cells. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 163–169. [Google Scholar] [CrossRef]
- Hejr, H.; Ghareghani, M.; Zibara, K.; Ghafari, M.; Sadri, F.; Salehpour, Z.; Hamedi, A.; Negintaji, K.; Azari, H.; Ghanbari, A. The ratio of 1/3 linoleic acid to alpha linolenic acid is optimal for oligodendrogenesis of embryonic neural stem cells. Neurosci. Lett. 2017, 651, 216–225. [Google Scholar] [CrossRef]
- Das, S.K.; Mondal, A.K.; Elbein, S.C. Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J. Lipid Res. 2010, 51, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesga-Jiménez, D.J.; Martin, C.; Barreto, G.E.; Aristizábal-Pachón, A.F.; Pinzón, A.; González, J. Fatty Acids: An Insight into the Pathogenesis of Neurodegenerative Diseases and Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 2577. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, D.; Zhang, Q.; Wang, J.; Zhan, J.; Xian, X.; Du, Z.; Wang, X.; Hao, A. Palmitic acid affects proliferation and differentiation of neural stem cells in vitro. J. Neurosci. Res. 2014, 92, 574–586. [Google Scholar] [CrossRef]
- Ardah, M.T.; Parween, S.; Varghese, D.S.; Emerald, B.S.; Ansari, S.A. Saturated fatty acid alters embryonic cortical neurogenesis through modulation of gene expression in neural stem cells. J. Nutr. Biochem. 2018, 62, 230–246. [Google Scholar] [CrossRef]
- Kandel, P.; Semerci, F.; Mishra, R.; Choi, W.; Bajic, A.; Baluya, D.; Ma, L.; Chen, K.; Cao, A.C.; Phongmekhin, T.; et al. Oleic acid is an endogenous ligand of TLX/NR2E1 that triggers hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2023784119. [Google Scholar] [CrossRef]
- Murai, K.; Qu, Q.; Sun, G.; Ye, P.; Li, W.; Asuelime, G.; Sun, E.; Tsai, G.E.; Shi, Y. Nuclear receptor TLX stimulates hippocampal neurogenesis and enhances learning and memory in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2014, 111, 9115–9120. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.A.; Baker, P.R.; Schopfer, F.J.; Woodcock, S.R.; Napolitano, A.; d’Ischia, M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008, 283, 15515–15519. [Google Scholar] [CrossRef] [Green Version]
- Pereckova, J.; Pekarova, M.; Szamecova, N.; Hoferova, Z.; Kamarytova, K.; Falk, M.; Perecko, T. Nitro-Oleic Acid Inhibits Stemness Maintenance and Enhances Neural Differentiation of Mouse Embryonic Stem Cells via STAT3 Signaling. Int. J. Mol. Sci. 2021, 22, 9981. [Google Scholar] [CrossRef]
- Lindqvist, A.; Mohapel, P.; Bouter, B.; Frielingsdorf, H.; Pizzo, D.; Brundin, P.; Erlanson-Albertsson, C. High-fat diet impairs hippocampal neurogenesis in male rats. Eur. J. Neurol. 2006, 13, 1385–1388. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, M.O.; Kim, Y.H.; Kim, J.S.; Han, H.J. Linoleic acid induces mouse embryonic stem cell proliferation via Ca2+/PKC, PI3K/Akt, and MAPKs. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2009, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sakayori, N.; Osumi, N. Polyunsaturated Fatty Acids and their Metabolites in Neural Development and Implications for Psychiatric Disorders. Curr. Psychopharmacol. 2013, 2, 73–83. [Google Scholar] [CrossRef]
- Ghareghani, M.; Zibara, K.; Azari, H.; Hejr, H.; Sadri, F.; Jannesar, R.; Ghalamfarsa, G.; Delaviz, H.; Nouri, E.; Ghanbari, A. Safflower Seed Oil, Containing Oleic Acid and Palmitic Acid, Enhances the Stemness of Cultured Embryonic Neural Stem Cells through Notch1 and Induces Neuronal Differentiation. Front. Neurosci. 2017, 11, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoudi, R.; Ghareghani, M.; Zibara, K.; Tajali Ardakani, M.; Jand, Y.; Azari, H.; Nikbakht, J.; Ghanbari, A. Alyssum homolocarpum seed oil (AHSO), containing natural alpha linolenic acid, stearic acid, myristic acid and β-sitosterol, increases proliferation and differentiation of neural stem cells in vitro. BMC Complementary Altern. Med. 2019, 19, 113. [Google Scholar] [CrossRef]
- Kawakita, E.; Hashimoto, M.; Shido, O. Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 2006, 139, 991–997. [Google Scholar] [CrossRef]
- He, C.; Qu, X.; Cui, L.; Wang, J.; Kang, J.X. Improved spatial learning performance of fat-1 mice is associated with enhanced neurogenesis and neuritogenesis by docosahexaenoic acid. Proc. Natl. Acad. Sci. USA 2009, 106, 11370–11375. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.N.; Hylton, N.K.; Wang, J.; Chindavong, P.S.; Alural, B.; Kurtser, I.; Subramanian, A.; Mazitschek, R.; Perlis, R.H.; Haggarty, S.J. Activation of WNT and CREB signaling pathways in human neuronal cells in response to the Omega-3 fatty acid docosahexaenoic acid (DHA). Mol. Cell Neurosci. 2019, 99, 103386. [Google Scholar] [CrossRef]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.Y. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Takashima, N.; Matsumata, M.; Ikegami, S.; Kontani, M.; Hara, Y.; Kawashima, H.; Owada, Y.; Kiso, Y.; Yoshikawa, T.; et al. Arachidonic acid drives postnatal neurogenesis and elicits a beneficial effect on prepulse inhibition, a biological trait of psychiatric illnesses. PLoS ONE 2009, 4, e5085. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, H.; Kontani, M.; Kawashima, H.; Kiso, Y.; Shibata, H.; Osumi, N. Differential effect of arachidonic acid and docosahexaenoic acid on age-related decreases in hippocampal neurogenesis. Neurosci. Res. 2014, 88, 58–66. [Google Scholar] [CrossRef]
- Sakayori, N.; Maekawa, M.; Numayama-Tsuruta, K.; Katura, T.; Moriya, T.; Osumi, N. Distinctive effects of arachidonic acid and docosahexaenoic acid on neural stem/progenitor cells. Genes Cells 2011, 16, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Coti Bertrand, P.; O’Kusky, J.R.; Innis, S.M. Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J. Nutr. 2006, 136, 1570–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltz, B.S.; Tlusty, M.F.; Benton, J.L.; Sandeman, D.C. Omega-3 fatty acids upregulate adult neurogenesis. Neurosci. Lett. 2007, 415, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Sun, W.; Fu, H.; Dong, H.; Xia, L.; Lu, Y.; Deckelbaum, R.J.; Qi, K. Dietary Ratios of N-6/N-3 Polyunsaturated Fatty Acids during Maternal Pregnancy Affect Hippocampal Neurogenesis and Apoptosis in Mouse Offspring. Nutr. Hosp. 2015, 32, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, M.; Cai, H.; Li, H.; Jiang, P.; Dang, R.; Liu, Y.; He, X.; Xue, Y.; Cao, L.; et al. Maternal diet of polyunsaturated fatty acid altered the cell proliferation in the dentate gyrus of hippocampus and influenced glutamatergic and serotoninergic systems of neonatal female rats. Lipids Health Dis. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrzljak, L.; Uylings, H.B.; Kostovic, I.; Van Eden, C.G. Prenatal development of neurons in the human prefrontal cortex: I. A qualitative Golgi study. J. Comp. Neurol. 1988, 271, 355–386. [Google Scholar] [CrossRef] [PubMed]
- Mrzljak, L.; Uylings, H.B.; Kostovic, I.; van Eden, C.G. Prenatal development of neurons in the human prefrontal cortex. II. A quantitative Golgi study. J. Comp. Neurol. 1992, 316, 485–496. [Google Scholar] [CrossRef]
- Kostovic, I.; Judas, M. The development of the subplate and thalamocortical connections in the human foetal brain. Acta Paediatr. 2010, 99, 1119–1127. [Google Scholar] [CrossRef]
