
Consumption of Non-Nutritive Sweetener, Acesulfame Potassium Exacerbates Atherosclerosis through Dysregulation of Lipid Metabolism in ApoE−/− Mice

 , ,
, ,  and
and 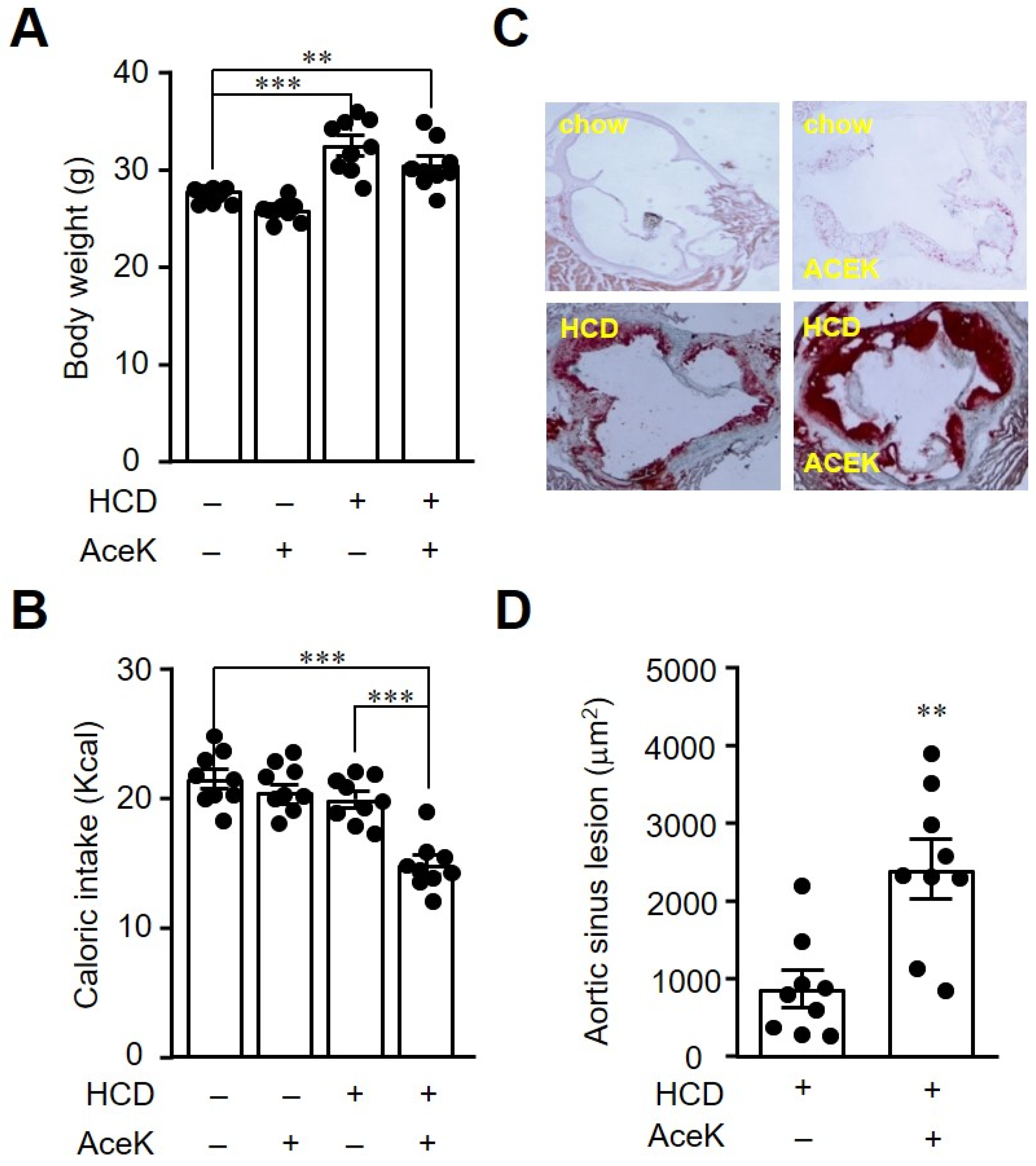{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Experimental Animals
2.2. Morphological Analysis
2.3. Biochemical Analysis
2.4. Cell Culture
2.5. Real-Time Polymerase Chain Reactions
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
3.1. AceK Exacerbated Atherosclerosis in High Cholesterol Diet Fed ApoE−/− Mice
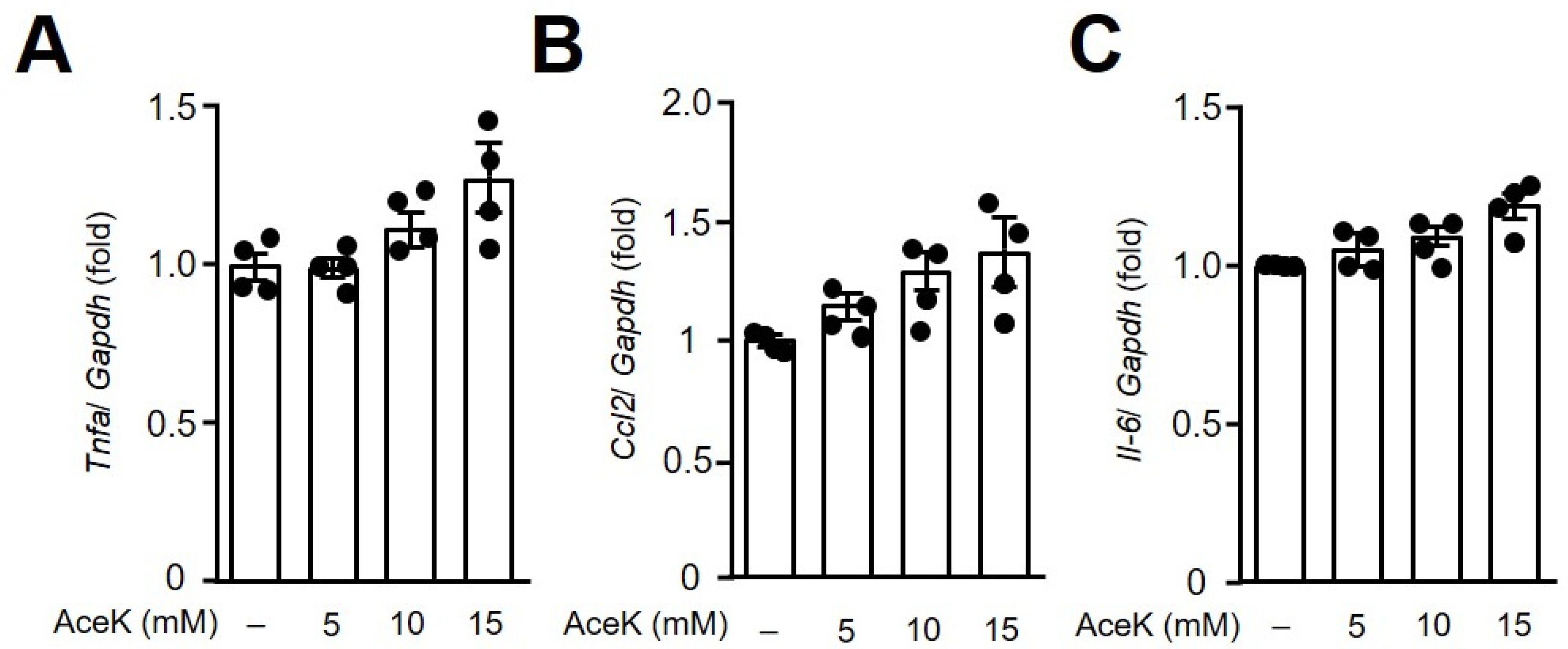
3.2. AceK Showed No Significant Effects on Proinflammatory Cytokine Expressions in RAW264.7 Macrophages
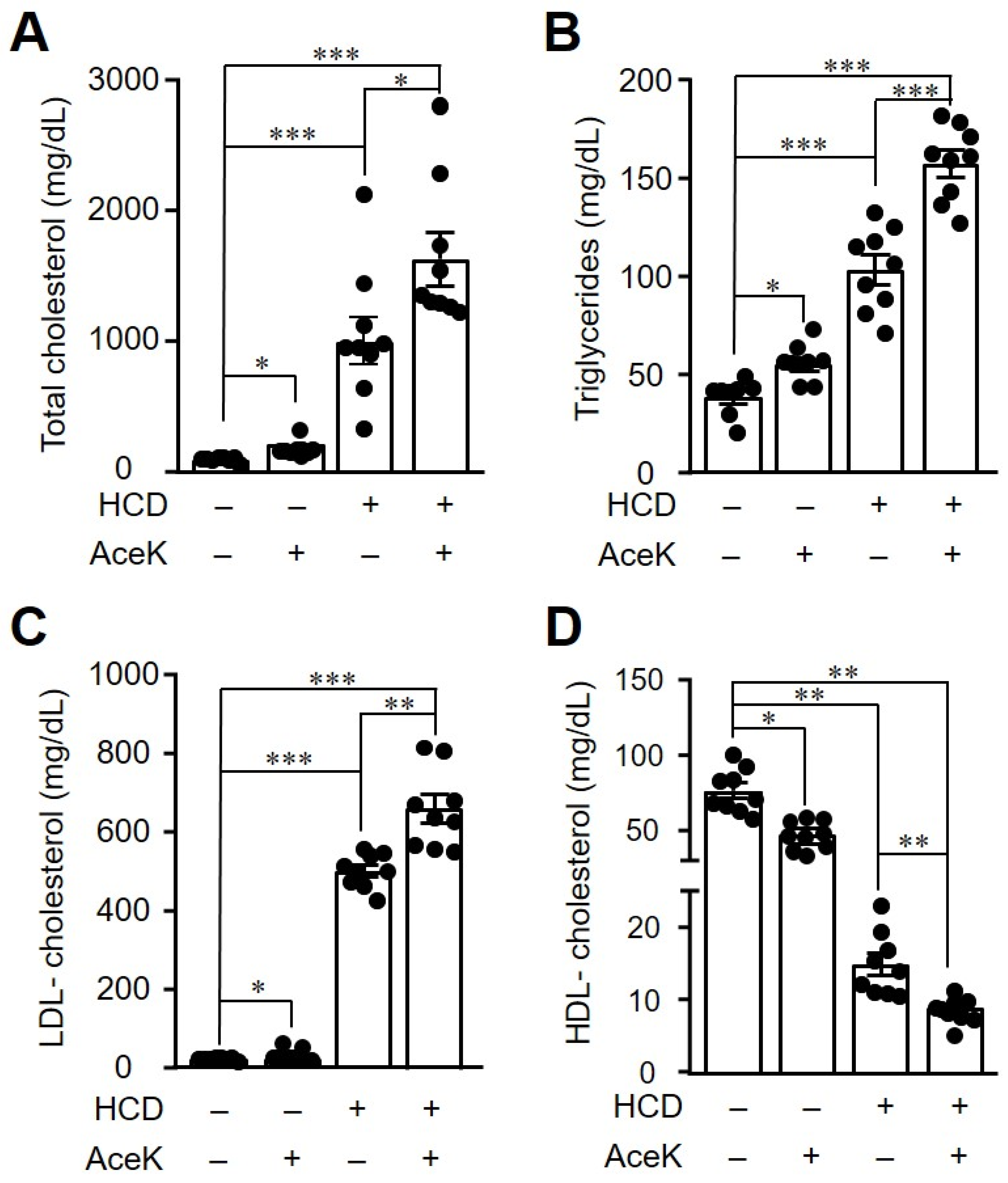
3.3. AceK Worsened Dyslipidemia in High Cholesterol Diet-Fed ApoE−/− Mice
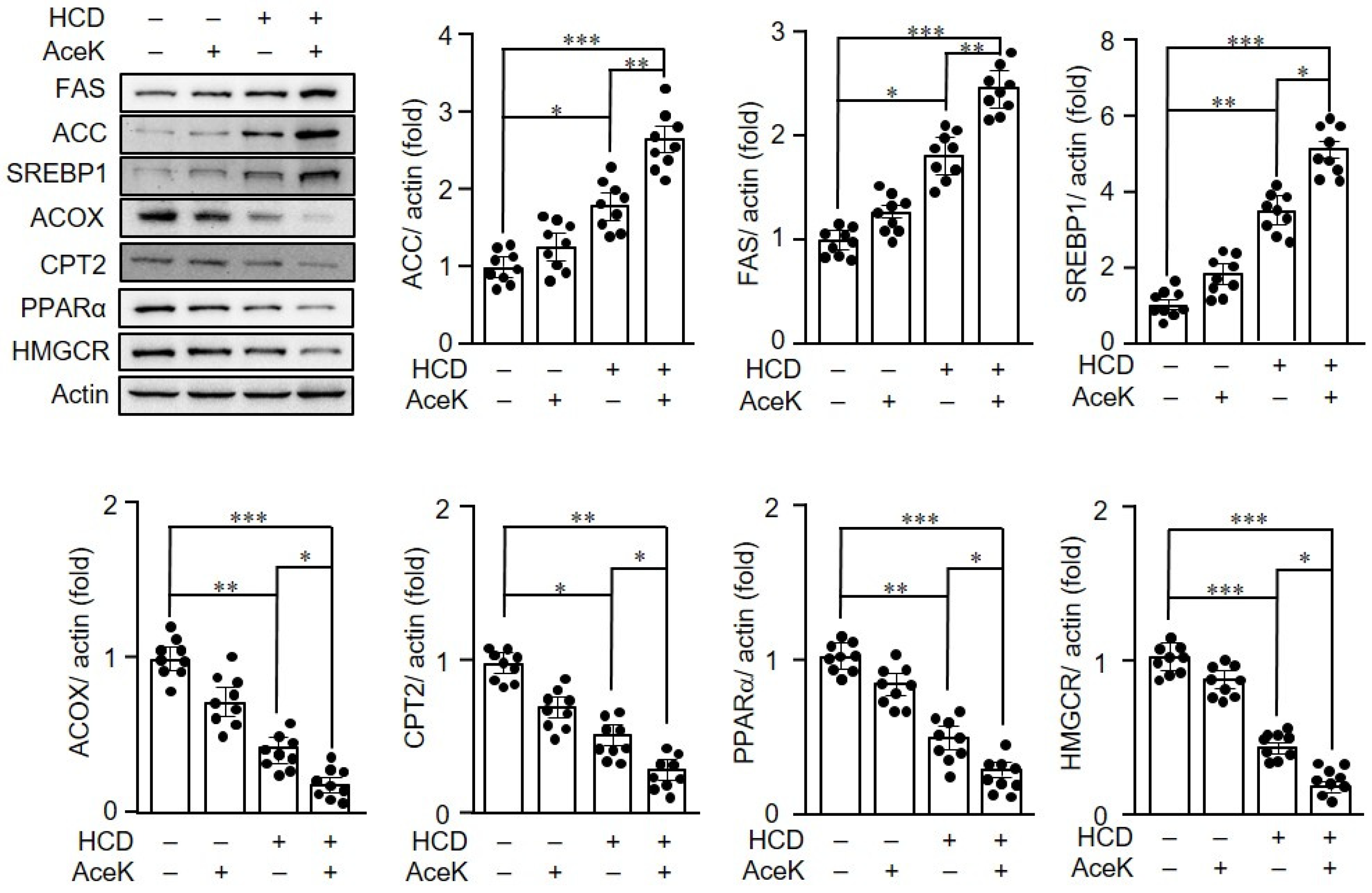
3.4. AceK Impaired Lipid Homeostasis in ApoE−/− Mice
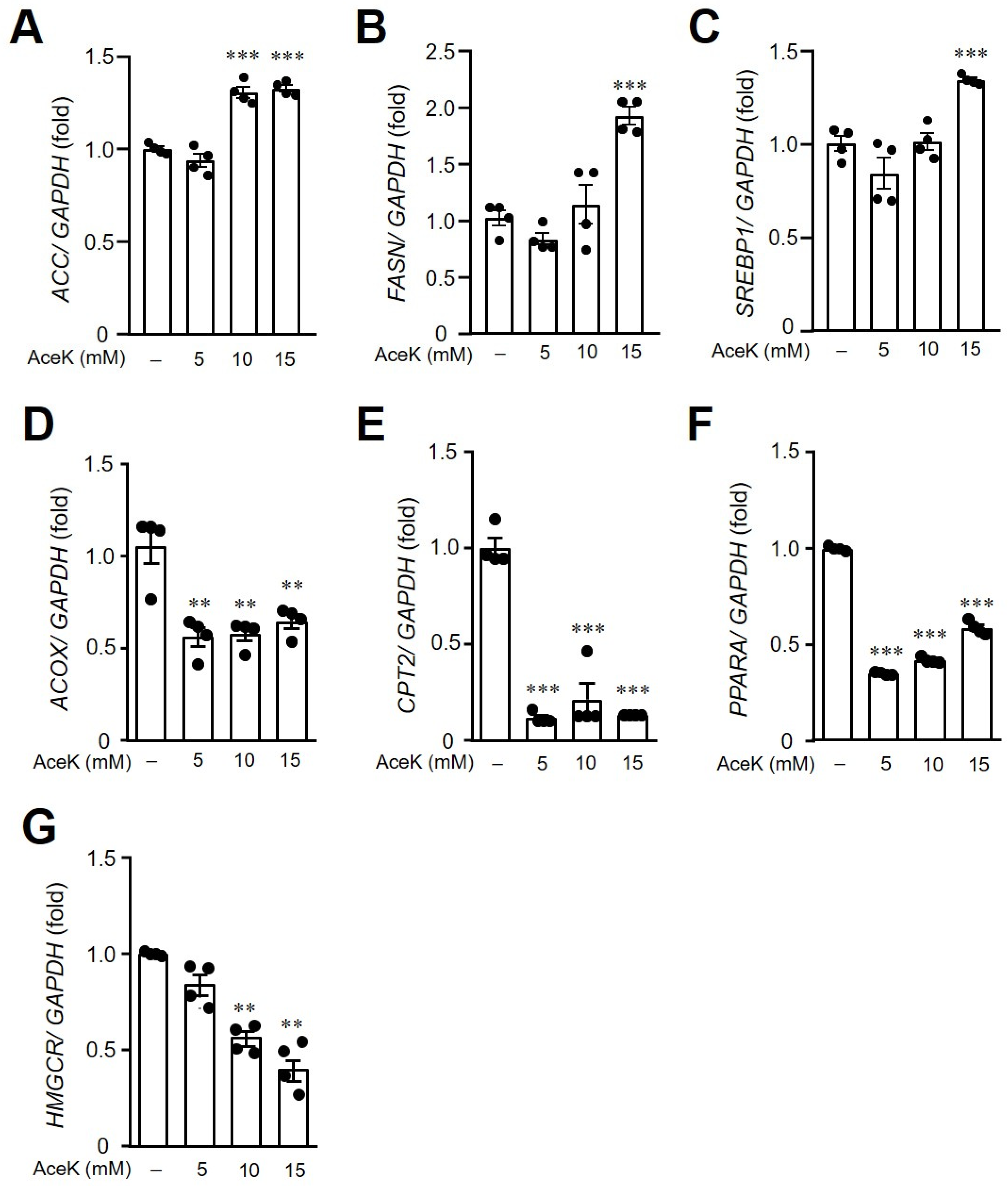
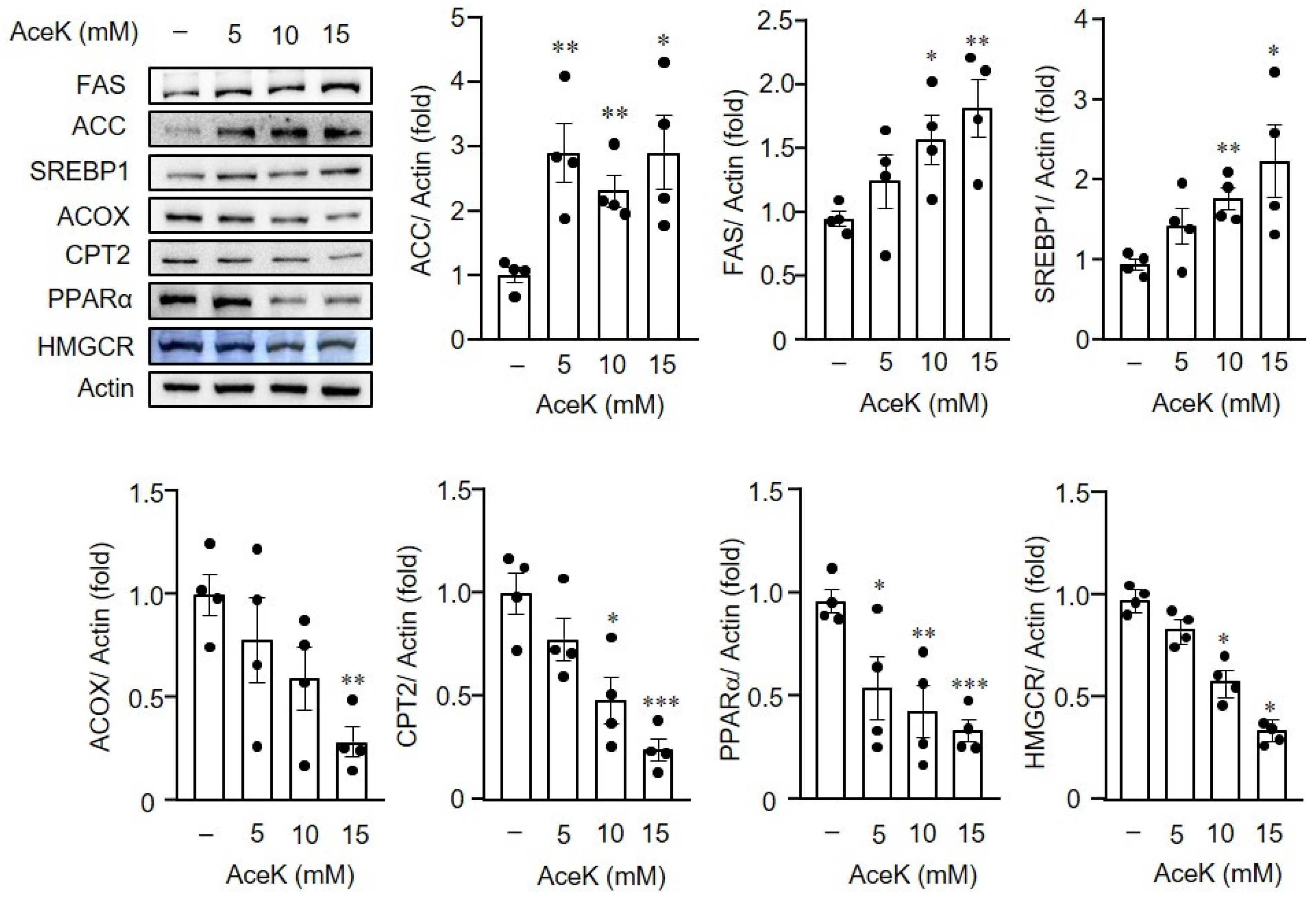
3.5. AceK-Induced Dysregulation of Lipid Homeostasis in HepG2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AceK | Acesulfame potassium |
| ACC | Acetyl-coA carboxylase |
| ApoE−/− | Apolipoprotein E deficient |
