
Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis



1
Department of Interdisciplinary Medicine, University of Bari “Aldo Moro”, 70124 Bari, Italy
2
Department of Biomedical Sciences and Human Oncology, University of Bari “Aldo Moro”, 70124 Bari, Italy
3
Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari “Aldo Moro”, 70124 Bari, Italy
4
INBB, National Institute for Biostructures and Biosystems, 00136 Rome, Italy
5
National Cancer Center, IRCCS Istituto Tumori Giovanni Paolo II, 70124 Bari, Italy
*
Author to whom correspondence should be addressed.
Nutrients 2021, 13(6), 2101; https://doi.org/10.3390/nu13062101
Submission received: 25 May 2021
/
Revised: 14 June 2021
/
Accepted: 16 June 2021
/
Published: 19 June 2021
(This article belongs to the Special Issue Human Nutrition and Cancer Prevention)
Abstract
:The prevalence of being overweight and obese has been expanded dramatically in recent years worldwide. Obesity usually occurs when the energetic introit overtakes energy expenditure from metabolic and physical activity, leading to fat accumulation mainly in the visceral depots. Excessive fat accumulation represents a risk factor for many chronic diseases, including cancer. Adiposity, chronic low-grade inflammation, and hyperinsulinemia are essential factors of obesity that also play a crucial role in tumor onset. In recent years, several strategies have been pointed toward boundary fat accumulation, thus limiting the burden of cancer attributable to obesity. While remodeling fat via adipocytes browning seems a tempting prospect, lifestyle interventions still represent the main pathway to prevent cancer and enhance the efficacy of treatments. Specifically, the Mediterranean Diet stands out as one of the best dietary approaches to curtail visceral adiposity and, therefore, cancer risk. In this Review, the close relationship between obesity and cancer has been investigated, highlighting the biological mechanisms at the basis of this link. Finally, strategies to remodel fat, including browning and lifestyle interventions, have been taken into consideration as a major perspective to limit excess body weight and tumor onset.
1. Obesity and Cancer
Overweight and obesity are defined by an abnormal and excessive fat accumulation that develop when caloric intake by meals exceeds energy expenditure from physical activity and cells metabolism. In clinical practice, obesity is commonly assessed by expressing body weight as a function of height, according to World Health Organization (WHO) classification of Body Max Index (BMI). BMI between 25 kg/m2 and 29.9 kg/m2 is categorized as overweight, whereas greater values define obesity condition [1]. According to data published by the WHO, 39% of adults were overweight and 13% were obese in 2016 [2]. Consequently, being obese is a well-known risk factor for cardiovascular and metabolic diseases, the burden of obesity in health systems is becoming more relevant every day. Moreover, metabolic dysregulations associated to obesity and, more specifically, to visceral adiposity has been found to play a crucial role in tumor biology, affecting cells proliferation and spreading, as well as response to therapy [3], and therefore are studied as cancer-related conditions in term of risk, prognosis and mortality [4].
1.1. Incidence: When Obesity Promotes Cancer
In 2012, about 3.6% of all new cancer cases in adults were associated with high BMI assessed ten years before cancer diagnosis. Corpus uteri, postmenopausal breast and colon cancers accounted for 63.6% of tumors linked to high BMI [5]. Renehan et al. [6] systematically reported that a 5 kg/m2 increase in BMI can be considered as a risk factor for several solid and non-solid cancer (cancer of thyroid, kidney and colon, esophageal adenocarcinoma, multiple myeloma, leukemia and non-Hodgkin lymphoma) in both sexes. In men, this increment in BMI has been associated with a greater risk also for rectal cancer and melanoma, whereas in women it raises the possibility of developing gallbladder, pancreatic, endometrium neoplasms, and breast cancers, especially in post-menopausal patients. Gender difference was nevertheless significant only in colorectal cancer. This finding is supported by a Mendelian randomization study [7] on the association between BMI and colorectal cancer incidence: risk was 22% higher among men per 4.2 kg/m2 increase in BMI and 9% higher among women per 5.2 kg/m2. In 2016, the International Agency for Research on Cancer (IARC) Working Group added hepatocellular carcinoma, meningioma, gastric cardia and ovarian cancers to the aforementioned obesity-associated tumors [8]. Obesity has a potential role in the etiology of gastric and esophageal cancers [9], and abdominal obesity has been associated with an increased risk of breast cancer [10] (Table 1).
It is noteworthy that BMI-cancer association seems to be specific only for some histological types of tumors considering a specific site [11]. For instance, obesity is more strongly associated with type I endometrial cancer than type II [12], to papillary subtype of thyroid carcinoma [13], cardia gastric cancer [14], and esophagus adenocarcinoma [15] among other histological types.
However, several studies depicted and inverse obesity-cancer association for lung cancer, a phenomenon called “obesity paradox”. Importantly, the misinterpretation that obesity might be protective against cancer has to be avoided, since potential confounders such as smoking habit and its influence on body weight [16], in addition to possible methodological flaws [17], must be taken into consideration when we are discussing the obesity paradox.
Albeit increased BMI is the most common studied obesity-linked condition, other biomarkers have been detected as hallmarks of an increased susceptibility to cancer. BMI and fasting insulin have both been identified as risk factors for breast cancer and are associated with late-stage disease and poor prognosis [18]. Moreover, a higher serum level of C-peptide, a measure of insulin secretion usually elevated in insulin resistance status, has been indicated as a risk factor for colorectal cancer development [19].
Finally, the hormonal changes that also occur in obesity can be associated with cancer onset. In this view, particular emphasis has been put on estrogens and menopausal status since they are strictly related with higher incidence of female specific cancers. Post-menopausal status in obese women increases risk of developing breast cancer, whereas ovarian cancer incidence is greater in pre-menopausal ones [15]. In addition, menopausal hormone replacement therapy positively affects the association between BMI and post-menopausal breast and endometrial cancer [11] while it reduces colorectal cancer rates by 37% [20].
1.2. Obesity and Cancer: Prognosis and Mortality
Although many efforts have been made in the past, identifying prognostic factors in cancer is still an important goal of ongoing studies. BMI, body weight, adult weight gain, and fat distribution, separately or taken in combination, may be considered as risk factors for poor cancer prognosis [21]. Obesity affects the development of aggressive and nonaggressive cancer differently; for instance, BMI ≥30 kg/m2 has been associated with a 29% increased risk of high-grade cancer in prostate [22]. In 2010, 3.9% of cancer mortality was linked to high BMI measured ten years earlier [23], and 15% to 20% of deaths in cancer patients have been related to overweight and obesity conditions [24]. Albeit outside of breast cancer the relationship between BMI and cancer survival is not so consistent [25], probably because of remarkable variations among studies, emerging data suggest that obesity can also represent an increased risk of cancer recurrence [24].
Calle et al. [26] studied the association between overweight and cancer-related deaths during 16 years of follow up, in a cohort of more than 900,000 U.S. adults who were free of cancer at the enrolment. The study revealed that the risk of dying of cancer was 50% to 62% higher in people with highest BMI. Obese male subjects had elevated mortality rates of liver cancer (the highest one), stomach cancer, non-Hodgkin’s lymphoma, and prostate cancer. On the contrary, in female subjects a positive association between weight and cancer death was detected for uterus, cervix, breast, liver, and ovarian cancers. Both high BMI men and women showed high mortality rates for cancers affecting the gastrointestinal system (pancreas, esophagus, gallbladder, colon, and rectum) as well as kidney cancer and multiple myeloma (Figure 1). By contrast, obesity displayed no significant associations with mortality due to brain cancer, bladder cancer, and melanoma in both sexes. In addition to this direct obesity-mortality association, a large amount of epidemiological evidence suggests a close association between obesity comorbidities, such as cardiovascular diseases and diabetes, and non-cancer related deaths in cancer survivors [27]. These observations highlight that the importance of weight management and healthy lifestyle not only prevent cancer, but also prolong the disease-free survival state.
Particularly in a large population study, diabetes was associated with increased hazard-ratio for cancer specific death [28]. Notably, insulin resistance in women affected by breast cancer displayed a two-fold increased risk of distance recurrence and a three-fold decreased survival [29], to the point that C-peptide has been proposed as prognostic factor in breast cancer [30].
Furthermore, the metabolic syndrome [31], a well-known obesity associated condition [32], increases the risk of developing [33] and having relapse in colorectal cancer [34,35].
Finally, obesity can interfere with therapies and contributes to morbidity from chemotherapy toxicities, thus promoting worst prognosis and mortality [27]. On the other hand, physicians usually under-dose cancer therapies when doses—calculated according to body surface area—seem to be too high [36], despite that no evidence has found for this practice of dose-capping [37]. Furthermore, obese patients undergoing oncologic surgery usually required longer operative and anesthetic times [36] and, on account of technical challenges, are offered open surgery more frequently than laparoscopic, although the latter has a better overall survival rate [38]. Finally, obesity represents a risk factor for poor wound healing, postoperative infections, and long-term surgical complications, like lymphedema [27].
2. Obesity and Cancer: Deepening the Relationship
The complexity of epidemiological evidence about the relationship between obesity and cancer reflects the wide interplay among metabolic pathways involved in cancer development, progression, and pharmacological responsiveness. To date, a plethora of circulating factors and molecular alterations, whereby obesity can lead to carcinogenesis, have been detected. Despite this, primarily relevant mechanisms are not well understood. Here, we provide an overview of the principal identified factors that link altered metabolism to tumor onset and progression, focusing on three typical and deeply interplaying cornerstones of obesity which are also possible early diagnostic biomarkers and strong predictors of cancer risk: adiposity, chronic low-grade inflammation, and hyperinsulinemia (Figure 2).
2.1. Adiposity
In humans, two main kinds of adipose tissue (AT) exist: white (WAT) and brown (BAT). Recently, the existence of a third kind has been proposed, i.e., “brite” or “beige” AT, described later in this manuscript [39]. BAT is mainly located in interscapular, perirenal, and deep neck regions of newborns [40]. In human adults, BAT is only present in particular conditions, such as the exposition to low temperature. Indeed, the main function of BAT is the thermogenesis—the dissipation of energy to produce heat. Differently from BAT, WAT is located in subcutaneous and visceral depots, and is present all lifelong. The primary role of WAT is storing the excessive amount of energy disposal as fat, mainly derived from serum triglycerides (TAG) and de novo lipogenesis (DNL) from exceeding carbohydrates or other non-fat precursors. Indeed, a low-fat, high-carbohydrate diet increases DNL in liver [41] and in WAT [42]. Notably, DNL has been found to be exacerbated in several cancer tissues and correlates with poorer prognosis [43]. Fatty acid synthase (FASN), the key enzyme of DNL process, is overexpressed and hyperactivated in cancers with a high risk of disease recurrence and death [44]. This observation shed a light on cancer metabolism, revealing that exacerbated lipogenesis and FFAs metabolism are crucial for cancer onset and progression, along with the well-established glycolytic and glutaminergic pathways [45,46].
By functioning as a fat depot, WAT prevents lipotoxicity, a detrimental process determined by excess of FFAs that can alter cell membrane structure and functions, creating an inflammatory environment, which finally leads to cell dysfunction and death. In obesity state, lipotoxicity has been found to contribute to increase macrophage infiltration into AT, insulin resistance, and hepatic steatosis [47].
During prolonged calorie abundance, adipocytes hyperplasia (recruitment of preadipocytes and differentiation to mature cells) and hypertropia (enlargement of existing cells) occur in healthy subjects to store the increase amount of fat. The differentiation process from mesenchymal stem cells to mature adipocytes is called adipogenesis and is elegantly regulated by transcription factors, among which peroxisome proliferator-activated receptor γ (PPARγ) is the most critical for the process. Notably, PPARγ is the target of new and highly efficacious class of insulin-sensitizing drugs known as thiazolidinediones [48]. The adipocytes commitment and recruitment in subcutaneous adipose tissue (SAT), the largest WAT depot, avoid fat accumulation in ectopic tissues (i.e., liver where fat leads to non-alcoholic fatty liver disease (NAFLD), heart and muscles), as well as in visceral adipose tissue (VAT), which is mainly distributed in omental (hangs off the stomach), mesenteric (associated with the intestine), retroperitoneal, gonadal, mammary, and pericardial depots. When SAT maximum expansibility (maximum hypertrophy of pre-existing adipocytes and failure in adipogenesis) is achieved, calories surplus, due to excess energy intake and/or reduced energy expenditure, accumulates as fat in VAT, leading to its expansion. A marker of this ectopic fat accumulation in humans is the increased abdominal obesity [4]. Men are more prone to visceral adiposity with respect to women, who tend to accumulate fat in the lower part of the body [49]. The different distribution of fat between the two sexes may explain the diverse metabolic risk, higher in men, [50] and gender incidence in some types of cancer [51]. Differently from the SAT, VAT is closer to internal organs and directly connected to portal vein [52]; it is more metabolically active [53] and produces more adipokines [54]. Importantly, behind its well-known associations to glucose intolerance, dyslipidemia, and hypertension [55], VAT disproportionate expansion is deleterious because its amount positively correlates with tumorigenesis. For instance, omental adipocytes displayed significant more efficiency in promoting invasion of ovarian cancer cells than subcutaneous ones [56]. VAT accumulation and central (not general) obesity are specifically associated to cancer risk and prognosis through hyperinsulinemia [57,58], whose pathogenesis could reside in adipogenesis dysregulation which occurs when excessive AT develops [59]. Indeed, the dysfunction and enlargement of adipocytes, the decrease expression of lipogenic genes, and the expansion of VAT are all potential mechanisms underlying the obesity-related insulin resistance [60]. Additionally, the excess of adiposity and the dysregulation in adipogenesis lead to adipocytes hypoperfusion and hypoxia, which explain the impaired production of adipocytokines and metabolic syndrome development in obesity [41]. Moreover, hypoxia increases adipocytes death, leading to a persistent state of damage that enhances macrophages infiltration and causes low grade chronic inflammation [61], which is an hallmark of obesity [62]. Nevertheless, WAT is more than a fat depot, since it provides paracrine signals, secretes adipokines as a large endocrine organ, and is involved in insulin sensitivity and thermogenesis.