- Becker, L.E.; Armstrong, D.L.; Chan, F.; Wood, M.M. Dendritic development in human occipital cortical neurons. Brain Res. 1984, 315, 117–124. [Google Scholar] [CrossRef]
- Lu, D.; He, L.; Xiang, W.; Ai, W.-M.; Cao, Y.; Wang, X.-S.; Pan, A.; Luo, X.-G.; Li, Z.; Yan, X.-X. Somal and Dendritic Development of Human CA3 Pyramidal Neurons F rom Midgestation to Middle Childhood: A Quantitative Golgi Study. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2013, 296, 123–132. [Google Scholar] [CrossRef]
- Uguagliati, B.; Stagni, F.; Emili, M.; Giacomini, A.; Russo, C.; Guidi, S.; Bartesaghi, R. Early Appearance of Dendritic Alterations in Neocortical Pyramidal Neurons of the Ts65Dn Model of Down Syndrome. Dev. Neurosci. 2022, 44, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.; Yamamoto, T. Postnatal ontogenesis of hippocampal CA1 area in rats. I. Development of dendritic arborisation in pyramidal neurons. Brain Res. Bull. 1981, 7, 113–120. [Google Scholar] [CrossRef]
- Juraska, J.M.; Fifkova, E. A Golgi study of the early postnatal development of the visual cortex of the hooded rat. J. Comp. Neurol. 1979, 183, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Uguagliati, B.; Al-Absi, A.R.; Stagni, F.; Emili, M.; Giacomini, A.; Guidi, S.; Nyengaard, J.R.; Bartesaghi, R. Early appearance of developmental alterations in the dendritic tree of the hippocampal granule cells in the Ts65Dn model of Down syndrome. Hippocampus 2021, 31, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Yuste, R.; Bonhoeffer, T. Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 2004, 5, 24–34. [Google Scholar] [CrossRef]
- Meller, K.; Breipohl, W.; Glees, P. Ontogeny of the mouse motor cortex. The polymorph layer or layer VI. A Golgi and electronmicroscopical study. Z Zellforsch. Mikrosk. Anat. 1969, 99, 443–458. [Google Scholar] [CrossRef]
- Romand, S.; Wang, Y.; Toledo-Rodriguez, M.; Markram, H. Morphological development of thick-tufted layer v pyramidal cells in the rat somatosensory cortex. Front. Neuroanat. 2011, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Medina, J.M.; Tabernero, A. Astrocyte-synthesized oleic acid behaves as a neurotrophic factor for neurons. J. Physiol. Paris 2002, 96, 265–271. [Google Scholar] [CrossRef]
- Tabernero, A.; Lavado, E.M.; Granda, B.; Velasco, A.; Medina, J.M. Neuronal differentiation is triggered by oleic acid synthesized and released by astrocytes. J. Neurochem. 2001, 79, 606–616. [Google Scholar] [CrossRef]
- Bento-Abreu, A.; Tabernero, A.; Medina, J.M. Peroxisome proliferator-activated receptor-alpha is required for the neurotrophic effect of oleic acid in neurons. J. Neurochem. 2007, 103, 871–881. [Google Scholar] [CrossRef]
- Polo-Hernández, E.; De Castro, F.; García-García, A.G.; Tabernero, A.; Medina, J.M. Oleic acid synthesized in the periventricular zone promotes axonogenesis in the striatum during brain development. J. Neurochem. 2010, 114, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.; Tabernero, A.; Medina, J.M. Role of oleic acid as a neurotrophic factor is supported in vivo by the expression of GAP-43 subsequent to the activation of SREBP-1 and the up-regulation of stearoyl-CoA desaturase during postnatal development of the brain. Brain Res. 2003, 977, 103–111. [Google Scholar] [CrossRef]
- Rumora, A.E.; LoGrasso, G.; Hayes, J.M.; Mendelson, F.E.; Tabbey, M.A.; Haidar, J.A.; Lentz, S.I.; Feldman, E.L. The Divergent Roles of Dietary Saturated and Monounsaturated Fatty Acids on Nerve Function in Murine Models of Obesity. J. Neurosci. 2019, 39, 3770–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Xue, R.; Xu, J.; Liu, Z. Effects of docosahexaenoic acid on the survival and neurite outgrowth of rat cortical neurons in primary cultures. J. Nutr. Biochem. 2005, 16, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Mayanagi, T.; Ichijo, H.; Fukumoto, K.; Otsuka, K.; Sakai, A.; Sobue, K. Docosahexaenoic Acid Promotes Axon Outgrowth by Translational Regulation of Tau and Collapsin Response Mediator Protein 2 Expression. J. Biol. Chem. 2016, 291, 4955–4965. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, R.A.; Tabernero, A.; Velasco, A.; Lavado, E.M.; Medina, J.M. The neurotrophic effect of oleic acid includes dendritic differentiation and the expression of the neuronal basic helix-loop-helix transcription factor NeuroD2. J. Neurochem. 2004, 88, 1041–1051. [Google Scholar] [CrossRef]
- Calderon, F.; Kim, H.Y. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004, 90, 979–988. [Google Scholar] [CrossRef]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.S.; Jun, S.B.; Lovinger, D.; Kim, H.Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef] [Green Version]
- Calderon, F.; Kim, H.Y. Role of RXR in neurite outgrowth induced by docosahexaenoic acid. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Kan, I.; Melamed, E.; Offen, D.; Green, P. Docosahexaenoic acid and arachidonic acid are fundamental supplements for the induction of neuronal differentiation. J. Lipid Res. 2007, 48, 513–517. [Google Scholar] [CrossRef] [Green Version]
- Robson, L.G.; Dyall, S.; Sidloff, D.; Michael-Titus, A.T. Omega-3 polyunsaturated fatty acids increase the neurite outgrowth of rat sensory neurones throughout development and in aged animals. Neurobiol. Aging 2010, 31, 678–687. [Google Scholar] [CrossRef]
- Ikemoto, A.; Kobayashi, T.; Watanabe, S.; Okuyama, H. Membrane fatty acid modifications of PC12 cells by arachidonate or docosahexaenoate affect neurite outgrowth but not norepinephrine release. Neurochem. Res. 1997, 22, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Almaguel, F.G.; Bu, L.; De Leon, D.D.; De Leon, M. Expression of E-FABP in PC12 cells increases neurite extension during differentiation: Involvement of n-3 and n-6 fatty acids. J. Neurochem. 2008, 106, 2015–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dotti, C.G.; Esteban, J.A.; Ledesma, M.D. Lipid dynamics at dendritic spines. Front. Neuroanat. 2014, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Polo-Hernández, E.; Tello, V.; Arroyo, A.A.; Domínguez-Prieto, M.; de Castro, F.; Tabernero, A.; Medina, J.M. Oleic acid synthesized by stearoyl-CoA desaturase (SCD-1) in the lateral periventricular zone of the developing rat brain mediates neuronal growth, migration and the arrangement of prospective synapses. Brain Res. 2014, 1570, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Cansev, M.; Marzloff, G.; Sakamoto, T.; Ulus, I.H.; Wurtman, R.J. Giving uridine and/or docosahexaenoic acid orally to rat dams during gestation and nursing increases synaptic elements in brains of weanling pups. Dev. Neurosci. 2009, 31, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Cansev, M.; Wurtman, R.J. Oral supplementation with docosahexaenoic acid and uridine-5’-monophosphate increases dendritic spine density in adult gerbil hippocampus. Brain Res. 2007, 1182, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cansev, M.; Wurtman, R.J. Chronic administration of docosahexaenoic acid or eicosapentaenoic acid, but not arachidonic acid, alone or in combination with uridine, increases brain phosphatide and synaptic protein levels in gerbils. Neuroscience 2007, 148, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Flachs, P.; Horakova, O.; Brauner, P.; Rossmeisl, M.; Pecina, P.; Franssen-van Hal, N.; Ruzickova, J.; Sponarova, J.; Drahota, Z.; Vlcek, C.; et al. Polyunsaturated fatty acids of marine origin upregulate mitochondrial biogenesis and induce beta-oxidation in white fat. Diabetologia 2005, 48, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Jeng, J.Y.; Yeh, T.S.; Chiu, Y.H.; Lee, Y.C.; Cheng, H.H.; Hsieh, R.H. Linoleic acid promotes mitochondrial biogenesis and maintains mitochondrial structure for prevention of streptozotocin damage in RIN-m5F cells. Biosci. Biotechnol. Biochem. 2009, 73, 1262–1267. [Google Scholar] [CrossRef]