| CPT2 | Carnitine palmitoyltransferase-2 |
| FAS | Fatty acid synthase |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| HCD | High cholesterol diet |
| HUVECs | Human umbilical vein endothelial cell line |
| HDL-c | High-density lipoprotein cholesterol |
| IL-6 | Interleukin 6 |
| LDL-c | Low-density lipoprotein cholesterol |
| CCL2 | C-C motif chemokine ligand 2 |
| NNS | Non-nutritive sweetener |
| ACOX | Peroxisomal acyl-coenzyme A oxidase |
| PPARα | Peroxisome proliferator-activated receptor-α |
| SREBP1 | Sterol regulatory element binding protein-1 |
| TC | Total cholesterol |
| TG | Triglycerides |
| TNF-α | Tumor necrosis factor-α |
References
- Eman, E.G.H.; Abdelaziz, M.A.; Taha, N.M.; El-Gama, M.S. The influence of acesulfame-k and aspartame on some physiological parameters in male albino rats. Egypt. J. Hosp. Med. 2019, 75, 1976–1981. [Google Scholar] [CrossRef]
- Henry, F.J. Obesity prevention: The key to non-communicable disease control. West Indian Med. J. 2011, 60, 446–451. [Google Scholar]
- Hruby, A.; Hu, F.B. The epidemiology of obesity: A big picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Malik, V.S.; Willet, W.C.; Hu, F.B. Nearly a decade on—Trends, risk factors and policy implications in global obesity. Nat. Rev. Endocrinol. 