2.2. Adipokines and Chronic Low-Grade Inflammation
Chronic inflammation is a cornerstone in both obesity and tumorigenesis [63,64], thereby dysfunction in adipokines production represents a possible biological explanation for cancer promotion in obesity [65]. Adipokines [e.g., adiponectin and leptin, Interleukin-6 (IL-6) and Tumor Necrosis Factor-α (TNF-α)] are biologically active polypeptides secreted by WAT. Upon being released in the bloodstream by visceral fat, adiponectin promotes fatty acid oxidation and protects against insulin resistance [66], since it decreases glucose and insulin levels by increasing tissues insulin sensitivity and glucose uptake in feeding conditions. Also, adiponectin plays a pivotal role in regulation of endothelial function [67] and tumor growth [68]. Furthermore, adiponectin is important in preventing inflammation [69], as demonstrated by the increased association with pro-inflammatory effects due to enhance IL-6 and TNF-α production in hypo-adiponectinemia [70]. Notably, adiponectin serum level inversely correlates with BMI, probably because insulin resistance status, associated to AT inflammation [71], reduces its release. According to recent meta-analysis, adiponectin levels are inversely associated to breast [72] and colorectal [73] cancers. Contrarily to adiponectin, circulating level of leptin displays linear positive correlation with BMI, fat cell volume, and plasma insulin [74], which positively regulates leptin genes expression [57]. Besides its well-known action as anorexigenic hormone, leptin stimulates pro-inflammatory cytokines production [75], promotes angiogenesis [76] and acts as a growth factor in tumorigenesis, thus contributing to development of more aggressive cancers [77]. Moreover, leptin plays a key role in breast cancer development and treatment response, particularly in obese women [78]. However, leptin does not affect metastatic spread so its prognostic effect in cancer is not so consistent [79]. Also IL-6, which positively correlates with body mass and FFAs concentration [80], has been proposed as a prognostic factor in breast cancer, since it promotes cell migration and the increase of aromatase activity [81]. Overproduction of IL-6 and TNF-α in obesity is mainly due to the augmented infiltration of macrophages [i.e., Adipose tissue macrophages (ATM)] within WAT [82]. Both M1 and M2 infiltrating macrophages are present in fat: M2 macrophages display anti-inflammatory properties, whereas M1 macrophages enhance inflammation. Diet-induced obesity leads to a shift from a M2-polarized state to a M1 proinflammatory state, thus highlighting once more the close association between accumulation of fat and inflammation [83]. Importantly, increased IL-6 levels have been found also in subjects fed with high-sugar, high-fat and pro-inflammatory diet [84], opening new perspectives about dietary ability in modulating inflammation. Given that IL-6 and soluble tumor necrosis factor receptor 2 (sTNF-R2) together may be useful markers to predict cancer development [85], these findings corroborate the evidence that chronic low-grade inflammation related to central obesity increases risk of malignancy.
Beside this, it is fundamental to underscore that macrophages implicated in tumorigenesis are triggered by chronic inflammation in AT and act by different pathways from tumor-associated macrophages (TAMs), which instead are responsible for progression and metastasis via their interplay with tumoral microenvironment [86]. The paracrine and autocrine mechanism, whereby chemokines sustain the growth and development of the tumor, has been shown in prostatic cancer [87] and remarks the microenvironment involvement in tumorigenesis [88]. In the contest of this pro-tumor environment, adipocytes possess a pivotal role. For instance, mammary AT influences breast cancer promotion and invasion because fat tissue surrounding cancer possesses high levels of aromatase enzyme activity [89].
2.3. Hyperinsulinemia
In healthy conditions, insulin promotes fat storing in WAT by enhancing adipogenesis, stimulating glucose cells uptake and lipogenesis, while inhibiting lipolysis. Insulin also increases the uptake of fatty acids derived from circulating lipoproteins [90]. Obesity causes insulin resistance and chronic hyperinsulinemia, which seem to be critical in the association between cancer and adiposity [91]. Chronic hyperinsulinemia might lead to neoplasm development directly acting as a mitogenic factor itself [92], but also through its deep interplay with sex hormones and other growth factors pathways, e.g., insulin growth factor 1 (IGF-1) [93]. IGF-1 is a master regulator of cell proliferation, differentiation, and apoptosis [94], being pro-mitotic and anti-apoptotic [95]. IGF-1 bioavailability depends on IGF-binding protein-1 (IGFBP-1), to which it is bound to circulate in blood. Consequently, alterations in IGF-1 and IGFBP-1 levels affect the balance between cell proliferation and apoptosis, finally leading to tumor development. The majority of the circulating IGF-1 production take place in the liver, and it is regulated by the Growth Hormone (GH). Of note, GH is also likely to promote adipogenesis [96]. Although GH levels are reduced in obesity, hyperinsulinemia raises density and sensitivity of hepatic GH-receptors in diabetic and obese patients [97], leading to overproduction of IGF-1 [98] and increasing its availability by decreasing IGFBP-1 levels [99]. Thus, tumor growth is enhanced in response to increased signaling by insulin and IGF-1, conferring selective advantage to cancer cells, especially in hyperinsulinemia associated conditions. Insulin receptors are overexpressed in malignant epithelial cells of breast carcinoma [92] and IGF-1 receptors are overexpressed in many tumor cell types [19,95]. Notably, IGF-1 circulating level are positively associated with an increased risk of colorectal and prostate cancers, as well as with premenopausal breast cancer, although the strength of these associations varies by cancer site [100,101,102]. However, it is important to note that in some tissues IGF-1 synthesis is regulated by other hormones apart from GH [95]. Particularly, the estrogens are involved in IGF-1 signaling by increasing number and affinity of IGF1-receptors and decreasing IGFBP-1 production. Consequently, similarly to insulin, the estrogens promote cell proliferation and inhibit apoptosis. The ovaries are the major source of estrogens in premenopausal women. Instead, in post menopause subjects, estrogens mainly derive from the peripheral conversion of adrenal androgens via adipose aromatase, whose levels depend on fat amount as well as on TNF-α and IL-6 stimulation [103]. Notably, the peripheral conversion of androstenedione to oestrone observed in obese subjects is not the only responsible for the increase estrogens bioavailability. Indeed, insulin also downregulates sex-hormone-binding globulin (SHBG) hepatic production, thus increasing the estrogen activity [98]. Therefore, obese subjects and post-menopausal women are characterized by higher levels of circulating estrogens than normal individuals. In men, obesity-associated low levels of testosterone and SHBG, coupled with increased estrogens, have been proposed to be with high-grade prostate cancer [22]. Similarly, since in post-menopause ovarian sex hormones synthesis is suppressed, increased peripheral estrogens production in obese women is the main proposed explanation for higher breast cancer incidence [104]. As a consequence of high estrogen levels, aromatase inhibitors in chemotherapy may be less effective when administrated at normal doses in obese patients, thus affecting the prognosis of breast cancer [105]. Contrarily, progesterone opposes estrogen-related actions, reducing inflammation, enhancing apoptosis and cell differentiation, and increasing IGFBP-1 production. Differently from estrogens, progesterone levels are reduced in obese individuals. Therefore, an “unopposed estrogen” action may explain why cancer risk is increased also in pre-menopause obese women, as it occurred for endometrial cancer [106].
3. Perspectives into Visceral Adiposity and Cancer
Since obesity affects cancer incidence and prognosis, accurately identifying subjects with higher risk via both anthropometric and metabolic assessment is overriding. In this view, it could be intriguing to study if central obesity per se is sufficient to increase cancer risk also in non-metabolic patients. Indeed, regardless of adiposity, a high cancer risk can be determined by insulin resistance [57], dyslipidemia [107], and predicted basal metabolic rate [108], being indicative of the whole-body energy metabolism. Therefore, characterizing the obese state by systemic and tissue-specific measures could provide a more reliable identification of high-risk populations and represents a fascinating approach for the development of preventive and therapeutic strategies [109]. Additionally, the right anthropometric assessment may be crucial to better identify subjects at high risk of developing cancer among apparently healthy individuals just before they turn into patients. Strategies aimed at counteracting obesity epidemic represent a way of primary cancer prevention. Moreover, they may be a viable complementary option to improve prognosis in addition to canonic and unavoidable pharmacological treatments. Overall, under the assumption that the amount of WAT increases the risk of cancer and affects prognosis and mortality rates, reducing fat mass and enhancing energy expenditure may represent a fundamental non-pharmacological way to prevent cancer and improve survival.
3.1. Anthropometric Assessment: How to Identify Visceral Obesity
Since visceral fat is crucial in the obesity-cancer association, an ideal assessment of adiposity would consider both the amount and the site of deposition of adipose tissue. For instance, people who carry most of the fatty tissue in the abdominal region is 70% more prone to develop pancreatic cancer compared to those who bear it around the hips [110].
Computed tomography (CT) and magnetic resonance imaging (MRI) can accurately distinguish between SAT and VAT, assessing adiposity with a single image slice at pelvic level [111]. Unfortunately, they are expensive and complex instruments to be used routinely in clinical practice and cannot be easily applied to large datasets because of a time requirement and human resources [112]. To overcome these limitations, deep learning systems have been developing to reach a fully automated segmentation method [113], but the fascinating field of innovative work being done in this area, with technology advancing quickly to improve body compartment estimates, is beyond the aim of this review.
On the other hand, using BMI as a metric of adiposity in adults may lead to some methodological bias. Firstly, BMI is only a ratio of weight to height and does not distinguish fat mass from lean body mass or among SAT and VAT, nor apple- from pear-shaped body. Secondly, the timing and method of BMI assessment seem to be critical [114,115]. When BMI assessment is not anthropometrically measured, it is usually self-reported by patients who tend to tell lower weight and higher height [116]. Moreover, lower BMI values should not be considered healthy when due to unintentional weight loss during cancer development, cachexia, and undernutrition resulting from chemotherapy-induced nausea and vomiting [114]. Indeed, in some circumstances weight loss may be a predictor of poor survival as for colon cancer [115]. Other anthropometric measures may instead better reflect adiposity and could be useful in clinical practice to individuate patients with higher risk of cancer. For instance, Waist Circumference (WC) is measured horizontally midway in the distance of the superior iliac crest and the lower margin of the last rib, so far it better reflects deep visceral adiposity [117]. Indeed, according to International Diabetes Federation definition, essential criteria to diagnose Metabolic Syndrome is adiposity assessed by WC [31]. Furthermore, WC is a sensitive predictor for the risk of obesity-related cancer [6], since it has been associated with increased incidence in endometrial [106] and colon [118] cancers, without the gender difference highlighted when adiposity was assessed by BMI [119]. In the future, it may be intriguing to assess the risk of cancer in patients with increased WC who are not classified as obese according to their BMI [120]. A study on 1,564,218 participants showed that high WC and high Waist-hip ratio (WHR) correlate with increased pancreatic ductal adenocarcinoma mortality, independently from BMI [121]. WHR is the ratio between WC and hip circumference. Adding WHR to a multivariable model increases the diagnostic accuracy for detecting prostate cancer [122]. Moreover, WHR has been associated with the worst subtypes of breast cancer [123]. Also, high WHR and WC, rather than general obesity measured by BMI, were associated with an increased risk of developing pancreatic cancer [124] in a large prospective study. This different accuracy in detecting cancer risk according to anthropometric assessment is additionally highlighted by evidence about colorectal cancer risk: while the association of BMI is stronger in men than in women, WHR raises the risk in both sexes [118]. Lastly, a recent prospective study concluded that body shape, assessed by WC, WHR, and A Body Shape Index (ABSI), is positively associated with lung cancer risk [125], reverting the aforementioned obesity paradox according to which higher BMI may be protective. Additionally, adult weight variations have been studied as a better metric. Adult weight gain is a dynamic measure and reflects better than BMI continuous and cumulative influence on carcinogenesis that excessive adiposity may exert. A meta-analysis by Keum et al. [126] concluded that each 5 kg increase in weight is associated with an approximately 11% increased risk of breast cancer, 13% for ovarian cancer, and 39% for endometrial cancer, among non-HRT users post-menopausal women, and increases the risk of colon and kidney cancers as well.