- Kitajka, K.; Puskás, L.G.; Zvara, A.; Hackler, L., Jr.; Barceló-Coblijn, G.; Yeo, Y.K.; Farkas, T. The role of n-3 polyunsaturated fatty acids in brain: Modulation of rat brain gene expression by dietary n-3 fatty acids. Proc. Natl. Acad. Sci. USA 2002, 99, 2619–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, B.; Lee, H.K.; Querfurth, H.W. Oleate prevents palmitate-induced mitochondrial dysfunction, insulin resistance and inflammatory signaling in neuronal cells. Biochim. Biophys. Acta 2014, 1843, 1402–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenna, J.T.; Diau, G.Y. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 fatty acids in neurodegenerative diseases: Focus on mitochondria. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Manabe, S.; Wada, O.; Crawford, M.A. Rapid incorporation of docosahexaenoic acid from dietary sources into brain microsomal, synaptosomal and mitochondrial membranes in adult mice. Int. J. Vitam. Nutr. Research. Int. Z. Fur Vitam.-Und Ernahrungsforschung. J. Int. De Vitaminol. Et De Nutr. 1997, 67, 272–278. [Google Scholar]
- Harper, M.E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331. [Google Scholar] [CrossRef]
- Müller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease--therapeutic aspects. Mol. Neurobiol. 2010, 41, 159–171. [Google Scholar] [CrossRef]
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000, 468, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, F.M.; Cafagna, F.; Petruzzella, V.; Gadaleta, M.N.; Quagliariello, E. Lipid composition in synaptic and nonsynaptic mitochondria from rat brains and effect of aging. J. Neurochem. 1992, 59, 487–491. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Cini, C.; Bellia, F.; Cambria, M.T.; Cornelius, C.; Butterfield, D.A.; et al. Redox proteomics in aging rat brain: Involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J. Neurosci. Res. 2010, 88, 3498–3507. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visioli, F.; Ingram, A.; Beckman, J.S.; Magnusson, K.R.; Hagen, T.M. Strategies to protect against age-related mitochondrial decay: Do natural products and their derivatives help? Free Radic Biol. Med. 2022, 178, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.J.; Pamplona, R.; Ramirez-Tortosa, M.C.; Granados-Principal, S.; Perez-Lopez, P.; Naudí, A.; Portero-Otin, M.; López-Frías, M.; Battino, M.; Quiles, J.L. Age-related changes in brain mitochondrial DNA deletion and oxidative stress are differentially modulated by dietary fat type and coenzyme Q10. Free Radic Biol. Med. 2011, 50, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.M.; Ding, L.; Wen, M.; Che, H.X.; Huang, J.Q.; Zhang, T.T.; Xue, C.H.; Mao, X.Z.; Wang, Y.M. Mechanisms of DHA-enriched phospholipids in improving cognitive deficits in aged SAMP8 mice with high-fat diet. J. Nutr. Biochem. 2018, 59, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.I.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Lonergan, P.E.; Martin, D.S.; Horrobin, D.F.; Lynch, M.A. Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J. Biol. Chem. 2002, 277, 20804–20811. [Google Scholar] [CrossRef] [Green Version]
- Delerive, P.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Zou, X.; Jia, H.; Li, X.; Zhu, Z.; Liu, X.; Bucheli, P.; Ballevre, O.; Hou, Y.; Zhang, W.; et al. Maternal docosahexaenoic acid feeding protects against impairment of learning and memory and oxidative stress in prenatally stressed rats: Possible role of neuronal mitochondria metabolism. Antioxid. Redox Signal. 2012, 16, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Sampath, H.; Ntambi, J.M. Polyunsaturated fatty acid regulation of gene expression. Nutr. Rev. 2004, 62, 333–339. [Google Scholar] [CrossRef]
- Basak, S.; Vilasagaram, S.; Duttaroy, A.K. Maternal dietary deficiency of n-3 fatty acids affects metabolic and epigenetic phenotypes of the developing fetus. Prostaglandins Leukot. Essent. Fat. Acids 2020, 158, 102109. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Duttaroy, A.K. Maternal PUFAs, Placental Epigenetics, and Their Relevance to Fetal Growth and Brain Development. Reprod Sci. 2022, 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.O.; Taha, A.Y.; Tock, J.L.; Totah, N.K.; Cheon, Y.; Torres, G.E.; Rapoport, S.I.; Moghaddam, B. Adolescent behavior and dopamine availability are uniquely sensitive to dietary omega-3 fatty acid deficiency. Biol. Psychiatry 2014, 75, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Sánchez, A.; Koletzko, B.; Larqué, E. Current understanding of placental fatty acid transport. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 265–272. [Google Scholar] [CrossRef]
- Helland, I.B.; Smith, L.; Saarem, K.; Saugstad, O.D.; Drevon, C.A. Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children’s IQ at 4 years of age. Pediatrics 2003, 111, e39–e44. [Google Scholar] [CrossRef] [Green Version]
- Cheruku, S.R.; Montgomery-Downs, H.E.; Farkas, S.L.; Thoman, E.B.; Lammi-Keefe, C.J. Higher maternal plasma docosahexaenoic acid during pregnancy is associated with more mature neonatal sleep-state patterning. Am. J. Clin. Nutr. 2002, 76, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Colombo, J.; Kannass, K.N.; Shaddy, D.J.; Kundurthi, S.; Maikranz, J.M.; Anderson, C.J.; Blaga, O.M.; Carlson, S.E. Maternal DHA and the development of attention in infancy and toddlerhood. Child Dev. 2004, 75, 1254–1267. [Google Scholar] [CrossRef]
- Freemantle, E.; Vandal, M.; Tremblay-Mercier, J.; Tremblay, S.; Blachère, J.C.; Bégin, M.E.; Brenna, J.T.; Windust, A.; Cunnane, S.C. Omega-3 fatty acids, energy substrates, and brain function during aging. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 213–220. [Google Scholar] [CrossRef]
- Montgomery, P.; Burton, J.R.; Sewell, R.P.; Spreckelsen, T.F.; Richardson, A.J. Low blood long chain omega-3 fatty acids in UK children are associated with poor cognitive performance and behavior: A cross-sectional analysis from the DOLAB study. PLoS ONE 2013, 8, e66697. [Google Scholar] [CrossRef]