2020, 16, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Saris, W.H.M.; Foster, G.D. Simple carbohydrates and obesity: Fact, fiction and future. Int. J. Obes. 2006, 30, S1–S3. [Google Scholar] [CrossRef]
- Khaodhiar, L.; McCowen, K.C.; Blackburn, G.L. Obesity and its comorbid conditions. Clin. Cornerstone 1999, 2, 17–31. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Tandel, K.R. Sugar substitutes: Health controversy over perceived benefits. J. Pharmacol. Pharmacother. 2011, 2, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walbolt, J.; Koh, Y. Non-nutritive sweeteners and their associations with obesity and type 2 diabetes. J. Obes. Metab. Syndr. 2020, 29, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Rother, K.I. Non-Nutritive sweeteners and their role in the gastrointestinal Tract. J. Clin. Endocrinol. Metab. 2012, 97, 2597–2605. [Google Scholar] [CrossRef] [Green Version]
- Pang, M.D.; Goossens, G.H.; Blaak, E.E. The impact of artificial sweeteners on body weight control and glucose homeostasis. Front. Nutr. 2021, 7, 598340. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, M.; Obert, J.; Casey, L. The association between artificial sweeteners and obesity. Curr. Gastroenterol. Rep. 2017, 19, 64. [Google Scholar] [CrossRef]