3.2. Browning of Adipose Tissue: Remodeling Fat Amount
Brown adipocytes differ from white ones since they contain multilocular lipid droplets, more abundant cristae-rich mitochondria, and higher levels of uncoupling protein-1 (UCP-1), which reduces the proton gradient across inner mitochondrial membrane producing heat rather than producing ATP, as instead occurs in white adipocytes. In this manner, fatty acids and glucose are burned as substrates in BAT through a process known as adaptive non-shivering thermogenesis [127], which occurs under specific stimuli and significantly contributes to energy expenditure [128]. Initially identified only in newborns, recent studies have shown that a tissue with cold-induced activity is still present in adults [129] and its amount is inversely correlated with BMI [48], percentage of body fat and visceral fat [130]. In overweight or obese subjects this tissue is not only reduced, but is also less metabolically active [131]. It remains unclear if this reduced activity is a predisposing factor for obesity or an adaptative trait since weight loss in obese subjects has been associated with BAT recruitment [132]. Further studies have identified this cold-induced tissue be made of brite adipocytes [133]. As suggested by the name “brite”, literally brown-in-white, these cells share some characteristics of both brown and white adipocytes. Indeed, brite adipocytes are typical white adipocytes residing in WAT that, upon specific stimuli, switch their phenotype into brown-like adipocytes, in a process called browning [134]. After browning, brite adipocytes express thermogenic genes, specifically UCP-1, and increase their mitochondrial content, although they never reach the thermogenic capability of classical brown adipocytes [135]. It has not yet been fully elucidated if brite adipocytes derive from de novo differentiation of precursor cells or from trans-differentiation of mature white adipocytes [136], but probably both mechanisms may contribute to brite fat biogenesis [137], since roughly 40% of pre-adipocytes isolated in SAT has brite cells features [138]. Brown adipocytes consume glucose [131] and uptake FFAs from circulating lipoproteins [139]. Thus, increasing the amount of brite cells and upregulating pre-existing BAT not just increase energy expenditure [140], but may also improve glucose tolerance [141] and dyslipidemia, finally contributing to adiposity control [130]. Notably, also adipokines are involved in BAT metabolism. Leptin increases UCP-1 expression, and therefore may activate thermogenesis and stimulate fat oxidation [142]. Moreover, acting together with insulin on hypothalamic POMC neurons, leptin promotes WAT browning [143]. Conversely, adiponectin [144] and TNF-α [145] reduce BAT activity.
It is important to note that the recruitment of brite adipocytes from WAT reduces the net number of white adipocytes and, in conclusion, the fat amount. Indeed, mice with increased brite fat mass gain significantly less body weight and adiposity when fed with a high fat diet [146]. Since just visceral adiposity has been related to more metabolically active BAT [130] and given that is possible to activate BAT and pre-brite cells in obese people [131], pharmacological and non-pharmacological methods to stimulate browning [147] have been studied as possible strategies to reduce fat amount and potentially cancer risk. To achieve this goal, acting on physiological stimuli of browning may represent a powerful approach. For instance, stimulating adaptive thermogenesis in response to high calorie or high fat diets [148] or cold exposure might be a way to prevent or treat obesity [149]. As adrenergic stimulation by epinephrine and norepinephrine [150] have been detected among cold-induced pathways, also beta-3 adrenergic agonists, as mirabegron, can potentially enhance browning [151]. Furthermore, physical exercise, through irisin production, has been proposed as a possible way to increase BAT thermogenesis and browning [152]. Particularly, irisin induction as a consequence of PPAR-α activation likely represent the way by which fenofibrate induces browning in subcutaneous WAT [153]. Also PPAR- γ induction by rosiglitazone is potentially involved in browning [154], while liraglutide leads to body weight loss by activating BAT thermogenesis and WAT browning, acting on hypothalamic GLP-1 receptors, independently from food intake [155]. Among nutrients, fish oil intake has been proposed to increase thermogenesis-associated genes expression [156], while beige remodeling of SAT may be one of factors conferring beneficial anti-obesity effects to resveratrol [157]. Lastly, the discovery that depletion [158] or transplantation of every-other-day fasting treated mice microbiota [159] enhances browning of WAT, thus shedding a light on a new potential role of diet in shaping microbiota to reduce adiposity.
3.3. Lifestyle Interventions—Let Food Be Your Medicine
Since it is undoubtedly clear that obesity is one modifiable risk factor for tumorigenesis, lifestyle interventions addressed to reduce adiposity, including dietary changes and physical activity, play a pivotal role in cancer prevention [160]. For instance, pancreatic cancer risk was inversely associated with a healthy lifestyle assessed considering diet quality, physical activity, smoking status, alcohol consumption and anthropometry [161].
Intensive lifestyle intervention (diet modification and physical activity) also improves glycemic control and leads to weight loss [162] and to a significative reduction of obesity-related cancer risk [163].
Physical activity reduces body fat [164], and regular moderate exercise, even with no dietary intervention, reduced preferentially visceral fat in nonobese healthy women [165]. Additionally endurance training induced fat loss, particularly VAT, in obese women with MS, also reducing BMI and WC [166,167]. These findings are consistent with a meta-analysis by Vissers et al. [168] and with the results of a study in rats fed with a high-fat diet in which the beneficial effects of exercise training were more pronounced on VAT than on SAT [169]. Furthermore, physical activity decreases appetite, particularly in obese individuals, and ameliorates lipid profile since it increases HDL cholesterol and reduces TAG blood levels [170].
Consequently, thanks to its role in reducing VAT, physical activity should be recommended not only in preventing cancer, but also as a complementary approach to reduce adverse events and ameliorate pharmacological strategies in survivors [171].
Of note, not only weight loss per se, but also the kind, the amount and the timing of meals influence those pathogenetic mechanisms that are at the basis of the close connection between cancer and obesity. The European Prospective Investigation into Cancer and Nutrition (EPIC) study, a multicentre prospective cohort study with the aim of investigating the relationship between nutrition and cancer, has assumed a paramount importance in this research area. The World Cancer Research Fund/American Institute for Cancer Research (WCRF/AICR) [164] strongly recommends being a healthy weight, physically active, eat a diet rich in wholegrains, vegetables, fruit, and beans, limiting consumption of red and processed meat, sugar sweetened drinks, and alcohol. Although these recommendations are addressed to single individuals, policymakers should encourage and sensibilize about healthy behaviors and also provide economic means to give everyone access to healthy food, in consideration of the relative low cost of unhealthy foods. [172].
Although some foods (e.g., allium and broccoli) and micronutrients (e.g., selenium, vitamin D, carotenoids) have been studied for their anti-cancer properties [173], it has emerged that studying dietary patterns as a whole is more beneficial than considering single nutrients. Among various dietary patterns, the traditional Mediterranean diet (MD) seems to produce substantial health benefits [174]. The core of this diet is mainly vegetarian, lower in meat and dairy products, with moderate alcohol consumption, mainly in the form of wine. However, MD is not only a combination of foods, but a regular lifestyle and a traditional way of interacting with environment.
Analyzing MD composition led to consider the importance of fat total intake. It has been demonstrated that a low-fat dietary intervention significantly reduces cancer incidence, as seen in pancreas [175], whereas high fat diet (60% of total energy intake derived from fat) may increase cancer risk [176]. Paradoxically, MD displays a high fat content (30–40% of total energy intake), but with a higher percentage of unsaturated than saturated FAs, given that extra-virgin olive oil is the major source of lipid in this diet [177]. The ratio between unsaturated and saturated FAs is important to determine the effect of this lipid source. On molecular level, high content of saturated FAs is more prone to create an inflammatory environment and perpetrate damage to cell membranes. This is consistent with the findings that extra-virgin olive oil exerts beneficial effects on chronic inflammatory disorders that may eventually lead to cancer [178]. Moreover, a high-unsaturated FAs diet improves adiponectin levels [179], thus limiting the detrimental process. Furthermore, it has been observed that replacing lard (rich in saturated FAs) with soybean oil (high content of unsaturated FAs) in high-fat diet alleviates obesity-related inflammation and insulin resistance by reducing macrophage infiltration into AT [180].
As discussed before, chronic inflammation, insulin resistance, and dysregulation in adiponectin levels characterize obesity, and specifically are associated with VAT. By eliciting the reshape of VAT, MD may prevent these metabolic alterations, therefore limiting the metabolic disorders and associated diseases.
Indeed, MD has been associated with a significant reduction in central obesity [181] and people with greater adherence to MD showed significantly lower WC [182]. Of note, this inverse association has been attributed to VAT and not to SAT [183]. Furthermore, some RCTs studied the MD effects for 3 months [184], 1-year [185], and 2-years [186] interventions in obese and overweight patients showing reduction in body weight, BMI, WC, and body fat. These effects are probably more pronounced in long-term interventions. Indeed, a Spanish RCT showed that a long-term high-vegetable-fat MD, even with an unrestricted-calorie, was associated with less gain in central adiposity compared with a control diet. [187]. Although further studies are needed to clarify if the effects of a MD pattern on obesity are specifically due to reduction of visceral fat, since VAT is highly associated with most of the metabolic effects of obesity [188] and obesity represents a risk factor for cancer, we speculate that the MD anticancer role may be related to its beneficial effects on visceral adiposity.
Several evidences highlight that MD reduces overall risk of cancer, in a dose-response manner [189]. More specifically, MD has been associated with reduced incidence of colorectal [190], gastric [191], high aggressive prostate [192] and breast cancer [193], and particularly receptor negative breast cancer in post-menopausal women [194]. Moreover, MD has been favorably associated with reduced cancer mortality [195]. In this contest, there are two hurdles to overcome. Firstly, these evidences are mainly based on many prospective and fewer interventional studies. Secondly, there is no unique index to assess adherence to MD regimen. Furthermore, it rests unclear which component of the MD mostly contributes to its beneficial effects, although the benefits of MD are mainly driven by higher intake of fruits, vegetables, and whole grain [196]. For instance, olive oil polyphenols, red wine resveratrol, and tomato lycopene are able to reduce colorectal cancer initiation and progression [197]. Moreover, high dietary fiber intake, as consequent of whole-cereals meals, a cornerstone of MD, is inversely associated with colorectal [198], liver and stomach cancer [199].
One of the proposed mechanisms by which MD displays a protective role against cancer relies on its antioxidant effects, which mainly reduce DNA and molecular damages that are implicated in tumorigenesis [200]. For instance, the antioxidant phenolic compounds present in olive oil are powerful inhibitors of free radical generation [201]. Moreover, MD displays anti-inflammatory and anti-aggregating MD properties [202], thus contraposing to Western-type diet, rich in red meat, high saturated-fat dairy products and refined grains, which has been related to increased inflammatory markers [84]. In the future, the identification of the metabolites influenced by a given nutritional intervention and the characterization of the complex metabolic effects of nutrients or foods through metabolomics may be of substantial help to identify novel risk factor for tumor [203], as it has occurred for prostate cancer [204].
Dietary characteristics of MD may also explain its protective role described in diabetes [205]. Indeed, a typical MD composition improves insulin sensitivity in patients without preexisting diabetes and this effect is probably linked to the increased amount of unsaturated FAs intake [179]. Therefore, given that MS, hyperinsulinemia, and obesity are risk factors for tumor initiation and progression, and since the MD-style pattern reduces the development of MS and central obesity by acting on lipids levels and glucose metabolism, it is reliable argument that MD may be protective against cancer onset and growth [182,206].
Lastly, MD features are associated with positive modulation in microbiome [207]. Gut microbiota has nowadays been identified as a true organ, hidden in the host, which dynamically responds to environmental factors, among which the diet plays an essential role. Dietary contents modulates microbiome composition [208], which in turn influences nutrients absorption, impairs host energy homeostasis regulating lipid and glucose metabolism, and provides immunomodulatory effects, through a crosstalk with the host [209,210]. Dysbiosis of the commensal microbiota is implicated in the pathogenesis of several diseases, including obesity and cancer, probably by chronic inflammation [211]. A possible explanation for this low-grade chronic inflammation also comes from animal models of diet-induced obesity in which dysbiosis increased gut permeability letting a low grade metabolic endotoxemia [212]. Notably, dysbiosis has been associated with colorectal cancer and with tumors arising in organs distant from the gut. Therefore, it is easy to infer that by modifying microbiome, MD intervention would be beneficial to counteract cancer progression.
Altogether, it appears quite clear the key role played by dietary interventions, not only in reducing adiposity per se, but also in reverting the inflammation and metabolic impairment that connects obesity to cancer. Nonetheless, in addition to the source and the properties of some food groups, and to their combination in dietary-pattern, the timing of meals and the length of fasting and feeding periods have also been studied as modulators of metabolic pathways and oxidative-stress. These hypotheses take origins from the findings that calorie restriction (CR), a reduction in energy daily intake with maintained number and frequency of meals, delays age-related pathologies and increases lifespan. Intriguingly, the implementation of a CR approach in mice has been shown to stimulate browning in VAT, resulting in loss of weight and visceral adiposity mainly due to a decrease of adipocytes size, which dampens the inflammatory processes overall [213,214]. By contrast, a CR approach for 8 weeks in obese individuals results in a negative regulation of browning in SAT [215].
In general, the weight decrease observed in overweight subjects after a CR regimen is associated with a great extent to VAT reduction rather than the SAT one [216,217]. This correlated also with a decrease of some biochemical parameters, including total cholesterol, triglycerides, and fasting glycaemia [216]. In line with this, two different studies on obese individuals underwent CR diet for 14 days demonstrated a significant reduction of the VAT, but not the SAT, probably due to a dissimilar responsiveness and physiological characteristic of the adipose cells in the two compartments: differently from the SAT, VAT cells display induction of lipid metabolism related genes in response to fasting [218,219]. Interestingly, when the effect of CR on body fat distribution was investigated independently or in combination with regular exercise, it emerged that the body weight reduction and the VAT loss were not statistically different between the different groups [220,221], therefore highlighting the cruciality of CR in the reduction of visceral adiposity.