- Tanabe, Y.; Hashimoto, M.; Sugioka, K.; Maruyama, M.; Fujii, Y.; Hagiwara, R.; Hara, T.; Hossain, S.M.; Shido, O. Improvement of spatial cognition with dietary docosahexaenoic acid is associated with an increase in Fos expression in rat CA1 hippocampus. Clin. Exp. Pharm. Physiol. 2004, 31, 700–703. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, W.; Liu, T.; Xiao, H.; Liang, W.; Xia, W.; Zhang, J. Perinatal supplementation with omega-3 polyunsaturated fatty acids improves sevoflurane-induced neurodegeneration and memory impairment in neonatal rats. PLoS ONE 2013, 8, e70645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, B.A.; Shaikh, I.A.; Khateeb, M.M.; Habeeb, S.M. Omega 3 polyunsaturated fatty acids enhance the protective effect of levetiracetam against seizures, cognitive impairment and hippocampal oxidative DNA damage in young kindled rats. Pharm. Biochem. Behav. 2015, 135, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Choi, J.M.; Lee, J.; Lee, M.H.; Lee, S.; Cho, E.J. Effects of Vegetable Oils with Different Fatty Acid Compositions on Cognition and Memory Ability in Aβ(25–35)-Induced Alzheimer’s Disease Mouse Model. J. Med. Food 2016, 19, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, D. Functional and Structural Benefits Induced by Omega-3 Polyunsaturated Fatty Acids During Aging. Curr. Neuropharmacol. 2017, 15, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Cutuli, D.; Pagani, M.; Caporali, P.; Galbusera, A.; Laricchiuta, D.; Foti, F.; Neri, C.; Spalletta, G.; Caltagirone, C.; Petrosini, L.; et al. Effects of Omega-3 Fatty Acid Supplementation on Cognitive Functions and Neural Substrates: A Voxel-Based Morphometry Study in Aged Mice. Front. Aging Neurosci. 2016, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, S.; Carasso, R.L. Modulation of learning, pain thresholds, and thermoregulation in the rat by preparations of free purified alpha-linolenic and linoleic acids: Determination of the optimal omega 3-to-omega 6 ratio. Proc. Natl. Acad. Sci. USA 1993, 90, 10345–10349. [Google Scholar] [CrossRef] [Green Version]
- Brenna, J.T. Animal studies of the functional consequences of suboptimal polyunsaturated fatty acid status during pregnancy, lactation and early post-natal life. Matern. Child Nutr. 2011, 7, 59–79. [Google Scholar] [CrossRef]
- Brainard, J.S.; Jimoh, O.F.; Deane, K.H.O.; Biswas, P.; Donaldson, D.; Maas, K.; Abdelhamid, A.S.; Hooper, L. Omega-3, Omega-6, and Polyunsaturated Fat for Cognition: Systematic Review and Meta-analysis of Randomized Trials. J. Am. Med. Dir. Assoc. 2020, 21, 1439–1450.e1421. [Google Scholar] [CrossRef]
- Jasbi, P.; Shi, X.; Chu, P.; Elliott, N.; Hudson, H.; Jones, D.; Serrano, G.; Chow, B.; Beach, T.G.; Liu, L.; et al. Metabolic Profiling of Neocortical Tissue Discriminates Alzheimer’s Disease from Mild Cognitive Impairment, High Pathology Controls, and Normal Controls. J. Proteome Res. 2021, 20, 4303–4317. [Google Scholar] [CrossRef]
- Mett, J. The Impact of Medium Chain and Polyunsaturated ω-3-Fatty Acids on Amyloid-β Deposition, Oxidative Stress and Metabolic Dysfunction Associated with Alzheimer’s Disease. Antioxidants 2021, 10, 1991. [Google Scholar] [CrossRef]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Tully, A.M.; Roche, H.M.; Doyle, R.; Fallon, C.; Bruce, I.; Lawlor, B.; Coakley, D.; Gibney, M.J. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: A case-control study. Br. J. Nutr. 2003, 89, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; García-Gómez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Loef, M.; Walach, H. The omega-6/omega-3 ratio and dementia or cognitive decline: A systematic review on human studies and biological evidence. J. Nutr. Gerontol. Geriatr. 2013, 32, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191–192. [Google Scholar] [CrossRef]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann. Neurol. 1997, 42, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Barberger-Gateau, P.; Letenneur, L.; Deschamps, V.; Pérès, K.; Dartigues, J.F.; Renaud, S. Fish, meat, and risk of dementia: Cohort study. BMJ 2002, 325, 932–933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, J.; Qiu, J.; Li, Y.; Wang, J.; Jiao, J. Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: A dose-response meta-analysis of 21 cohort studies. Am. J. Clin. Nutr. 2016, 103, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Su, K.P.; Cheng, T.C.; Liu, H.C.; Chang, C.J.; Dewey, M.E.; Stewart, R.; Huang, S.Y. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: A preliminary randomized double-blind placebo-controlled study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1538–1544. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxén-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.O.; Palmblad, J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: A randomized double-blind trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, S.; Sakaguchi, E.; Warashina, S.; Matsukawa, N.; Ishikura, Y.; Kiso, Y.; Sakakibara, M.; Yoshimoto, T.; Guo, J.; Yamashima, T. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci. Res. 2006, 56, 159–164. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; Van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Diet and risk of dementia: Does fat matter?: The Rotterdam Study. Neurology 2002, 59, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Kröger, E.; Verreault, R.; Carmichael, P.H.; Lindsay, J.; Julien, P.; Dewailly, E.; Ayotte, P.; Laurin, D. Omega-3 fatty acids and risk of dementia: The Canadian Study of Health and Aging. Am. J. Clin. Nutr. 2009, 90, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R., Jr.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef]
- Yanai, H. Effects of N-3 Polyunsaturated Fatty Acids on Dementia. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dierssen, M.; Fructuoso, M.; Martínez de Lagrán, M.; Perluigi, M.; Barone, E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020, 14, 670. [Google Scholar] [CrossRef]
- Yu, Q.; He, Z.; Zubkov, D.; Huang, S.; Kurochkin, I.; Yang, X.; Halene, T.; Willmitzer, L.; Giavalisco, P.; Akbarian, S.; et al. Lipidome alterations in human prefrontal cortex during development, aging, and cognitive disorders. Mol. Psychiatry 2020, 25, 2952–2969. [Google Scholar] [CrossRef] [Green Version]
- de Almeida, E.W.; Greguol, M. Lipid profile in people with Down syndrome: A literature review. J. Hum. Growth Dev. 2020, 30, 197–208. [Google Scholar] [CrossRef]
- Seven, M.; Cengiz, M.; Tüzgen, S.; Iscan, M.Y. Plasma carnitine levels in children with Down syndrome. Am. J. Hum. Biol. Off. J. Hum. Biol. Counc. 2001, 13, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.S.; Kim, S.H.; Fountoulakis, M.; Lubec, G. Heart type fatty acid binding protein (H-FABP) is decreased in brains of patients with Down syndrome and Alzheimer’s disease. J. Neural. Transmission. Suppl. 2003, 67, 225–234. [Google Scholar] [CrossRef]
- Tall, A.R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J. Intern. Med. 2008, 263, 256–273. [Google Scholar] [CrossRef]
- Lorkowski, S.; Rust, S.; Engel, T.; Jung, E.; Tegelkamp, K.; Galinski, E.A.; Assmann, G.; Cullen, P. Genomic sequence and structure of the human ABCG1 (ABC8) gene. Biochem. Biophys. Res. Commun. 2001, 280, 121–131. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, J.; Ren, C.; Chen, H.; Zhang, J. DYRK1A inhibitors for disease therapy: Current status and perspectives. European journal of medicinal chemistry 2022, 229, 114062. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.; Benhaddou, S.; Dard, R.; Tolu, S.; Hamzé, R.; Vialard, F.; Movassat, J.; Janel, N. Metabolic Diseases and Down Syndrome: How Are They Linked Together? Biomedicines 2021, 9, 221. [Google Scholar] [CrossRef]
- Pinto, J.; Almeida, L.M.; Martins, A.S.; Duarte, D.; Domingues, M.R.; Barros, A.S.; Galhano, E.; Pita, C.; Almeida Mdo, C.; Carreira, I.M.; et al. Impact of fetal chromosomal disorders on maternal blood metabolome: Toward new biomarkers? Am. J. Obstet. Gynecol. 2015, 213, e841. [Google Scholar] [CrossRef]