- Bian, X.; Chi, L.; Gao, B.; Tu, P.; Ru, H.; Lu, K. The artificial sweetener acesulfame potassium affects the gut microbiome and body weight gain in CD-1 mice. PLoS ONE 2017, 12, e0178426. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sarr, M.G. Effect of the artificial sweetener, acesulfame potassium, a sweet taste receptor agonist, on glucose uptake in small intestinal cell lines. J. Gastrointest. Surg. 2012, 17, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Ghoshal, S.; Mukherjee, A. Genotoxicity testing of low-calorie sweeteners: Aspartame, Acesulfame-K and Saccharin. Drug Chem. Toxicol. 2008, 31, 447–457. [Google Scholar] [CrossRef]
- Cong, W.-N.; Wang, R.; Cai, H.; Daimon, C.M.; Scheibye-Knudsen, M.; Bohr, V.A.; Turkin, R.; Wood, W.H.; Becker, K.; Moaddel, R.; et al. Long-Term Artificial Sweetener Acesulfame Potassium Treatment Alters Neurometabolic Functions in C57BL/6J Mice. PLoS ONE 2013, 8, e70257. [Google Scholar] [CrossRef]
- Mukherjee, A.; Chakrabarti, J. In vivo cytogenetic studies on mice exposed to acesulfame-k—A non-nutritive sweetener. Food Chem. Toxicol. 1997, 35, 1177–1179. [Google Scholar] [CrossRef]
- Mattes, R.D.; Popkin, B.M. Nonnutritive sweetener consumption in humans: Effects on appetite and food intake and their putative mechanisms. Am. J. Clin. Nutr. 2008, 89, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-H.; Yu, T.-Y.; Tsai, F.-C.; Weston, C.J.; Lin, M.-S.; Hung, C.-S.; Kao, H.-L.; Li, Y.-I.; Solé, M.; Unzeta, M.; et al. Inhibition of semicarbazide-sensitive amine oxidase reduces atherosclerosis in apolipoprotein E-deficient mice. Transl. Res. 2018, 197, 12–31. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients. 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ma, X.; Li, L.; Wang, L.; Chen, Y.; Liu, J.; Luo, Y.; Zhuang, Z.; Yang, W.; Zang, S.; et al. Berberine ameliorates non alcoholic steatohepatitis in ApoE−/− mice. Exp. Ther. Med. 2017, 14, 4134–4140. [Google Scholar] [CrossRef] [Green Version]
- Greaves, D.R.; Gordon, S. Immunity, atherosclerosis and cardiovascular disease. Trends Immunol. 2001, 22, 180–181. [Google Scholar] [CrossRef]