By boosting the regenerative capacity of stem cells as seen in multiple rodent tissues [222], decreasing metabolic rate and oxidative damage [223], CR plays also a protective role in tumorigenesis [224]. In rat models of colon cancer, CR decreased leptin level and concomitantly reduced tumor growth [83]. Unfortunately, a long-term CR regimen is difficultly feasible in clinical practice, because of adverse events. Consequently, other strategies have been studied to achieve the same goals with a better profile in patient safety and adherence. In this contest, starvation has been shown to promote stress resistance and longevity in mice and humans [225]. One general mechanism of action of fasting is that it triggers adaptative cellular stress response, which results in a major efficiency in counteracting diseases; in addition, among the major effects of fasting relevant to aging and disease there is decrease in IGF-1, IGFBP-1, glucose, and insulin levels. Different methods of fasting (Table 2) have been pinpointed to decrease weight, delay aging, reduce tumorigenesis, and protect mice from chemotherapy drugs when fasting cycles were associated with treatments [226].
Moreover, in humans, fasting seems to reduce common chemotherapy associated side effects [227]. Intermitting fasting (IF) usually refers to a water-only or very low-calorie period lasting less than 24 h, followed by a normal feeding period of 1 or 2 days. In animal models, IF ameliorates lifespan and mitigates a wide range of chronic diseases, including obesity, insulin resistance, diabetes, and cancer, through inducing a metabolic switching and improving cellular stress resistance [228]. Every other day feeding (EODF) [229] is a kind of IF program that extends lifespan, stimulates browning, and reduces obesity through its interactions with gut microbiota, as mentioned before [159]. Time-restricted feeding (TRF) requires restricting the timing of meals, without regard to their caloric content, to a time window of few hours. Studies on TRF have also highlighted the importance of respecting circadian rhythm to maintain optimal metabolic function [230]. Proposing fasting program instead of CR is potentially more fruitful in clinical translation to prevent cancer and ameliorate therapy responsiveness and tolerance, since starvation seems to be more powerful in determining metabolic changes, such as IGF-1 and glucose dramatic decrease [231], and may be more suitable to be followed by patients [225].
Dietary regimens that provide a normal or high caloric content but are able to induce typical fasting metabolic pathways have been proposed, i.e., fasting mimicking diets (FMDs) [232]. Among these FMDs, ketogenic diet (KD) has been associated with weight loss and has been proposed to protect against cancer and as a therapeutic anti-cancer agent [233,234]. KD is a low carbohydrate (usually less than 50 g/day), high fat, and protein regimen that lowers insulin levels and induces ketone bodies over-production in the liver, mimicking the metabolic state of fasting. The acute metabolic benefits of KD principally relies on the metabolic switch from glucose metabolism towards fatty acid oxidation. High protein diets are considered a powerful strategy to improve body weight management and decrease fat mass in both normal and obese individuals [235,236]. Very-low calorie KD (VLCKD) have been generally associated with body weight loss, reduction of visceral adiposity, and improvement of lipid profile, as well as cardiac parameters [237,238,239]. Moreover, VLCKD induces a greater reduction in body weight and WC than a standard low-calories diet, mainly through a selective re-shaping of VAT. Intriguingly, these modifications are still effective after 2 years from diet intervention [240]. Similar results have been observed also in women with endometrial or ovarian cancer that underwent KD or a diet high in fiber and low in fat: change in visceral fat depots was grater in the KD group with retention of the lean mass and decrease of serum fasting insulin [241]. Of note, the remodeling of VAT caused by short time KD could be related to changes in the mix of innate immune cells in this fat depots, which result in a reduced inflammation and in metabolic health improvement [242]. However, whereas the short-term KD approach is beneficial, and does not display cytotoxic effect or increase oxidative stress, long-term continuous KD induces obesity and glucose intolerance, as well as inflammation in visceral fat stores, thus pointing at the need of a managed weighed control during this dietary regimen [237,242].
Brandhorst et al. have proposed a FMD with low protein, low sugar, and relatively high fat content, coupled with a periodic fasting (PF) program. PF is another form of starvation that lasts 2 or more days and is separated from the next cycle by at least 1 week of normal feeding, with well-established beneficial effects on inflammation. So far, this kind of FMD lasts 4 days and provides 10–50% of the normal caloric, showing potent effect on lifespan and health span, leading rats to weight loss by reducing visceral fat amount, reducing inflamed tissues, and decreasing neoplasms development. In a pilot trial on humans, similar FMD reduced fasting blood glucose, circulating IGF-1 and IGFBP-1 levels, and led to significant fat loss, without affecting lean body mass [243]. Therefore, PF and FMD have an exceptional potential also in enhancing disease treatment in patients at risk for cachexia. However, although the recent developments in this field have generated a lot of excitement, some doubts exist with regard to its translation in clinical practice, as more robust clinical experiments are needed to support findings in animal models. From a practical perspective, a first hurdle to overcome is the feasibility of such a revolutionary diet intervention in a western routine made of always available food, nighttime eating and hypercaloric snacks. Moreover, some fasting methods are unfeasible in the long run and many side effects could be reported if longer studies were performed in humans. Furthermore, each of us has a peculiar metabolic baseline, and dietary intervention should be individualized as much as possible. For instance, fasting should not be proposed to those with deficiencies in metabolic pathways or in those patients that require a nutritional support not consistent with the composition of a FMD. Furthermore, personal preferences and metabolic considerations might inform individualized tailoring of dietary interventions [186] to be more easily proposed to patients and ameliorate their compliance.
4. Conclusions and Recommendations
In the future, it will be unavoidable to summarize evidence from epidemiological data and experiments in animal models, with clinical trials and a deep knowledge in dysregulated metabolic pathways, in order to provide targeted interventions in lifestyle, nutrients, and drugs, able to prevent and reduce adiposity and to break down its link with cancer.
Dietary interventions provide an economically viable, non-pharmacological approach for eliciting beneficial adaptation in body composition, decreasing VAT and improving weight loss. A healthy lifestyle based on the combination of appropriate diet approach and physical activity represents a preferential way to dampen the negative sequalae of inflammation, and halt cancer onset and progression.
Findings about dietary extensive influence on metabolism and cancer let us speculate whether different habits in dietary regimens affect the effectiveness and tolerance of some drugs traditionally used to treat cancer. In keeping with this, although projecting drugs which elegantly affect a single molecule or a specific pathway may be intriguing, an integrated therapeutical approach is required.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
A.M. is funded by: Interreg V-A Greece-Italy 2014–2020—SILVER WELLBEING, MIS5003627; MIUR-PRIN 2017 n. 2017J3E2W2; HDHL-INTIMIC EuJPI-FATMAL; AIRC IG 2019 Id. 23239; MIUR PON “R&I” 2014–2020 n. ARS01_01220. E.P. is funded by PON AIM1853334—Attività 2, linea 1.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors acknowledge Smart Servier Medical Art (http://smart.servier.com/—accessed date: 17 May 2021) for providing comprehensive medical and biological figures and datasets of interest for the international scientific community.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Obesity: Preventing and Managing the Global Epidemic; WHO Technical Report Series 894; World Health Organisation: Geneva, Switzerland, 2000; 253p. [Google Scholar]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 31 December 2020).
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Tchernof, A.; Despres, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Bull, C.J.; Bell, J.A.; Murphy, N.; Sanderson, E.; Davey Smith, G.; Timpson, N.J.; Banbury, B.L.; Albanes, D.; Berndt, S.I.; Bezieau, S.; et al. Adiposity, metabolites, and colorectal cancer risk: Mendelian randomization study. BMC Med. 2020, 18, 396. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Hidayat, K.; Shi, B.M. Abdominal obesity and gastroesophageal cancer risk: Systematic review and meta-analysis of prospective studies. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.A.; Tigu, A.B.; Muntean, M.; Cenariu, D.; Slaby, O.; Berindan-Neagoe, I. Molecular Links between Central Obesity and Breast Cancer. Int. J. Mol. Sci. 2019, 20, 5364. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Soerjomataram, I.; Leitzmann, M.F. Interpreting the epidemiological evidence linking obesity and cancer: A framework for population-attributable risk estimations in Europe. Eur. J. Cancer 2010, 46, 2581–2592. [Google Scholar] [CrossRef]
- Evans, T.; Sany, O.; Pearmain, P.; Ganesan, R.; Blann, A.; Sundar, S. Differential trends in the rising incidence of endometrial cancer by type: Data from a UK population-based registry from 1994 to 2006. Br. J. Cancer 2011, 104, 1505–1510. [Google Scholar] [CrossRef] [Green Version]
- Leitzmann, M.F.; Brenner, A.; Moore, S.C.; Koebnick, C.; Park, Y.; Hollenbeck, A.; Schatzkin, A.; Ron, E. Prospective study of body mass index, physical activity and thyroid cancer. Int. J. Cancer 2010, 126, 2947–2956. [Google Scholar] [CrossRef] [Green Version]
- Abnet, C.C.; Freedman, N.D.; Hollenbeck, A.R.; Fraumeni, J.F., Jr.; Leitzmann, M.; Schatzkin, A. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur. J. Cancer 2008, 44, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, D.L.; Chen, Z.Z.; Gou, B.F. Associations of body mass index with cancer incidence among populations, genders, and menopausal status: A systematic review and meta-analysis. Cancer Epidemiol. 2016, 42, 1–8. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The Obesity Paradox in Cancer: A Review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- Stephenson, G.D.; Rose, D.P. Breast cancer and obesity: An update. Nutr. Cancer 2003, 45, 1–16. [Google Scholar] [CrossRef]
- Ma, J.; Pollak, M.; Giovannucci, E.; Chan, J.M.; Tao, Y.; Hennekens, C.; Stampfer, M.J. A prospective study of plasma levels of insulin-like growth factor I (IGF-I) and IGF-binding protein-3, and colorectal cancer risk among men. Growth Horm. IGF Res. 2000, 10 (Suppl. A), S28–S29. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, L.; Zhou, Q.; Imam, M.U.; Cai, J.; Wang, Y.; Qi, M.; Sun, P.; Ping, Z.; Fu, X. Body mass index had different effects on premenopausal and postmenopausal breast cancer risks: A dose-response meta-analysis with 3,318,796 subjects from 31 cohort studies. BMC Public Health 2017, 17, 936. [Google Scholar] [CrossRef]
- Gong, Z.; Neuhouser, M.L.; Goodman, P.J.; Albanes, D.; Chi, C.; Hsing, A.W.; Lippman, S.M.; Platz, E.A.; Pollak, M.N.; Thompson, I.M.; et al. Obesity, diabetes, and risk of prostate cancer: Results from the prostate cancer prevention trial. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef] [Green Version]
- Ligibel, J.A.; Wollins, D. American Society of Clinical Oncology Obesity Initiative: Rationale, Progress, and Future Directions. J. Clin. Oncol. 2016, 34, 4256–4260. [Google Scholar] [CrossRef]
- Greenlee, H.; Unger, J.M.; LeBlanc, M.; Ramsey, S.; Hershman, D.L. Association between Body Mass Index and Cancer Survival in a Pooled Analysis of 22 Clinical Trials. Cancer Epidemiol. Biomark. Prev. 2017, 26, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligibel, J.A.; Alfano, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Burger, R.A.; Chlebowski, R.T.; Fabian, C.J.; Gucalp, A.; Hershman, D.L.; Hudson, M.M.; et al. American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 2014, 32, 3568–3574. [Google Scholar] [CrossRef]
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Madarnas, Y.; Hartwick, W.; Hoffman, B.; Hood, N. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J. Clin. Oncol. 2002, 20, 42–51. [Google Scholar] [CrossRef]
- Pollak, M.N.; Chapman, J.W.; Shepherd, L.; Meng, D.; Richardson, P.; Wilson, C.; Orme, B.; Pritchard, K.I. Insulin resistance, estimated by serum C-peptide level, is associated with reduced event-free survival for postmenopausal women in NCIC CTG MA.14 adjuvant breast cancer trial. J. Clin. Oncol. 2006, 24, 524. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef]