- Brooksbank, B.W.; Martinez, M.; Balazs, R. Altered composition of polyunsaturated fatty acyl groups in phosphoglycerides of Down’s syndrome fetal brain. J. Neurochem. 1985, 44, 869–874. [Google Scholar] [CrossRef]
- Tabernero, A.; Velasco, A.; Granda, B.; Lavado, E.M.; Medina, J.M. Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J. Biol. Chem. 2002, 277, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Møllgård, K.; Dziegielewska, K.M.; Saunders, N.R.; Zakut, H.; Soreq, H. Synthesis and localization of plasma proteins in the developing human brain. Integrity of the fetal blood-brain barrier to endogenous proteins of hepatic origin. Dev. Biol. 1988, 128, 207–221. [Google Scholar] [CrossRef]
- Hijazi, M.; Medina, J.M.; Velasco, A. Restrained Phosphatidylcholine Synthesis in a Cellular Model of Down’s Syndrome is Associated with the Overexpression of Dyrk1A. Mol. Neurobiol. 2017, 54, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.S.; Bannon, F.J. Serum albumin in Down Syndrome with and without Alzheimer’s Disease. Ir. J. Med. Sci. 2005, 174, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.L. Serum protein and lipoprotein fractions in mongolism. Am. J. Dis. Child. 1961, 102, 369–374. [Google Scholar] [CrossRef]
- Shah, S.N. Fatty acid composition of lipids of human brain myelin and synaptosomes: Changes in phenylketonuria and Down’s syndrome. Int. J. Biochem. 1979, 10, 477–482. [Google Scholar] [CrossRef]
- Murphy, E.J.; Schapiro, M.B.; Rapoport, S.I.; Shetty, H.U. Phospholipid composition and levels are altered in Down syndrome brain. Brain Res. 2000, 867, 9–18. [Google Scholar] [CrossRef]
- Brooksbank, B.W.; Martinez, M. Lipid abnormalities in the brain in adult Down’s syndrome and Alzheimer’s disease. Mol. Chem. Neuropathol. 1989, 11, 157–185. [Google Scholar] [CrossRef]
- Hamilton, L.K.; Dufresne, M.; Joppé, S.E.; Petryszyn, S.; Aumont, A.; Calon, F.; Barnabé-Heider, F.; Furtos, A.; Parent, M.; Chaurand, P.; et al. Aberrant Lipid Metabolism in the Forebrain Niche Suppresses Adult Neural Stem Cell Proliferation in an Animal Model of Alzheimer’s Disease. Cell Stem. Cell 2015, 17, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Cerro, S.; Rueda, N.; Vidal, V.; Puente, A.; Campa, V.; Lantigua, S.; Narcis, O.; Velasco, A.; Bartesaghi, R.; Martinez-Cue, C. Prenatal Administration of Oleic Acid or Linolenic Acid Reduces Neuromorphological and Cognitive Alterations in Ts65dn Down Syndrome Mice. J. Nutr. 2020, 150, 1631–1643. [Google Scholar] [CrossRef]
- García-García, A.G.; Polo-Hernández, E.; Tabernero, A.; Medina, J.M. Alpha-fetoprotein (AFP) modulates the effect of serum albumin on brain development by restraining the neurotrophic effect of oleic acid. Brain Res. 2015, 1624, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Kronquist, K.E.; Dreazen, E.; Keener, S.L.; Nicholas, T.W.; Crandall, B.F. Reduced fetal hepatic alpha-fetoprotein levels in Down’s syndrome. Prenat. Diagn. 1990, 10, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Brooksbank, B.W.; Walker, D.; Balazs, R.; Jorgensen, O.S. Neuronal maturation in the foetal brain in Down’s syndrome. Early Hum. Dev. 1989, 18, 237–246. [Google Scholar] [CrossRef]
- Fukaya, M.; Watanabe, M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP90 protein family by protease section pretreatment: A study in the adult mouse brain. J. Comp. Neurol. 2000, 426, 572–586. [Google Scholar] [CrossRef]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, V.; House, D.R.; Bredt, D.S.; Nicoll, R.A. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci. 2003, 23, 5503–5506. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cue, C.; Delatour, B.; Potier, M.C. Treating enhanced GABAergic inhibition in Down syndrome: Use of GABA alpha5-selective inverse agonists. Neurosci. Biobehav. Rev. 2014, 46, 218–227. [Google Scholar] [CrossRef]
- Vidal, V.; García-Cerro, S.; Rueda, N.; Puente, A.; Bartesaghi, R.; Martínez-Cué, C. Early postnatal oleic acid administration enhances synaptic development and cognitive abilities in the Ts65Dn mouse model of Down syndrome. Nutr. Neurosci. 2020, 25, 1400–1412. [Google Scholar] [CrossRef]
- Giacomini, A.; Stagni, F.; Emili, M.; Guidi, S.; Salvalai, M.E.; Grilli, M.; Vidal-Sanchez, V.; Martinez-Cue, C.; Bartesaghi, R. Treatment with corn oil improves neurogenesis and cognitive performance in the Ts65Dn mouse model of Down syndrome. Brain Res. Bull. 2018, 140, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Ciaccio, M.; Piccione, M.; Giuffrè, M.; Macaione, V.; Vocca, L.; Bono, A.; Corsello, G. Aminoacid profile and oxidative status in children affected by Down syndrome before and after supplementary nutritional treatment. Ital. J. Biochem. 2003, 52, 72–79. [Google Scholar]
- Vacca, R.A.; Valenti, D. Green tea EGCG plus fish oil omega-3 dietary supplements rescue mitochondrial dysfunctions and are safe in a Down’s syndrome child. Clin. Nutr. 2015, 34, 783–784. [Google Scholar] [CrossRef]
- Scala, I.; Valenti, D.; Scotto D’Aniello, V.; Marino, M.; Riccio, M.P.; Bravaccio, C.; Vacca, R.A.; Strisciuglio, P. Epigallocatechin-3-Gallate Plus Omega-3 Restores the Mitochondrial Complex I and F(0)F(1)-ATP Synthase Activities in PBMCs of Young Children with Down Syndrome: A Pilot Study of Safety and Efficacy. Antioxidants 2021, 10, 469. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; de Lagran, M.M.; Farre, M.; Fito, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Stringer, M.; LaCombe, J.; Patel, R.; Wallace, J.M.; Roper, R.J. Evaluation of the therapeutic potential of Epigallocatechin-3-gallate (EGCG) via oral gavage in young adult Down syndrome mice. Sci. Rep. 2020, 10, 10426. [Google Scholar] [CrossRef] [PubMed]
- Jamal, R.; LaCombe, J.; Patel, R.; Blackwell, M.; Thomas, J.R.; Sloan, K.; Wallace, J.M.; Roper, R.J. Increased dosage and treatment time of Epigallocatechin-3-gallate (EGCG) negatively affects skeletal parameters in normal mice and Down syndrome mouse models. PLoS ONE 2022, 17, e0264254. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, K.; Jing, N.; Zhao, Y.; Yang, X. EGCG regulates fatty acid metabolism of high-fat diet-fed mice in association with enrichment of gut Akkermansia muciniphila. J. Funct. Foods 2020, 75, 104261. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, P.A.; Gao, L.Y.; Saxena, A.R.; Chavannes, N.K.; Hushmendy, S.F.; Bhoiwala, D.L.; Crawford, D.R. Fish oil improves gene targets of Down syndrome in C57BL and BALB/c mice. Nutr. Res. 2015, 35, 440–448. [Google Scholar] [CrossRef]
Figure 1.
Summary of the major neurodevelopmental alterations in Down syndrome. See text for explanation and references. Abbreviations: AD, Alzheimer’s disease; NPC, neural progenitor cells. The upward pointing red arrows indicates an increase. The downward pointing blue arrows indicates a reduction.
Figure 1.
Summary of the major neurodevelopmental alterations in Down syndrome. See text for explanation and references. Abbreviations: AD, Alzheimer’s disease; NPC, neural progenitor cells. The upward pointing red arrows indicates an increase. The downward pointing blue arrows indicates a reduction.