- Simon, B.R.; Parlee, S.D.; Learman, B.S.; Mori, H.; Scheller, E.; Cawthorn, W.; Ning, X.; Gallagher, K.; Tyrberg, B.; Assadi-Porter, F.M.; et al. Artificial sweeteners stimulate adipogenesis and suppress lipolysis independently of sweet taste receptors. J. Biol. Chem. 2013, 288, 32475–32489. [Google Scholar] [CrossRef] [Green Version]
- Servant, G.; Kenakin, T.; Zhang, L.; Williams, M.; Servant, N. The function and allosteric control of the human sweet taste receptor. Adv. Pharmacol. 2020, 88, 59–82. [Google Scholar] [CrossRef]
- Wauson, E.M.; Lorente-Rodríguez, A.; Cobb, M.H. Minireview: Nutrient sensing by G protein-coupled receptors. Mol. Endocrinol. 2013, 27, 1188–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator–activated receptor-α during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Simon, B.R.; Learman, B.S.; Parlee, S.D.; Scheller, E.L.; Mori, H.; Cawthorn, W.P.; Ning, X.; Krishnan, V.; Ma, Y.L.; Tyrberg, B.; et al. Sweet taste receptor deficient mice have decreased adiposity and increased bone mass. PLoS ONE 2014, 9, e86454. [Google Scholar] [CrossRef]
- Pol, T.; Held, C.; Westerbergh, J.; Lindbäck, J.; Alexander, J.H.; Alings, M.; Erol, C.; Goto, S.; Halvorsen, S.; Huber, K.; et al. Dyslipidemia and risk of cardiovascular events in patients with atrial fibrillation