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, K.S.; Kim, H.; Jeong, H.; Choi, M.J.; Yoo, H.W.; Han, T.H.; Lee, H. The relationship between metabolic syndrome and the incidence of colorectal cancer. Environ. Health Prev. Med. 2020, 25, 6. [Google Scholar] [CrossRef]
- Stocks, T.; Lukanova, A.; Bjorge, T.; Ulmer, H.; Manjer, J.; Almquist, M.; Concin, H.; Engeland, A.; Hallmans, G.; Nagel, G.; et al. Metabolic factors and the risk of colorectal cancer in 580,000 men and women in the metabolic syndrome and cancer project (Me-Can). Cancer 2011, 117, 2398–2407. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Moore, J.X.; Pisu, M.; Judd, S.E.; Goodman, M.; Shikany, J.M.; Howard, V.J.; Safford, M.; Gilchrist, S.C. A Prospective Study of Obesity, Metabolic Health, and Cancer Mortality. Obesity (Silver Spring) 2018, 26, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Slawinski, C.G.V.; Barriuso, J.; Guo, H.; Renehan, A.G. Obesity and Cancer Treatment Outcomes: Interpreting the Complex Evidence. Clin. Oncol. 2020, 32, 591–608. [Google Scholar] [CrossRef]
- Griggs, J.J.; Mangu, P.B.; Anderson, H.; Balaban, E.P.; Dignam, J.J.; Hryniuk, W.M.; Morrison, V.A.; Pini, T.M.; Runowicz, C.D.; Rosner, G.L.; et al. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2012, 30, 1553–1561. [Google Scholar] [CrossRef]
- Jayne, D.G.; Thorpe, H.C.; Copeland, J.; Quirke, P.; Brown, J.M.; Guillou, P.J. Five-year follow-up of the Medical Research Council CLASICC trial of laparoscopically assisted versus open surgery for colorectal cancer. Br. J. Surg. 2010, 97, 1638–1645. [Google Scholar] [CrossRef]
- Wu, J.; Cohen, P.; Spiegelman, B.M. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013, 27, 234–250. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Minehira, K.; Bettschart, V.; Vidal, H.; Vega, N.; Di Vetta, V.; Rey, V.; Schneiter, P.; Tappy, L. Effect of carbohydrate overfeeding on whole body and adipose tissue metabolism in humans. Obes. Res. 2003, 11, 1096–1103. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Warburg, O. Iron, the Oxygen-Carrier of Respiration-Ferment. Science 1925, 61, 575–582. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef] [Green Version]
- Geer, E.B.; Shen, W. Gender differences in insulin resistance, body composition, and energy balance. Gend. Med. 2009, 6 (Suppl. S1), 60–75. [Google Scholar] [CrossRef] [Green Version]
- Kyrgiou, M.; Kalliala, I.; Markozannes, G.; Gunter, M.J.; Paraskevaidis, E.; Gabra, H.; Martin-Hirsch, P.; Tsilidis, K.K. Adiposity and cancer at major anatomical sites: Umbrella review of the literature. BMJ 2017, 356, j477. [Google Scholar] [CrossRef] [Green Version]
- Bjorntorp, P. “Portal” adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis 1990, 10, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, K.A.; Lonnroth, P.; Parkkola, R.; Peltoniemi, P.; Asola, M.; Viljanen, T.; Tolvanen, T.; Knuuti, J.; Ronnemaa, T.; Huupponen, R.; et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J. Clin. Endocrinol. Metab. 2002, 87, 3902–3910. [Google Scholar] [CrossRef]
- Lee, M.-J.; Fried, S.K. Depot-Specific Biology of Adipose Tissues: Links to Fat Distribution and Metabolic Risk. In Adipose Tissue in Health and Disease; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Hafidi, M.E.; Buelna-Chontal, M.; Sanchez-Munoz, F.; Carbo, R. Adipogenesis: A Necessary but Harmful Strategy. Int. J. Mol. Sci. 2019, 20, 3657. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.P.; Komninou, D.; Stephenson, G.D. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004, 5, 153–165. [Google Scholar] [CrossRef]
- Harvie, M.; Hooper, L.; Howell, A.H. Central obesity and breast cancer risk: A systematic review. Obes. Rev. 2003, 4, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Haase, J.; Weyer, U.; Immig, K.; Kloting, N.; Bluher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Targeting obesity-related adipose tissue dysfunction to prevent cancer development and progression. Semin. Oncol. 2016, 43, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, N.; Terauchi, Y.; Yamauchi, T.; Kubota, T.; Moroi, M.; Matsui, J.; Eto, K.; Yamashita, T.; Kamon, J.; Satoh, H.; et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J. Biol. Chem. 2002, 277, 25863–25866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999, 100, 2473–2476. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, S.; Piao, C.; Qiu, S.; Li, H.; Du, J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 2016, 55, 1796–1806. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Nakamura, T.; Nishida, M.; Kumada, M.; Okamoto, Y.; Ohashi, K.; Nagaretani, H.; Kishida, K.; et al. Reciprocal association of C-reactive protein with adiponectin in blood stream and adipose tissue. Circulation 2003, 107, 671–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Woo, Y.C.; Wang, Y.; Yeung, C.Y.; Xu, A.; Lam, K.S. Obesity, adipokines and cancer: An update. Clin. Endocrinol. (Oxf.) 2015, 83, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Sarjeant, K.; Stephens, J.M. Adipogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008417. [Google Scholar] [CrossRef] [Green Version]
- Macis, D.; Guerrieri-Gonzaga, A.; Gandini, S. Circulating adiponectin and breast cancer risk: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.K.; Kim, W.J.; Lee, S.A. Association between obesity-related adipokines and colorectal cancer: A case-control study and meta-analysis. World J. Gastroenterol. 2014, 20, 7941–7949. [Google Scholar] [CrossRef]
- Lonnqvist, F.; Nordfors, L.; Jansson, M.; Thorne, A.; Schalling, M.; Arner, P. Leptin secretion from adipose tissue in women. Relationship to plasma levels and gene expression. J. Clin. Invest. 1997, 99, 2398–2404. [Google Scholar] [CrossRef] [Green Version]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; Pereira, J.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front. Endocrinol. (Lausanne) 2019, 10, 841. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; Garcia-Cardena, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological action of leptin as an angiogenic factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delort, L.; Rossary, A.; Farges, M.C.; Vasson, M.P.; Caldefie-Chezet, F. Leptin, adipocytes and breast cancer: Focus on inflammation and anti-tumor immunity. Life Sci. 2015, 140, 37–48. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Ennis, M.; Fantus, I.G.; Pritchard, K.I.; Trudeau, M.E.; Koo, J.; Hood, N. Is leptin a mediator of adverse prognostic effects of obesity in breast cancer? J. Clin. Oncol. 2005, 23, 6037–6042. [Google Scholar] [CrossRef]
- Lazar, M.A. How obesity causes diabetes: Not a tall tale. Science 2005, 307, 373–375. [Google Scholar] [CrossRef] [Green Version]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Shivappa, N.; Hebert, J.R.; Rietzschel, E.R.; De Buyzere, M.L.; Langlois, M.; Debruyne, E.; Marcos, A.; Huybrechts, I. Associations between dietary inflammatory index and inflammatory markers in the Asklepios Study. Br. J. Nutr. 2015, 113, 665–671. [Google Scholar] [CrossRef] [Green Version]
- Yeung, C.Y.; Tso, A.W.; Xu, A.; Wang, Y.; Woo, Y.C.; Lam, T.H.; Lo, S.V.; Fong, C.H.; Wat, N.M.; Woo, J.; et al. Pro-inflammatory adipokines as predictors of incident cancers in a Chinese cohort of low obesity prevalence in Hong Kong. PLoS ONE 2013, 8, e78594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Cassol, E.; Poli, G. Macrophage polarization in health and disease. Sci. World J. 2011, 11, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Bulun, S.E.; Mahendroo, M.S.; Simpson, E.R. Aromatase gene expression in adipose tissue: Relationship to breast cancer. J. Steroid Biochem. Mol. Biol. 1994, 49, 319–326. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Invest. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Druta, M.; Shibata, D.; Coppola, D.; Boler, I.; Elahi, A.; Reich, R.R.; Siegel, E.; Extermann, M. Metabolic syndrome and colorectal cancer: Is hyperinsulinemia/insulin receptor-mediated angiogenesis a critical process? J. Geriatr. Oncol. 2014, 5, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Papa, V.; Belfiore, A. Insulin receptors in breast cancer: Biological and clinical role. J. Endocrinol. Invest. 1996, 19, 324–333. [Google Scholar] [CrossRef]
- Bianchini, F.; Kaaks, R.; Vainio, H. Weight control and physical activity in cancer prevention. Obes. Rev. 2002, 3, 5–8. [Google Scholar] [CrossRef]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Khandwala, H.M.; McCutcheon, I.E.; Flyvbjerg, A.; Friend, K.E. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr. Rev. 2000, 21, 215–244. [Google Scholar] [CrossRef]
- Kawai, M.; Namba, N.; Mushiake, S.; Etani, Y.; Nishimura, R.; Makishima, M.; Ozono, K. Growth hormone stimulates adipogenesis of 3T3-L1 cells through activation of the Stat5A/5B-PPARgamma pathway. J. Mol. Endocrinol. 2007, 38, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, N.E.; Key, T.J.; Appleby, P.N.; Travis, R.C.; Roddam, A.W.; Rinaldi, S.; Egevad, L.; Rohrmann, S.; Linseisen, J.; Pischon, T.; et al. Serum insulin-like growth factor (IGF)-I and IGF-binding protein-3 concentrations and prostate cancer risk: Results from the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Purohit, A.; Reed, M.J. Regulation of estrogen synthesis in postmenopausal women. Steroids 2002, 67, 979–983. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sestak, I.; Distler, W.; Forbes, J.F.; Dowsett, M.; Howell, A.; Cuzick, J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: An exploratory analysis from the ATAC trial. J. Clin. Oncol. 2010, 28, 3411–3415. [Google Scholar] [CrossRef]
- Shaw, E.; Farris, M.; McNeil, J.; Friedenreich, C. Obesity and Endometrial Cancer. In Recent Results in Cancer Research; Springer: Cham, Switzerland, 2016; Volume 208, pp. 107–136. [Google Scholar] [CrossRef]
- Furberg, A.S.; Veierod, M.B.; Wilsgaard, T.; Bernstein, L.; Thune, I. Serum high-density lipoprotein cholesterol, metabolic profile, and breast cancer risk. J. Natl. Cancer Inst. 2004, 96, 1152–1160. [Google Scholar] [CrossRef]
- Kliemann, N.; Murphy, N.; Viallon, V.; Freisling, H.; Tsilidis, K.K.; Rinaldi, S.; Mancini, F.R.; Fagherazzi, G.; Boutron-Ruault, M.C.; Boeing, H.; et al. Predicted basal metabolic rate and cancer risk in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2020, 147, 648–661. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Luo, J.; Margolis, K.L.; Adami, H.O.; LaCroix, A.; Ye, W.; Women’s Health Initiative Investigators. Obesity and risk of pancreatic cancer among postmenopausal women: The Women’s Health Initiative (United States). Br. J. Cancer 2008, 99, 527–531. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Punyanitya, M.; Wang, Z.; Gallagher, D.; St-Onge, M.P.; Albu, J.; Heymsfield, S.B.; Heshka, S. Total body skeletal muscle and adipose tissue volumes: Estimation from a single abdominal cross-sectional image. J. Appl. Physiol. (1985) 2004, 97, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Shin, Y.; Park, J.; Kim, H.; Lee, I.S.; Seo, D.W.; Huh, J.; Lee, T.Y.; Park, T.; Lee, J.; et al. Development and Validation of a Deep Learning System for Segmentation of Abdominal Muscle and Fat on Computed Tomography. Korean J. Radiol. 2020, 21, 88–100. [Google Scholar] [CrossRef]
- Paris, M.T. Body Composition Analysis of Computed Tomography Scans in Clinical Populations: The Role of Deep Learning. Lifestyle Genom. 2020, 13, 28–31. [Google Scholar] [CrossRef]
- Wu, S.; Liu, J.; Wang, X.; Li, M.; Gan, Y.; Tang, Y. Association of obesity and overweight with overall survival in colorectal cancer patients: A meta-analysis of 29 studies. Cancer Causes Control. 2014, 25, 1489–1502. [Google Scholar] [CrossRef]
- Campbell, P.T.; Newton, C.C.; Dehal, A.N.; Jacobs, E.J.; Patel, A.V.; Gapstur, S.M. Impact of body mass index on survival after colorectal cancer diagnosis: The Cancer Prevention Study-II Nutrition Cohort. J. Clin. Oncol. 2012, 30, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Charvat, H.; Freisling, H.; Noh, H.; Adami, H.O.; Soerjomataram, I.; Weiderpass, E. Adult Overweight and Survival from Breast and Colorectal Cancer in Swedish Women. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1518–1524. [Google Scholar] [CrossRef]
- Yumuk, V.; Tsigos, C.; Fried, M.; Schindler, K.; Busetto, L.; Micic, D.; Toplak, H. European Guidelines for Obesity Management in Adults. Obes. Facts 2015, 8, 402–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jacobs, E.J.; Patel, A.V.; Rodriguez, C.; McCullough, M.L.; Thun, M.J.; Calle, E.E. A prospective study of waist circumference and body mass index in relation to colorectal cancer incidence. Cancer Causes Control 2008, 19, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Pischon, T.; Lahmann, P.H.; Boeing, H.; Friedenreich, C.; Norat, T.; Tjonneland, A.; Halkjaer, J.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Body size and risk of colon and rectal cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). J. Natl. Cancer Inst. 2006, 98, 920–931. [Google Scholar] [CrossRef]