Figure 2.
Naming of unsaturated fatty acids based on the position of the double bonds. According to one classification (IUPAC classification), this position refers to the distance from the carboxyl terminus, C1 (C-x). Alternatively, numbering refers to the distance of the double bond from the C atom of the methyl group, which is designated as n or ω. In n minus x (ω minus x), a double bond of the fatty acid is located on the xth carbon–carbon bond counting from the terminal methyl carbon (designated as n or ω) toward the carboxyl terminus. An 18-carbon FA is reported here as an example. The representation is idealized because, unlike saturated fatty acids, which have a straight-chain structure, PUFAs are typically bent.
Figure 2.
Naming of unsaturated fatty acids based on the position of the double bonds. According to one classification (IUPAC classification), this position refers to the distance from the carboxyl terminus, C1 (C-x). Alternatively, numbering refers to the distance of the double bond from the C atom of the methyl group, which is designated as n or ω. In n minus x (ω minus x), a double bond of the fatty acid is located on the xth carbon–carbon bond counting from the terminal methyl carbon (designated as n or ω) toward the carboxyl terminus. An 18-carbon FA is reported here as an example. The representation is idealized because, unlike saturated fatty acids, which have a straight-chain structure, PUFAs are typically bent.

Figure 3.
Synthesis of n-3 and n-6 PUFAs. ALA produces EPA, DPA, and DHA. LA produces ARA. n-3 PUFA-derived lipid metabolites largely inhibit inflammation. These mediators include resolvins and protectins. By contrast, ARA produces inflammatory metabolites (eicosanoids), which tend to promote inflammation. They include prostaglandins, leukotrienes, and thromboxanes (see [56,58]). ARA, however, also produces lipoxins that promote the resolution of inflammation (see [59]). Abbreviations: ALA, α-linolenic acid; ARA, arachidonic acid; DPA, docosapentaenoic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; LA, linoleic acid.
Figure 3.
Synthesis of n-3 and n-6 PUFAs. ALA produces EPA, DPA, and DHA. LA produces ARA. n-3 PUFA-derived lipid metabolites largely inhibit inflammation. These mediators include resolvins and protectins. By contrast, ARA produces inflammatory metabolites (eicosanoids), which tend to promote inflammation. They include prostaglandins, leukotrienes, and thromboxanes (see [56,58]). ARA, however, also produces lipoxins that promote the resolution of inflammation (see [59]). Abbreviations: ALA, α-linolenic acid; ARA, arachidonic acid; DPA, docosapentaenoic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; LA, linoleic acid.