treated with oral anticoagulation therapy: Insights from the ARISTOTLE (apixaban for reduction in stroke and other thromboembolic events in atrial fibrillation) trial. J. Am. Heart Assoc. 2018, 7, e007444. [Google Scholar] [CrossRef] [Green Version]
- Veseli, B.E.; Perrotta, P.; De Meyer, G.R.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Kuipers, F.; Van Ree, J.M.; Hofker, M.H.; Wolters, H.; Veld, G.I.; Havinga, R.; Vonk, R.J.; Princen, H.M.; Havekes, L.M. Altered lipid metabolism in apolipoprotein E-deficient mice does not affect cholesterol balance across the liver. Hepatology 1996, 24, 241–247. [Google Scholar] [CrossRef]
- Slätis, K.; Gåfvels, M.; Kannisto, K.; Ovchinnikova, O.; Paulsson-Berne, G.; Parini, P.; Jiang, Z.-Y.; Eggertsen, G. Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J. Lipid Res. 2010, 51, 3289–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, W.; Jeoung, N.H.; Cho, K.-H. Modified apolipoprotein (apo) A-I by artificial sweetener causes severe premature cellular senescence and atherosclerosis with impairment of functional and structural properties of apoA-I in lipid-free and lipid-bound state. Mol. Cells 2011, 31, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse models for atherosclerosis research—Which is my line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.X.; Mallat, Z. Targeting the immune system in atherosclerosis: JASS state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 1691–1706. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-H.; Li, H.-Y.; Wang, S.-H.; Chen, Y.-H.; Chen, Y.-C.; Wu, H.-T. Consumption of Non-Nutritive Sweetener, Acesulfame Potassium Exacerbates Atherosclerosis through Dysregulation of Lipid Metabolism in ApoE−/− Mice. Nutrients 2021, 13, 3984. https://doi.org/10.3390/nu13113984
Lin C-H, Li H-Y, Wang S-H, Chen Y-H, Chen Y-C, Wu H-T. Consumption of Non-Nutritive Sweetener, Acesulfame Potassium Exacerbates Atherosclerosis through Dysregulation of Lipid Metabolism in ApoE−/− Mice. Nutrients. 2021; 13(11):3984. https://doi.org/10.3390/nu13113984
Chicago/Turabian StyleLin, Cheng-Hsin, Hung-Yuan Li, Shu-Huei Wang, Yue-Hwa Chen, Yang-Ching Chen, and Hung-Tsung Wu. 2021. "Consumption of Non-Nutritive Sweetener, Acesulfame Potassium Exacerbates Atherosclerosis through Dysregulation of Lipid Metabolism in ApoE−/− Mice" Nutrients 13, no. 11: 3984. https://doi.org/10.3390/nu13113984