- Pischon, T.; Boeing, H.; Hoffmann, K.; Bergmann, M.; Schulze, M.B.; Overvad, K.; van der Schouw, Y.T.; Spencer, E.; Moons, K.G.; Tjonneland, A.; et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 2008, 359, 2105–2120. [Google Scholar] [CrossRef] [Green Version]
- Arslan, A.A.; Helzlsouer, K.J.; Kooperberg, C.; Shu, X.O.; Steplowski, E.; Bueno-de-Mesquita, H.B.; Fuchs, C.S.; Gross, M.D.; Jacobs, E.J.; Lacroix, A.Z.; et al. Anthropometric measures, body mass index, and pancreatic cancer: A pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Arch. Intern. Med. 2010, 170, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Han, C.T.; Zhang, G.M.; Zhang, C.Z.; Yang, W.Y.; Shen, Y.; Vidal, A.C.; Freedland, S.J.; Zhu, Y.; Ye, D.W. Waist-hip Ratio (WHR), a Better Predictor for Prostate Cancer than Body Mass Index (BMI): Results from a Chinese Hospital-based Biopsy Cohort. Sci. Rep. 2017, 7, 43551. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, L.; Cui, S.; Tian, F.; Fan, Z.; Geng, C.; Cao, X.; Yang, Z.; Wang, X.; Liang, H.; et al. Distinct Effects of Body Mass Index and Waist/Hip Ratio on Risk of Breast Cancer by Joint Estrogen and Progestogen Receptor Status: Results from a Case-Control Study in Northern and Eastern China and Implications for Chemoprevention. Oncologist 2017, 22, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Berrington de Gonzalez, A.; Spencer, E.A.; Bueno-de-Mesquita, H.B.; Roddam, A.; Stolzenberg-Solomon, R.; Halkjaer, J.; Tjonneland, A.; Overvad, K.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Anthropometry, physical activity, and the risk of pancreatic cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol. Biomark. Prev. 2006, 15, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Ardesch, F.H.; Ruiter, R.; Mulder, M.; Lahousse, L.; Stricker, B.H.C.; Kiefte-de Jong, J.C. The Obesity Paradox in Lung Cancer: Associations With Body Size Versus Body Shape. Front. Oncol. 2020, 10, 591110. [Google Scholar] [CrossRef]
- Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult weight gain and adiposity-related cancers: A dose-response meta-analysis of prospective observational studies. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Ouellet, V.; Labbe, S.M.; Blondin, D.P.; Phoenix, S.; Guerin, B.; Haman, F.; Turcotte, E.E.; Richard, D.; Carpentier, A.C. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Invest. 2012, 122, 545–552. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Vijgen, G.H.; Bouvy, N.D.; Teule, G.J.; Brans, B.; Hoeks, J.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J. Clin. Endocrinol. Metab. 2012, 97, E1229–E1233. [Google Scholar] [CrossRef] [Green Version]
- Contreras, C.; Gonzalez, F.; Ferno, J.; Dieguez, C.; Rahmouni, K.; Nogueiras, R.; Lopez, M. The brain and brown fat. Ann. Med. 2015, 47, 150–168. [Google Scholar] [CrossRef] [Green Version]
- Nedergaard, J.; Cannon, B. The browning of white adipose tissue: Some burning issues. Cell Metab. 2014, 20, 396–407. [Google Scholar] [CrossRef] [Green Version]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2016, 17, 691–702. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest. 2013, 123, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Commins, S.P.; Watson, P.M.; Padgett, M.A.; Dudley, A.; Argyropoulos, G.; Gettys, T.W. Induction of uncoupling protein expression in brown and white adipose tissue by leptin. Endocrinology 1999, 140, 292–300. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Yoo, H.; Bosco, C.; Lee, B.; Feng, G.S.; Schaack, J.; Chi, N.W.; Shao, J. Adiponectin reduces thermogenesis by inhibiting brown adipose tissue activation in mice. Diabetologia 2014, 57, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Nisoli, E.; Briscini, L.; Giordano, A.; Tonello, C.; Wiesbrock, S.M.; Uysal, K.T.; Cinti, S.; Carruba, M.O.; Hotamisligil, G.S. Tumor necrosis factor alpha mediates apoptosis of brown adipocytes and defective brown adipocyte function in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 8033–8038. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest. 2011, 121, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Montanari, T.; Poscic, N.; Colitti, M. Factors involved in white-to-brown adipose tissue conversion and in thermogenesis: A review. Obes. Rev. 2017, 18, 495–513. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. A role for brown adipose tissue in diet-induced thermogenesis. Obes. Res. 1997, 5, 650–656. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Ning, G.; Gu, W.; Su, T.; Xu, M.; Li, B.; Wang, W. Brown adipose tissue in humans is activated by elevated plasma catecholamines levels and is inversely related to central obesity. PLoS ONE 2011, 6, e21006. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Franquet Elia, E.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Loprinzi, P.D.; Yang, L.; Liu, J.; Liu, S.; Zou, L. The Beneficial Effects of Traditional Chinese Exercises for Adults with Low Back Pain: A Meta-Analysis of Randomized Controlled Trials. Medicina (Kaunas) 2019, 55, 118. [Google Scholar] [CrossRef] [Green Version]
- Rachid, T.L.; Penna-de-Carvalho, A.; Bringhenti, I.; Aguila, M.B.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Mol. Cell. Endocrinol. 2015, 402, 86–94. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [Green Version]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villarroya, F.; Serrano, M.; Ferno, J.; Salvador, J.; Escalada, J.; et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Goto, T.; Yu, R.; Uchida, K.; Tominaga, M.; Kano, Y.; Takahashi, N.; Kawada, T. Fish oil intake induces UCP1 upregulation in brown and white adipose tissue via the sympathetic nervous system. Sci. Rep. 2015, 5, 18013. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liang, X.; Yang, Q.; Fu, X.; Rogers, C.J.; Zhu, M.; Rodgers, B.D.; Jiang, Q.; Dodson, M.V.; Du, M. Resveratrol induces brown-like adipocyte formation in white fat through activation of AMP-activated protein kinase (AMPK) alpha1. Int. J. Obes. (Lond.) 2015, 39, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Zamorano, N.; Fabbiano, S.; Chevalier, C.; Stojanovic, O.; Colin, D.J.; Stevanovic, A.; Veyrat-Durebex, C.; Tarallo, V.; Rigo, D.; Germain, S.; et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat. Med. 2015, 21, 1497–1501. [Google Scholar] [CrossRef]
- Li, G.; Xie, C.; Lu, S.; Nichols, R.G.; Tian, Y.; Li, L.; Patel, D.; Ma, Y.; Brocker, C.N.; Yan, T.; et al. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab. 2017, 26, 672–685.e4. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, F.; Biessy, C.; Ferrari, P.; Freisling, H.; Rinaldi, S.; Chajes, V.; Dahm, C.C.; Overvad, K.; Dossus, L.; Lagiou, P.; et al. Healthy Lifestyle and Risk of Cancer in the European Prospective Investigation Into Cancer and Nutrition Cohort Study. Medicine (Baltimore) 2016, 95, e2850. [Google Scholar] [CrossRef] [Green Version]
- Naudin, S.; Viallon, V.; Hashim, D.; Freisling, H.; Jenab, M.; Weiderpass, E.; Perrier, F.; McKenzie, F.; Bueno-de-Mesquita, H.B.; Olsen, A.; et al. Healthy lifestyle and the risk of pancreatic cancer in the EPIC study. Eur. J. Epidemiol. 2020, 35, 975–986. [Google Scholar] [CrossRef]
- Look, A.R.G.; Wing, R.R. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: Four-year results of the Look AHEAD trial. Arch. Intern. Med. 2010, 170, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Hendryx, M.; Manson, J.E.; Figueiredo, J.C.; LeBlanc, E.S.; Barrington, W.; Rohan, T.E.; Howard, B.V.; Reding, K.; Ho, G.Y.; et al. Intentional Weight Loss and Obesity-Related Cancer Risk. JNCI Cancer Spectr. 2019, 3, pkz054. [Google Scholar] [CrossRef]
- World Cancer Research Fund; American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective. Available online: https://www.wcrf.org/diet-and-cancer/ (accessed on 10 June 2021).
- Thomas, E.L.; Brynes, A.E.; McCarthy, J.; Goldstone, A.P.; Hajnal, J.V.; Saeed, N.; Frost, G.; Bell, J.D. Preferential loss of visceral fat following aerobic exercise, measured by magnetic resonance imaging. Lipids 2000, 35, 769–776. [Google Scholar] [CrossRef]
- Irving, B.A.; Davis, C.K.; Brock, D.W.; Weltman, J.Y.; Swift, D.; Barrett, E.J.; Gaesser, G.A.; Weltman, A. Effect of exercise training intensity on abdominal visceral fat and body composition. Med. Sci. Sports Exerc. 2008, 40, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Slentz, C.A.; Duscha, B.D.; Johnson, J.L.; Ketchum, K.; Aiken, L.B.; Samsa, G.P.; Houmard, J.A.; Bales, C.W.; Kraus, W.E. Effects of the amount of exercise on body weight, body composition, and measures of central obesity: STRRIDE—A randomized controlled study. Arch. Intern. Med. 2004, 164, 31–39. [Google Scholar] [CrossRef]
- Vissers, D.; Hens, W.; Taeymans, J.; Baeyens, J.P.; Poortmans, J.; Van Gaal, L. The effect of exercise on visceral adipose tissue in overweight adults: A systematic review and meta-analysis. PLoS ONE 2013, 8, e56415. [Google Scholar] [CrossRef] [Green Version]
- Gollisch, K.S.; Brandauer, J.; Jessen, N.; Toyoda, T.; Nayer, A.; Hirshman, M.F.; Goodyear, L.J. Effects of exercise training on subcutaneous and visceral adipose tissue in normal- and high-fat diet-fed rats. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E495–E504. [Google Scholar] [CrossRef]
- WHO. Weight Control and Physical Activity, 1st ed.; IARC Publications: Lyon, France, 2002; Volume 6. [Google Scholar]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Munt, A.E.; Partridge, S.R.; Allman-Farinelli, M. The barriers and enablers of healthy eating among young adults: A missing piece of the obesity puzzle: A scoping review. Obes. Rev. 2017, 18, 1–17. [Google Scholar] [CrossRef]
- Donaldson, M.S. Nutrition and cancer: A review of the evidence for an anti-cancer diet. Nutr. J. 2004, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Sofi, F.; Cesari, F.; Abbate, R.; Gensini, G.F.; Casini, A. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 337, a1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, L.; Chen, L.; White, D.L.; Tinker, L.; Chlebowski, R.T.; Van Horn, L.V.; Richardson, P.; Lane, D.; Sangi-Haghpeykar, H.; El-Serag, H.B. Low-fat Dietary Pattern and Pancreatic Cancer Risk in the Women’s Health Initiative Dietary Modification Randomized Controlled Trial. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Martinez-Gonzalez, M.A.; Tong, T.Y.; Forouhi, N.G.; Khandelwal, S.; Prabhakaran, D.; Mozaffarian, D.; de Lorgeril, M. Definitions and potential health benefits of the Mediterranean diet: Views from experts around the world. BMC Med. 2014, 12, 112. [Google Scholar] [CrossRef] [Green Version]
- De Santis, S.; Cariello, M.; Piccinin, E.; Sabba, C.; Moschetta, A. Extra Virgin Olive Oil: Lesson from Nutrigenomics. Nutrients 2019, 11, 2085. [Google Scholar] [CrossRef] [Green Version]
- Gadgil, M.D.; Appel, L.J.; Yeung, E.; Anderson, C.A.; Sacks, F.M.; Miller, E.R., 3rd. The effects of carbohydrate, unsaturated fat, and protein intake on measures of insulin sensitivity: Results from the OmniHeart trial. Diabetes Care 2013, 36, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cheng, M.; Zhao, M.; Ge, A.; Guo, F.; Zhang, M.; Yang, Y.; Liu, L.; Yang, N. Differential effects of high-fat-diet rich in lard oil or soybean oil on osteopontin expression and inflammation of adipose tissue in diet-induced obese rats. Eur. J. Nutr. 2013, 52, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Bendall, C.L.; Mayr, H.L.; Opie, R.S.; Bes-Rastrollo, M.; Itsiopoulos, C.; Thomas, C.J. Central obesity and the Mediterranean diet: A systematic review of intervention trials. Crit. Rev. Food Sci. Nutr. 2018, 58, 3070–3084. [Google Scholar] [CrossRef] [PubMed]
- Rumawas, M.E.; Meigs, J.B.; Dwyer, J.T.; McKeown, N.M.; Jacques, P.F. Mediterranean-style dietary pattern, reduced risk of metabolic syndrome traits, and incidence in the Framingham Offspring Cohort. Am. J. Clin. Nutr. 2009, 90, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, S.; Leone, A.; Vignati, L.; Bedogni, G.; Martinez-Gonzalez, M.A.; Bes-Rastrollo, M.; Spadafranca, A.; Vanzulli, A.; Battezzati, A. Adherence to the Mediterranean diet is inversely associated with visceral abdominal tissue in Caucasian subjects. Clin. Nutr. 2015, 34, 1266–1272. [Google Scholar] [CrossRef]
- Sofi, F.; Dinu, M.; Pagliai, G.; Cesari, F.; Gori, A.M.; Sereni, A.; Becatti, M.; Fiorillo, C.; Marcucci, R.; Casini, A. Low-Calorie Vegetarian Versus Mediterranean Diets for Reducing Body Weight and Improving Cardiovascular Risk Profile: CARDIVEG Study (Cardiovascular Prevention With Vegetarian Diet). Circulation 2018, 137, 1103–1113. [Google Scholar] [CrossRef]
- Brehm, B.J.; Lattin, B.L.; Summer, S.S.; Boback, J.A.; Gilchrist, G.M.; Jandacek, R.J.; D’Alessio, D.A. One-year comparison of a high-monounsaturated fat diet with a high-carbohydrate diet in type 2 diabetes. Diabetes Care 2009, 32, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Shai, I.; Schwarzfuchs, D.; Henkin, Y.; Shahar, D.R.; Witkow, S.; Greenberg, I.; Golan, R.; Fraser, D.; Bolotin, A.; Vardi, H.; et al. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N. Engl. J. Med. 2008, 359, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Estruch, R.; Martinez-Gonzalez, M.A.; Corella, D.; Salas-Salvado, J.; Fito, M.; Chiva-Blanch, G.; Fiol, M.; Gomez-Gracia, E.; Aros, F.; Lapetra, J.; et al. Effect of a high-fat Mediterranean diet on bodyweight and waist circumference: A prespecified secondary outcomes analysis of the PREDIMED randomised controlled trial. Lancet Diabetes Endocrinol. 2019, 7, e6–e17. [Google Scholar] [CrossRef]