Figure 4.
Summary of the effects of fatty acids on neurogenesis. Abbreviations: ALA, α-linolenic acid; ARA, arachidonic acid; As, astrocyte; DHA, docosahexaenoic acid; LA, linoleic acid; N, neuron; NPC, neural progenitor cell; OA, oleic acid; OL, oligodendrocyte; PA, palmitic acid. The double-headed horizontal arrow means no effect. The upward pointing arrow indicates an increase. The downward pointing arrow indicates a reduction. The double-headed horizontal arrow means no effect.
Figure 4.
Summary of the effects of fatty acids on neurogenesis. Abbreviations: ALA, α-linolenic acid; ARA, arachidonic acid; As, astrocyte; DHA, docosahexaenoic acid; LA, linoleic acid; N, neuron; NPC, neural progenitor cell; OA, oleic acid; OL, oligodendrocyte; PA, palmitic acid. The double-headed horizontal arrow means no effect. The upward pointing arrow indicates an increase. The downward pointing arrow indicates a reduction. The double-headed horizontal arrow means no effect.

Figure 5.
Summary of the effects of fatty acids on neuron maturation. Abbreviations: ARA, arachidonic acid; DHA, docosahexaenoic acid; OA, oleic acid. The upward pointing arrow indicates an increase. The downward pointing arrow indicates a reduction. The double-headed horizontal arrow means no effect.
Figure 5.
Summary of the effects of fatty acids on neuron maturation. Abbreviations: ARA, arachidonic acid; DHA, docosahexaenoic acid; OA, oleic acid. The upward pointing arrow indicates an increase. The downward pointing arrow indicates a reduction. The double-headed horizontal arrow means no effect.

Figure 6.
Summary of the effects of fatty acids on brain development in the Ts65Dn model of DS. The downward-pointing black arrows indicate trisomy-related impairment of the variables reported in each panel. The positive effects of treatment with fatty acids are indicated on the right by the upward-pointing arrows. See text for details. Abbreviations: CO, corn oil; LA, linolenic acid; n.a., not available; OA, oleic acid.
Figure 6.
Summary of the effects of fatty acids on brain development in the Ts65Dn model of DS. The downward-pointing black arrows indicate trisomy-related impairment of the variables reported in each panel. The positive effects of treatment with fatty acids are indicated on the right by the upward-pointing arrows. See text for details. Abbreviations: CO, corn oil; LA, linolenic acid; n.a., not available; OA, oleic acid.


{kind=link}
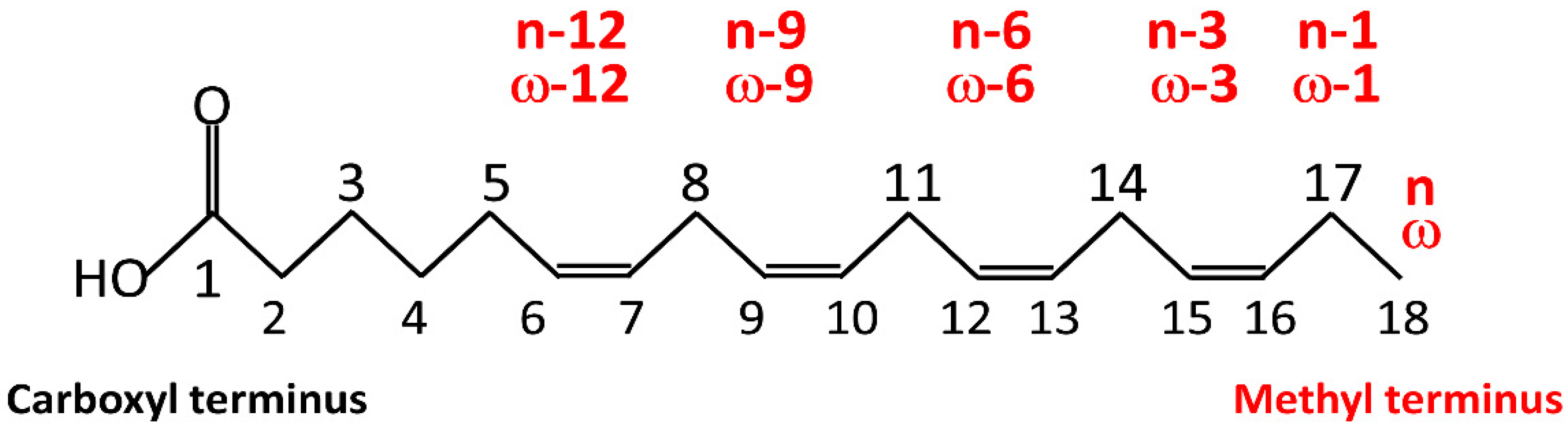{kind=link}
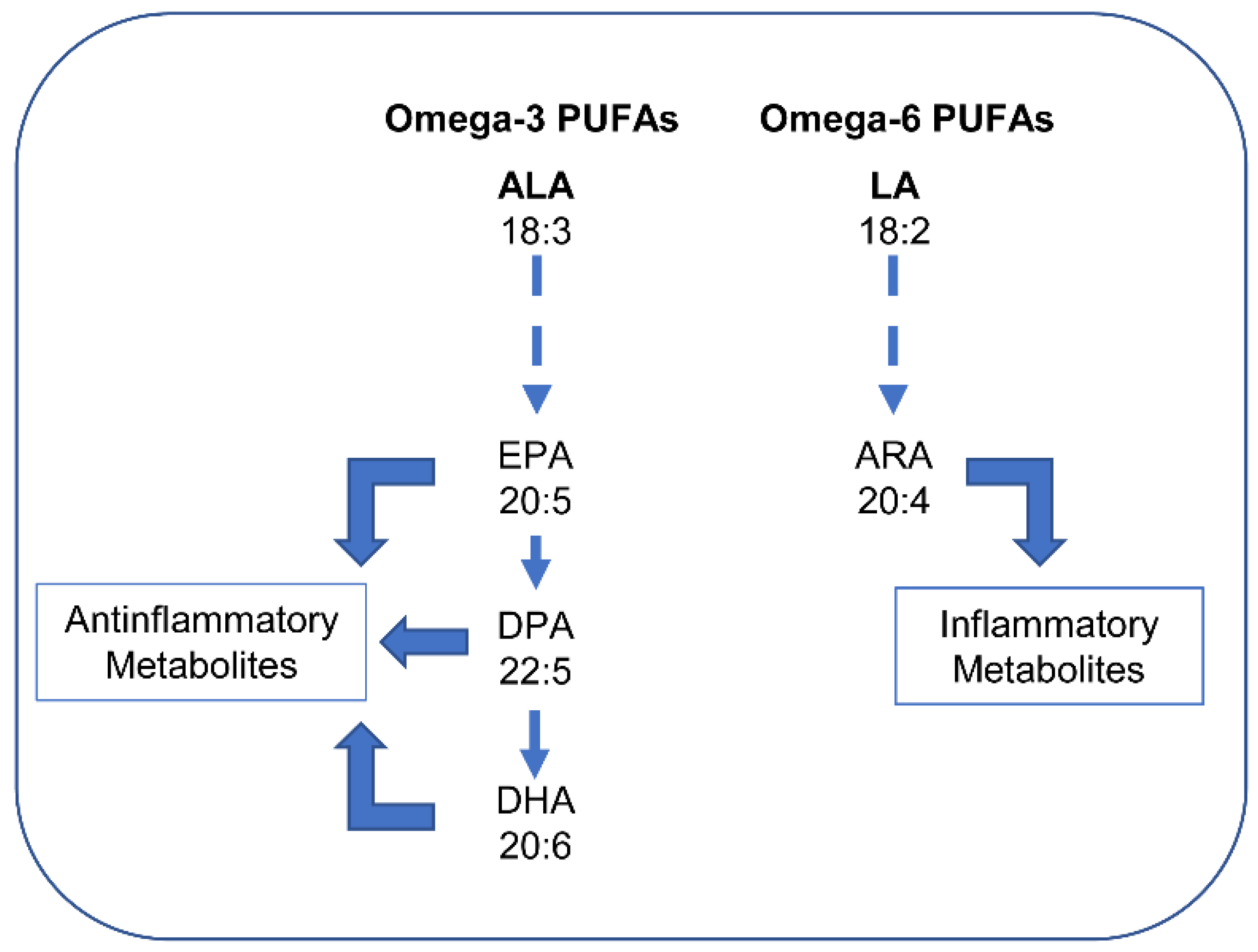{kind=link}
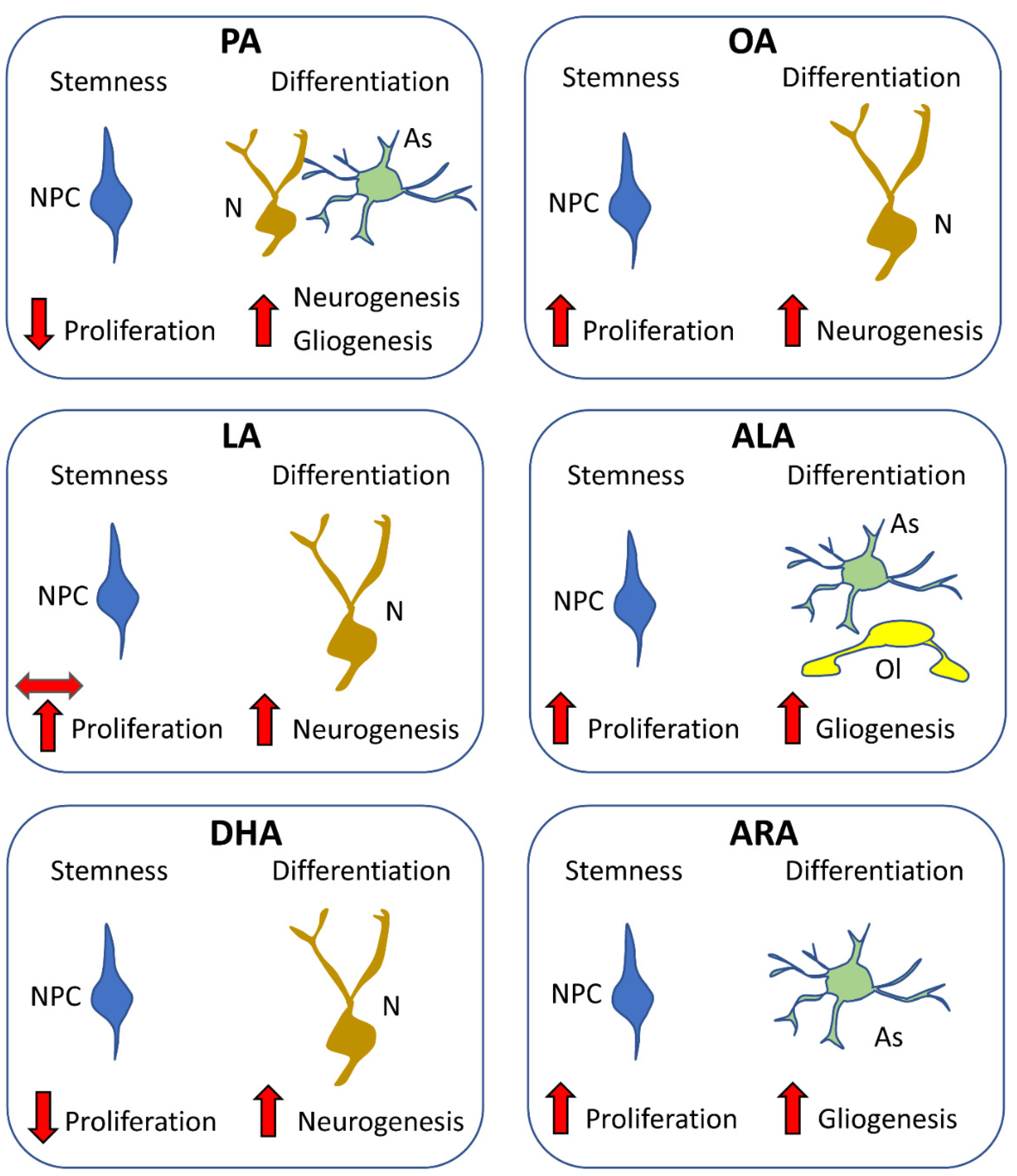{kind=link}
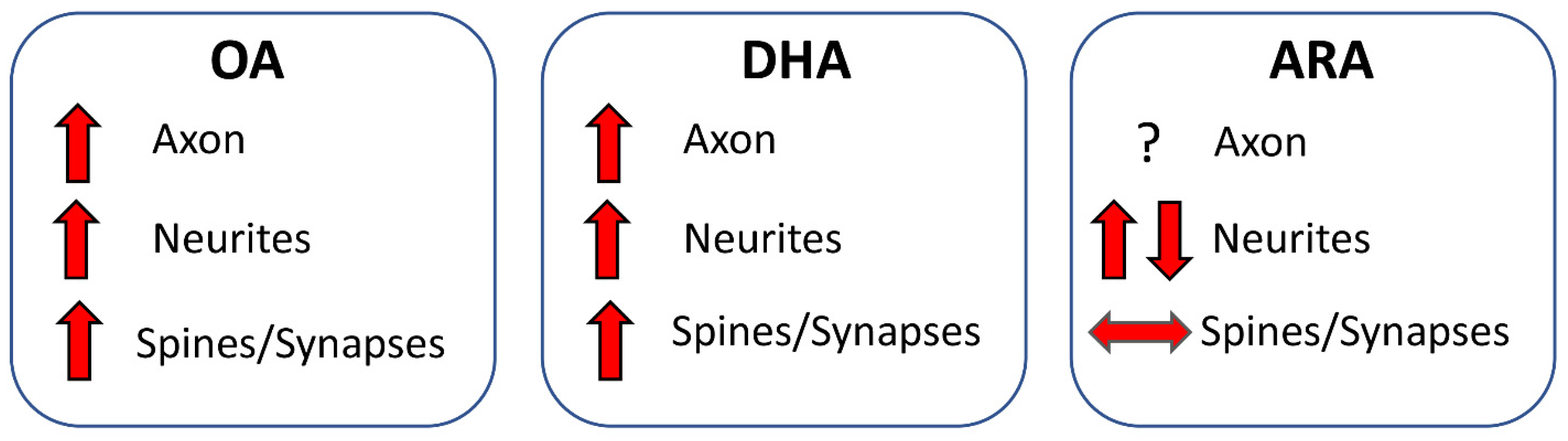{kind=link}
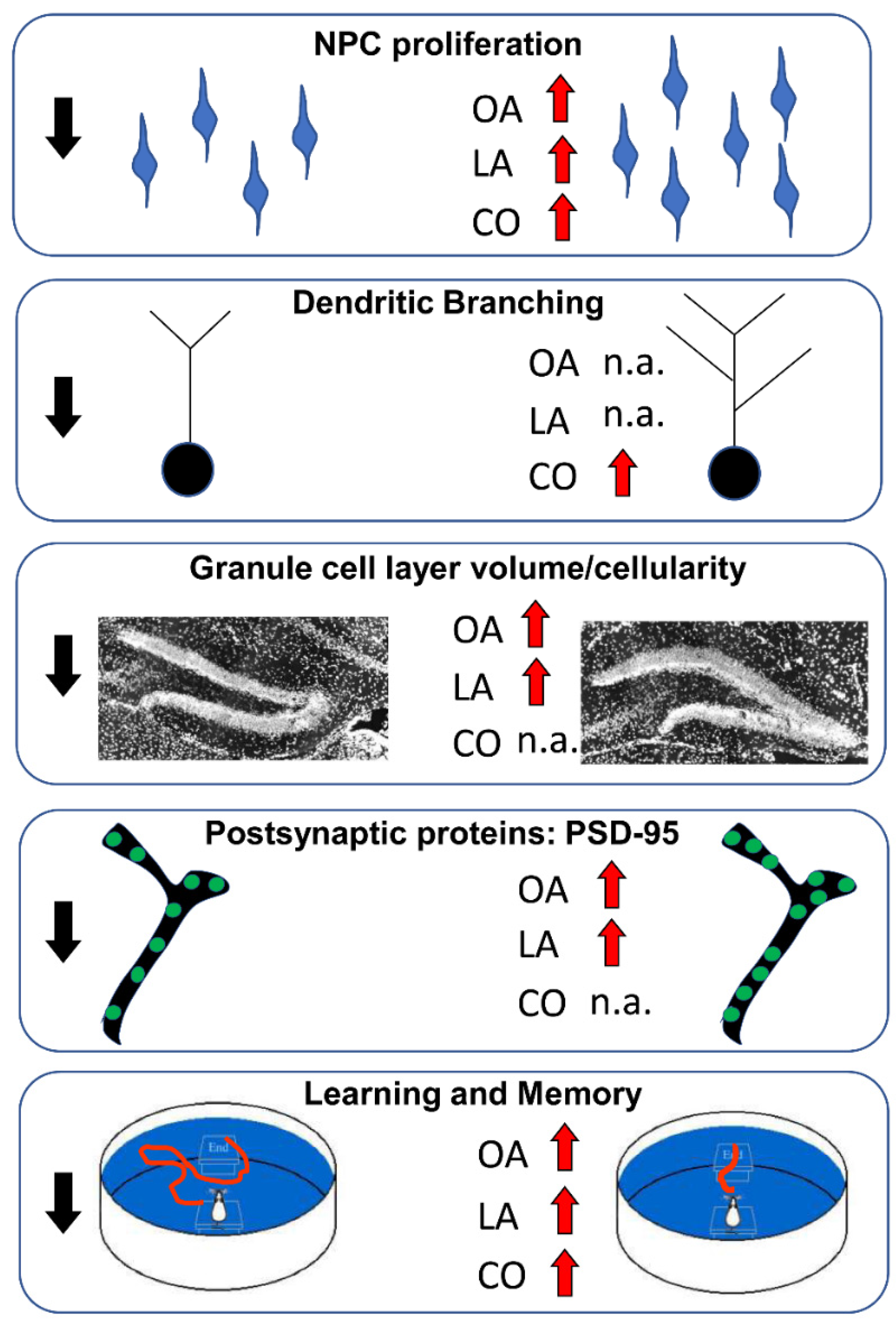{kind=link}
Table 1.
Classification of fatty acids based on the length of the hydrocarbon chain.
| Short-chain fatty acids | <8 carbons |
| Medium-chain fatty acids | 8–14 carbons |
| Long-chain fatty acid | >16 carbons |
| Very-long-chain fatty acids | >22 carbons |
Table 2.
Examples of FAs.
| Common Name | Abbreviation | C:D n-x |
|---|---|---|
| Saturated fatty acids (SFAs) | ||
| Butyric | 04:00 | |
| Caproic | 06:00 | |
| Caprylic | 08:00 | |
| Capric | 10:00 | |
| Lauric | 12:00 | |
| Myristic | 14:00 | |
| Palmitic | PA | 16:00 |
| Stearic | 18:00 | |
| Arachidic | 20:00 | |
| Mono-unsaturated fatty acids (MUFAs) | ||
| Palmitoleic | 16:01 n-7 | |
| Oleic | OA | 18:01 n-9 |
| Erucic | 22:01 n-9 | |
| Polyunsaturated fatty acids (PUFAs) | ||
| α-Linolenic * | ALA | 18:3 n-3 |
| Stearidonic | SDA | 18:4 n-3 |
| Eicosatetraenoic | ETE | 20:4 n-3 |
| Eicosapentaenoic | EPA | 20:5 n-3 |
| Docosapentaenoic | DPA | 22:5 n-3 |
| Docosahexaenoic | DHA | 22:6 n-3 |
| Linoleic * | LA | 18:2 n-6 |
| γ-Linolenic | GLA | 18:3 n-6 |
| Arachidonic | ARA | 20:4 n-6 |
Note: Number of carbon atoms in the fatty acid (C), number of double bonds (D), and the position of the first double bond counting from the terminal methyl carbon (n-x). EPA, DPA, and DHA derive from ALA and ARA derives from LA, in addition to being introduced with various types of food. ALA and LA (labeled with an asterisk) are essential FAs that must be introduced with the diet.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Martínez-Cué, C.; Bartesaghi, R. Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients 2022, 14, 2880. https://doi.org/10.3390/nu14142880
AMA Style
Martínez-Cué C, Bartesaghi R. Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome? Nutrients. 2022; 14(14):2880. https://doi.org/10.3390/nu14142880
Chicago/Turabian StyleMartínez-Cué, Carmen, and Renata Bartesaghi. 2022. "Fatty Acids: A Safe Tool for Improving Neurodevelopmental Alterations in Down Syndrome?" Nutrients 14, no. 14: 2880. https://doi.org/10.3390/nu14142880
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.