- Smith, S.R.; Zachwieja, J.J. Visceral adipose tissue: A critical review of intervention strategies. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto, E.; Boffetta, P.; Lagiou, P.; Ferrari, P.; Buckland, G.; Overvad, K.; Dahm, C.C.; Tjonneland, A.; Olsen, A.; Clavel-Chapelon, F.; et al. Mediterranean dietary pattern and cancer risk in the EPIC cohort. Br. J. Cancer 2011, 104, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamia, C.; Lagiou, P.; Buckland, G.; Grioni, S.; Agnoli, C.; Taylor, A.J.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjonneland, A.; et al. Mediterranean diet and colorectal cancer risk: Results from a European cohort. Eur. J. Epidemiol. 2013, 28, 317–328. [Google Scholar] [CrossRef]
- Buckland, G.; Agudo, A.; Lujan, L.; Jakszyn, P.; Bueno-de-Mesquita, H.B.; Palli, D.; Boeing, H.; Carneiro, F.; Krogh, V.; Sacerdote, C.; et al. Adherence to a Mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am. J. Clin. Nutr. 2010, 91, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Su, L.J.; Arab, L.; Bensen, J.T.; Farnan, L.; Fontham, E.T.H.; Song, L.; Hussey, J.; Merchant, A.T.; Mohler, J.L.; et al. Dietary patterns based on the Mediterranean diet and DASH diet are inversely associated with high aggressive prostate cancer in PCaP. Ann. Epidemiol. 2019, 29, 16–22.e11. [Google Scholar] [CrossRef]
- Turati, F.; Carioli, G.; Bravi, F.; Ferraroni, M.; Serraino, D.; Montella, M.; Giacosa, A.; Toffolutti, F.; Negri, E.; Levi, F.; et al. Mediterranean Diet and Breast Cancer Risk. Nutrients 2018, 10, 326. [Google Scholar] [CrossRef] [Green Version]
- Van den Brandt, P.A.; Schulpen, M. Mediterranean diet adherence and risk of postmenopausal breast cancer: Results of a cohort study and meta-analysis. Int. J. Cancer 2017, 140, 2220–2231. [Google Scholar] [CrossRef]
- Mitrou, P.N.; Kipnis, V.; Thiebaut, A.C.; Reedy, J.; Subar, A.F.; Wirfalt, E.; Flood, A.; Mouw, T.; Hollenbeck, A.R.; Leitzmann, M.F.; et al. Mediterranean dietary pattern and prediction of all-cause mortality in a US population: Results from the NIH-AARP Diet and Health Study. Arch. Intern. Med. 2007, 167, 2461–2468. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef]
- Farinetti, A.; Zurlo, V.; Manenti, A.; Coppi, F.; Mattioli, A.V. Mediterranean diet and colorectal cancer: A systematic review. Nutrition 2017, 43–44, 83–88. [Google Scholar] [CrossRef]
- Bingham, S.A.; Day, N.E.; Luben, R.; Ferrari, P.; Slimani, N.; Norat, T.; Clavel-Chapelon, F.; Kesse, E.; Nieters, A.; Boeing, H.; et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): An observational study. Lancet 2003, 361, 1496–1501. [Google Scholar] [CrossRef]
- Bradbury, K.E.; Appleby, P.N.; Key, T.J. Fruit, vegetable, and fiber intake in relation to cancer risk: Findings from the European Prospective Investigation into Cancer and Nutrition (EPIC). Am. J. Clin. Nutr. 2014, 100 (Suppl. S1), 394S–398S. [Google Scholar] [CrossRef] [Green Version]
- Pitsavos, C.; Panagiotakos, D.B.; Tzima, N.; Chrysohoou, C.; Economou, M.; Zampelas, A.; Stefanadis, C. Adherence to the Mediterranean diet is associated with total antioxidant capacity in healthy adults: The ATTICA study. Am. J. Clin. Nutr. 2005, 82, 694–699. [Google Scholar] [CrossRef]
- Owen, R.W.; Giacosa, A.; Hull, W.E.; Haubner, R.; Spiegelhalder, B.; Bartsch, H. The antioxidant/anticancer potential of phenolic compounds isolated from olive oil. Eur. J. Cancer 2000, 36, 1235–1247. [Google Scholar] [CrossRef]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Das, U.N.; Stefanadis, C. Adherence to the Mediterranean diet attenuates inflammation and coagulation process in healthy adults: The ATTICA Study. J. Am. Coll. Cardiol. 2004, 44, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Appleby, P.N.; Achaintre, D.; Gicquiau, A.; Gunter, M.J.; Ferrari, P.; Kaaks, R.; et al. Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3057 matched case-control sets from EPIC. Int. J. Cancer 2020, 146, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Turati, F.; Lagiou, P.; Trichopoulos, D.; Augustin, L.S.; La Vecchia, C.; Trichopoulou, A. Mediterranean diet and glycaemic load in relation to incidence of type 2 diabetes: Results from the Greek cohort of the population-based European Prospective Investigation into Cancer and Nutrition (EPIC). Diabetologia 2013, 56, 2405–2413. [Google Scholar] [CrossRef] [Green Version]
- Kastorini, C.M.; Milionis, H.J.; Esposito, K.; Giugliano, D.; Goudevenos, J.A.; Panagiotakos, D.B. The effect of Mediterranean diet on metabolic syndrome and its components: A meta-analysis of 50 studies and 534,906 individuals. J. Am. Coll. Cardiol. 2011, 57, 1299–1313. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J.; Pazienza, V. Impact of Different Types of Diet on Gut Microbiota Profiles and Cancer Prevention and Treatment. Medicina (Kaunas) 2019, 55, 84. [Google Scholar] [CrossRef] [Green Version]
- Vitaglione, P.; Mennella, I.; Ferracane, R.; Rivellese, A.A.; Giacco, R.; Ercolini, D.; Gibbons, S.M.; La Storia, A.; Gilbert, J.A.; Jonnalagadda, S.; et al. Whole-grain wheat consumption reduces inflammation in a randomized controlled trial on overweight and obese subjects with unhealthy dietary and lifestyle behaviors: Role of polyphenols bound to cereal dietary fiber. Am. J. Clin. Nutr. 2015, 101, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Frazier, T.H.; DiBaise, J.K.; McClain, C.J. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J. Parenter. Enteral. Nutr. 2011, 35, 14S–20S. [Google Scholar] [CrossRef]
- Fabbiano, S.; Suarez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef]
- Rodrigues, M.O.M.; Evangelista-Silva, P.H.; Neves, N.N.; Moreno, L.G.; Santos, C.S.; Rocha, K.L.S.; Ottone, V.O.; Batista-da-Silva, B.; Dias-Peixoto, M.F.; Magalhaes, F.C.; et al. Caloric restriction-induced weight loss with a high-fat diet does not fully recover visceral adipose tissue inflammation in previously obese C57BL/6 mice. Appl. Physiol. Nutr. Metab. 2020, 45, 1353–1359. [Google Scholar] [CrossRef]
- Barquissau, V.; Leger, B.; Beuzelin, D.; Martins, F.; Amri, E.Z.; Pisani, D.F.; Saris, W.H.M.; Astrup, A.; Maoret, J.J.; Iacovoni, J.; et al. Caloric Restriction and Diet-Induced Weight Loss Do Not Induce Browning of Human Subcutaneous White Adipose Tissue in Women and Men with Obesity. Cell Rep. 2018, 22, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Pinho, C.P.S.; Diniz, A.D.S.; Arruda, I.K.G.; Leite, A.; Rodrigues, I.G. Effects of weight loss on adipose visceral and subcutaneous tissue in overweight adults. Clin. Nutr. 2018, 37, 1252–1258. [Google Scholar] [CrossRef]
- Gasteyger, C.; Larsen, T.M.; Vercruysse, F.; Pedersen, D.; Toubro, S.; Astrup, A. Visceral fat loss induced by a low-calorie diet: A direct comparison between women and men. Diabetes Obes. Metab. 2009, 11, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, E.; Peirce, G.; Brown, S.; Freemyer, J.; Rice, W.; Lee, L.; Coviello, L.; Davis, K.G. A short-duration restrictive diet reduces visceral adiposity in the morbidly obese surgical patient. Am. J. Surg. 2016, 212, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bujo, H.; Takahashi, K.; Shibasaki, M.; Zhu, Y.; Yoshida, Y.; Otsuka, Y.; Hashimoto, N.; Saito, Y. Visceral fat: Higher responsiveness of fat mass and gene expression to calorie restriction than subcutaneous fat. Exp. Biol. Med. (Maywood) 2003, 228, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, E.; Kumahara, H.; Tobina, T.; Matsuda, T.; Ayabe, M.; Kiyonaga, A.; Anzai, K.; Higaki, Y.; Tanaka, H. Lifestyle intervention involving calorie restriction with or without aerobic exercise training improves liver fat in adults with visceral adiposity. J. Obes. 2014, 2014, 197216. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, T.; Paulsen, S.K.; Bruun, J.M.; Overgaard, K.; Ringgaard, S.; Pedersen, S.B.; Positano, V.; Richelsen, B. Comparable reduction of the visceral adipose tissue depot after a diet-induced weight loss with or without aerobic exercise in obese subjects: A 12-week randomized intervention study. Eur. J. Endocrinol. 2009, 160, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Sabatini, D.M.; Yilmaz, O.H. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell 2014, 14, 292–305. [Google Scholar] [CrossRef] [Green Version]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Longo, V.D. Fasting vs dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms arend clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Brandhorst, S.; Longo, V.D. Fasting and Caloric Restriction in Cancer Prevention and Treatment. In Recent Results in Cancer Research; Springer: Cham, Switzerland, 2016; Volume 207, pp. 241–266. [Google Scholar] [CrossRef]
- De Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Varady, K.A.; Hellerstein, M.K. Alternate-day fasting and chronic disease prevention: A review of human and animal trials. Am. J. Clin. Nutr. 2007, 86, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, R.E.; Laughlin, G.A.; LaCroix, A.Z.; Hartman, S.J.; Natarajan, L.; Senger, C.M.; Martinez, M.E.; Villasenor, A.; Sears, D.D.; Marinac, C.R.; et al. Intermittent Fasting and Human Metabolic Health. J. Acad. Nutr. Diet. 2015, 115, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Villareal, D.T.; Das, S.K.; Smith, S.R.; Meydani, S.N.; Pittas, A.G.; Klein, S.; Bhapkar, M.; Rochon, J.; Ravussin, E.; et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: A randomized clinical trial. Aging Cell 2016, 15, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhu, X.; Wang, H.; Wang, F.; Guan, W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: A systematic review and meta-analysis. PLoS ONE 2014, 9, e115147. [Google Scholar] [CrossRef]
- Zou, Y.; Fineberg, S.; Pearlman, A.; Feinman, R.D.; Fine, E.J. The effect of a ketogenic diet and synergy with rapamycin in a mouse model of breast cancer. PLoS ONE 2020, 15, e0233662. [Google Scholar] [CrossRef]
- Leidy, H.J.; Clifton, P.M.; Astrup, A.; Wycherley, T.P.; Westerterp-Plantenga, M.S.; Luscombe-Marsh, N.D.; Woods, S.C.; Mattes, R.D. The role of protein in weight loss and maintenance. Am. J. Clin. Nutr. 2015, 101, 1320S–1329S. [Google Scholar] [CrossRef] [Green Version]
- Wycherley, T.P.; Buckley, J.D.; Noakes, M.; Clifton, P.M.; Brinkworth, G.D. Comparison of the effects of weight loss from a high-protein versus standard-protein energy-restricted diet on strength and aerobic capacity in overweight and obese men. Eur. J. Nutr. 2013, 52, 317–325. [Google Scholar] [CrossRef]
- Valenzano, A.; Polito, R.; Trimigno, V.; Di Palma, A.; Moscatelli, F.; Corso, G.; Sessa, F.; Salerno, M.; Montana, A.; Di Nunno, N.; et al. Effects of Very Low Calorie Ketogenic Diet on the Orexinergic System, Visceral Adipose Tissue, and ROS Production. Antioxidants (Basel) 2019, 8, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.I.; Gomez-Arbelaez, D.; Crujeiras, A.B.; Granero, R.; Aguera, Z.; Jimenez-Murcia, S.; Sajoux, I.; Lopez-Jaramillo, P.; Fernandez-Aranda, F.; Casanueva, F.F. Effect of A Very Low-Calorie Ketogenic Diet on Food and Alcohol Cravings, Physical and Sexual Activity, Sleep Disturbances, and Quality of Life in Obese Patients. Nutrients 2018, 10, 1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilone, V.; Tramontano, S.; Renzulli, M.; Romano, M.; Cobellis, L.; Berselli, T.; Schiavo, L. Metabolic effects, safety, and acceptability of very low-calorie ketogenic dietetic scheme on candidates for bariatric surgery. Surg. Obes. Relat. Dis. 2018, 14, 1013–1019. [Google Scholar] [CrossRef]
- Moreno, B.; Crujeiras, A.B.; Bellido, D.; Sajoux, I.; Casanueva, F.F. Obesity treatment by very low-calorie-ketogenic diet at two years: Reduction in visceral fat and on the burden of disease. Endocrine 2016, 54, 681–690. [Google Scholar] [CrossRef]
- Cohen, C.W.; Fontaine, K.R.; Arend, R.C.; Alvarez, R.D.; Leath, C.A., III.; Huh, W.K.; Bevis, K.S.; Kim, K.H.; Straughn, J.M., Jr.; Gower, B.A. A Ketogenic Diet Reduces Central Obesity and Serum Insulin in Women with Ovarian or Endometrial Cancer. J. Nutr. 2018, 148, 1253–1260. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Shchukina, I.; Asher, J.L.; Sidorov, S.; Artyomov, M.N.; Dixit, V.D. Ketogenesis activates metabolically protective gammadelta T cells in visceral adipose tissue. Nat. Metab. 2020, 2, 50–61. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Overweight and obesity increased the risk for developing cancer in different sites. Body fat has been associated with increased risks for a number of cancers that occur in different sites according to sex. The cancer types depicted in the figure displayed increased mortality rate if in association with an obesity condition. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 1.
Overweight and obesity increased the risk for developing cancer in different sites. Body fat has been associated with increased risks for a number of cancers that occur in different sites according to sex. The cancer types depicted in the figure displayed increased mortality rate if in association with an obesity condition. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.

Figure 2.
Main biological mechanisms linking obesity and cancer risk. Obesity constitutes major determinants of the increasing incidence and prevalence of cancer. Several aspects underlying obesity, such as hyperinsulinemia, adiposity, and low grade inflammation, have been found as the major causes leading to cancer onset. Downward arrow indicates a decrease, whereas upward arrow indicates an increase. Abbreviations: GHR: Growth Hormone Receptor; IGFBP-1: Insulin-like growth factor-binding protein 1; SHBG: Sex Hormone Binding Globulin; TNF-alfa, Tumor Necrosis Factor-alfa; IL-6: Interleukin-6; IGF-1: Insulin Growth Factor-1. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Figure 2.
Main biological mechanisms linking obesity and cancer risk. Obesity constitutes major determinants of the increasing incidence and prevalence of cancer. Several aspects underlying obesity, such as hyperinsulinemia, adiposity, and low grade inflammation, have been found as the major causes leading to cancer onset. Downward arrow indicates a decrease, whereas upward arrow indicates an increase. Abbreviations: GHR: Growth Hormone Receptor; IGFBP-1: Insulin-like growth factor-binding protein 1; SHBG: Sex Hormone Binding Globulin; TNF-alfa, Tumor Necrosis Factor-alfa; IL-6: Interleukin-6; IGF-1: Insulin Growth Factor-1. Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.


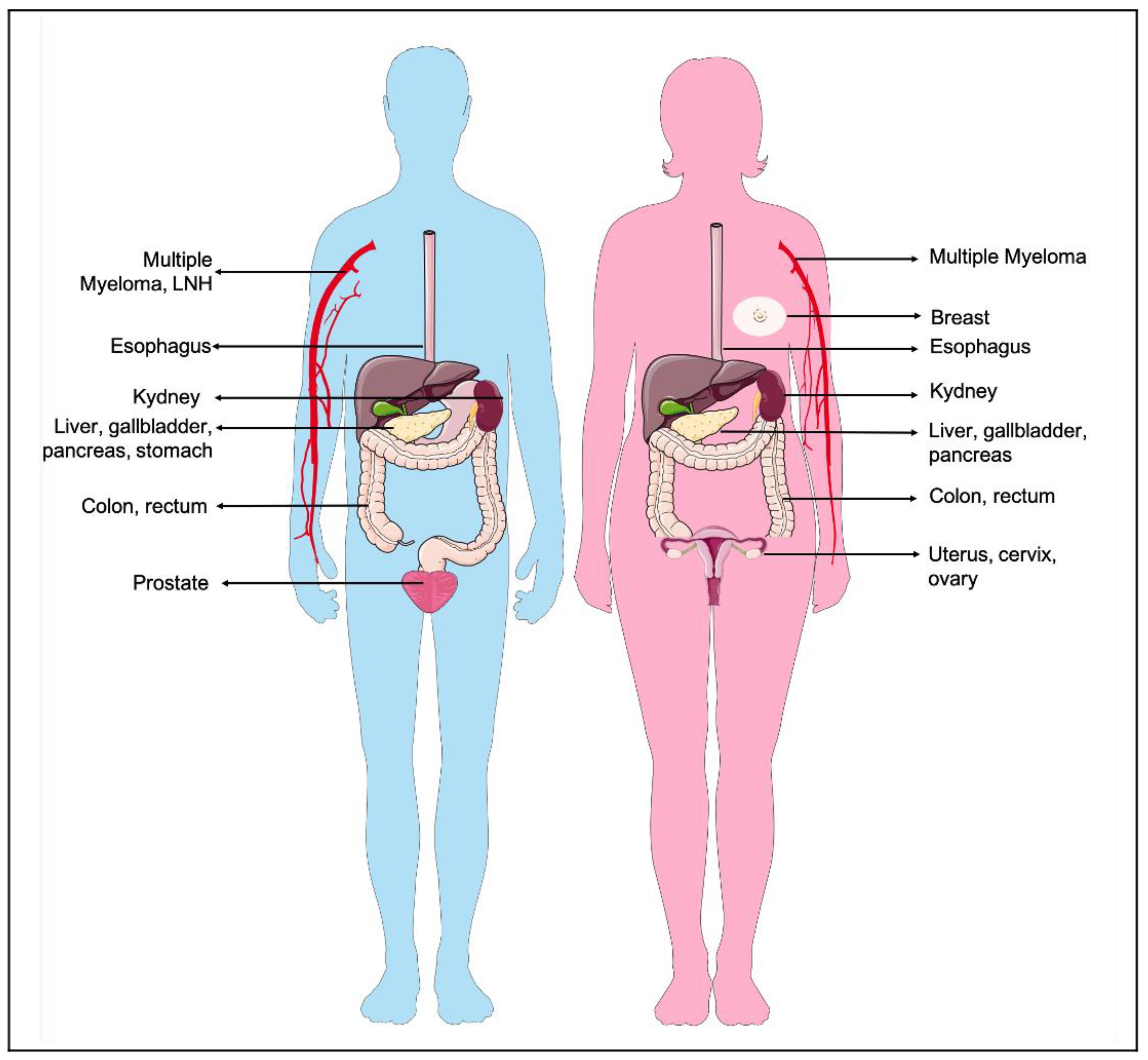{kind=link}
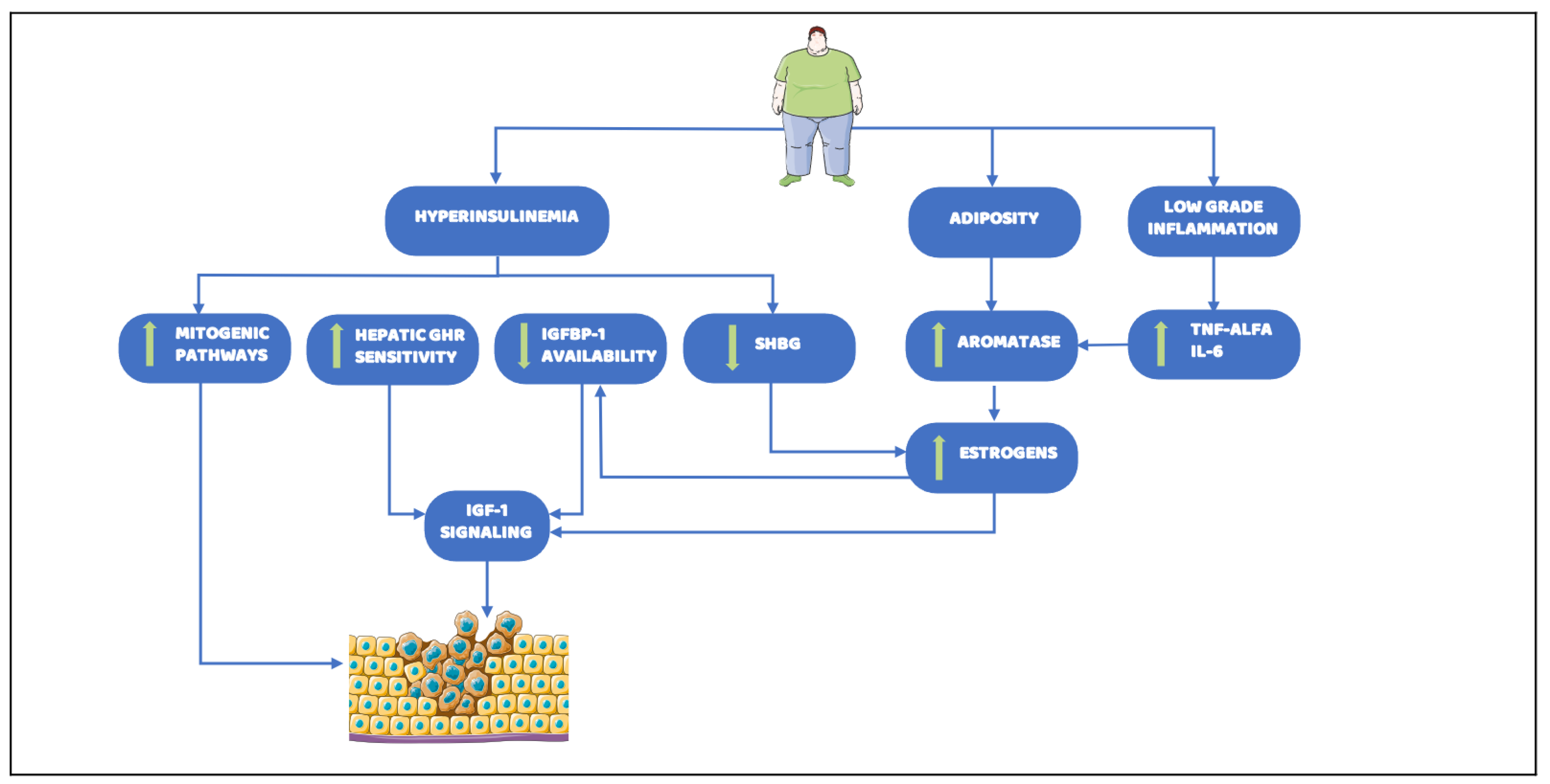{kind=link}
Table 1.
Summary of evidences for increased cancer risk in obesity.
| Cancer Site or Type | Renehan et al. 1 (2008) | IARC Working Group 2 (2006) | Wang et al. 3 (2016) |
|---|---|---|---|
| Thyroid | Men (p = 0.02) | Yes | Men (p < 0.0001) |
| Women (p = 0.001) | Women (p = 0.728) | ||
| Kidney | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | Women (p < 0.0001) | ||
| Colon | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | (colorectal) | Women (p = 0.005) | |
| Rectum | Men (p < 0.0001) | (colorectal) | |
| Esophagus | Men (p < 0.0001) | Yes | Men (p < 0.0001) |
| Women (p < 0.0001) | (adenocarcinoma) | Women (p = 0.041) | |
| (adenocarcinoma) | (esophagus and stomach) | ||
| Stomach | - | Yes | |
| Multiple Myeloma | Men (p < 0.001) | Yes | - |
| Women (p < 0.0001) | |||
| Leukemia | Men (p < 0.0001) | - | - |
| Women (p = 0.01) | |||
| Non Hodgkin Lymphoma | Men (p < 0.0001) | - | - |
| Women (p = 0.01) | |||
| Melanoma | Men (p = 0.04) | - | - |
| Gallbladder | Women (p = 0.04) | Yes | - |
| Pancreas | Women (p = 0.01) | Yes | Men (p < 0.0001) |
| Women (p = 0.014) | |||
| Liver | - | Yes | Men (p < 0.0001) |
| Women (p = 0.9) | |||
| Meningioma | - | Yes | - |
| Ovary | - | Yes | Women (p = 0.009) |
| Prostate | - | - | Men (p < 0.0001) |
| Endometrium | Women (p < 0.001) | Yes | - |
| (corpus uteri) | |||
| Postmenopausal Breast cancer | Women (p < 0.0001) | Yes | Women (p < 0.0001) |
1 Increased RR per 5 kg/m2 increase; 2 Increased Relative Risk of the highest BMI category evaluated vs Normal BMI (95% Confidence Interval), no gender difference; 3 Increased RR per 5 kg/m2 increase.
Table 2.
Proposed methods for fasting.
| Method | Description |
|---|---|
| Every-Other-Day Fasting | Food is withdrawn for 24 h on alternate days, with water provided ad libitum. Overall calorie intake need not be limited. |
| Time-Restricted Feeding | It restricts the timing of meals, without regard to their caloric content, to a time window of few hours in a day. |
| Periodic Fasting | It lasts 2 or more days and is separated from the next cycle by at least 1 week of normal feeding. |
| Brandhorst | It lasts 4 days and provides 10–50% of the normal caloric intake |
| Fasting Mimicking Diet | Periodic cycle of diets that provides a relatively high caloric content but mimics effects of fasting. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Crudele, L.; Piccinin, E.; Moschetta, A. Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients 2021, 13, 2101. https://doi.org/10.3390/nu13062101
AMA Style
Crudele L, Piccinin E, Moschetta A. Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis. Nutrients. 2021; 13(6):2101. https://doi.org/10.3390/nu13062101
Chicago/Turabian StyleCrudele, Lucilla, Elena Piccinin, and Antonio Moschetta. 2021. "Visceral Adiposity and Cancer: Role in Pathogenesis and Prognosis" Nutrients 13, no. 6: 2101. https://doi.org/10.3390/nu13062101
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.