Differential Effects of Sucrase-Isomaltase Mutants on Its Trafficking and Function in Irritable Bowel Syndrome: Similarities to Congenital Sucrase-Isomaltase Deficiency


Abstract
:1. Introduction
2. Materials and Methods
2.1. cDNA Construction
2.2. Transfection, Immunoprecipitation, SDS-PAGE and Western Blotting
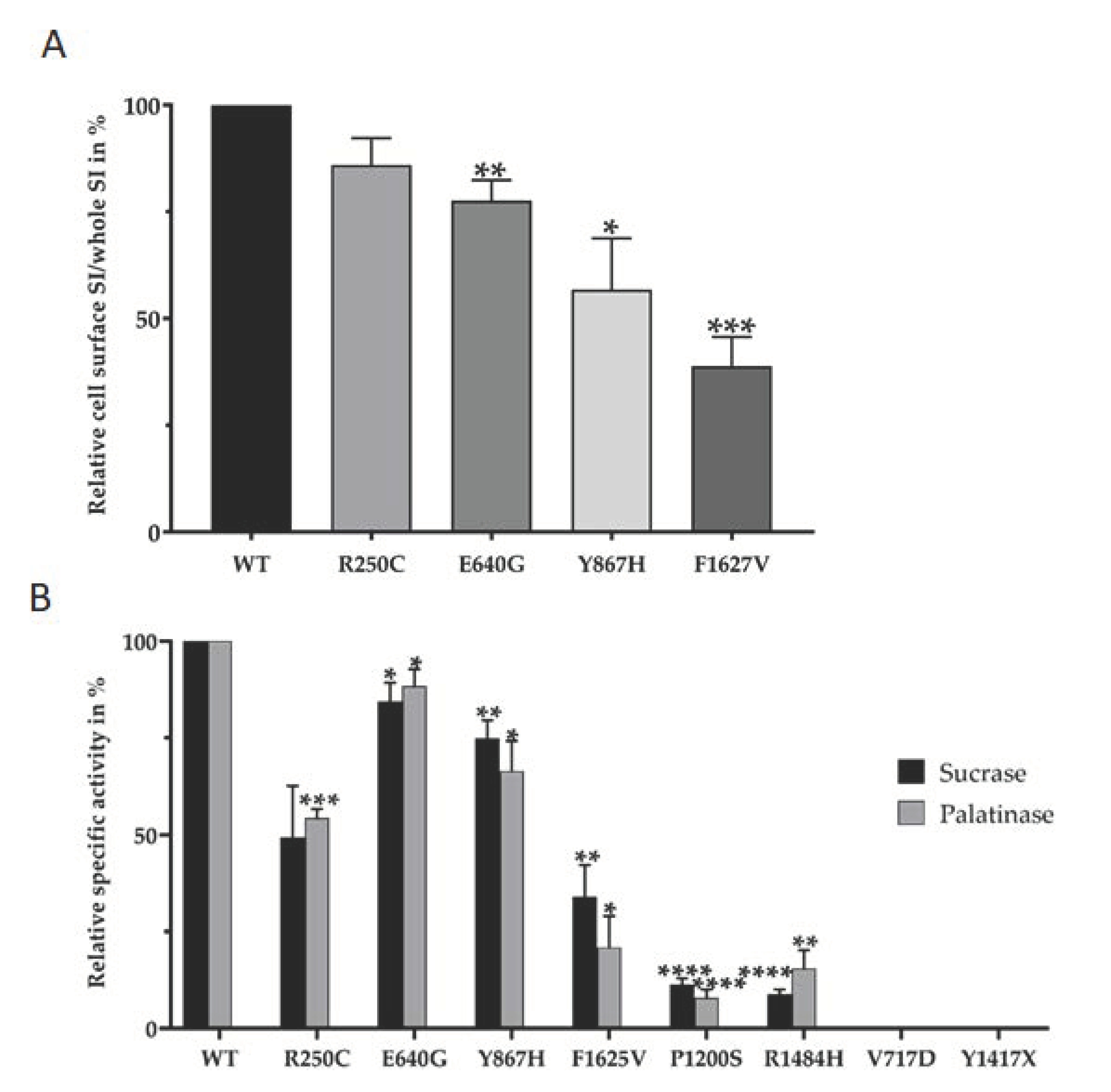
2.3. Biotinylation Assay
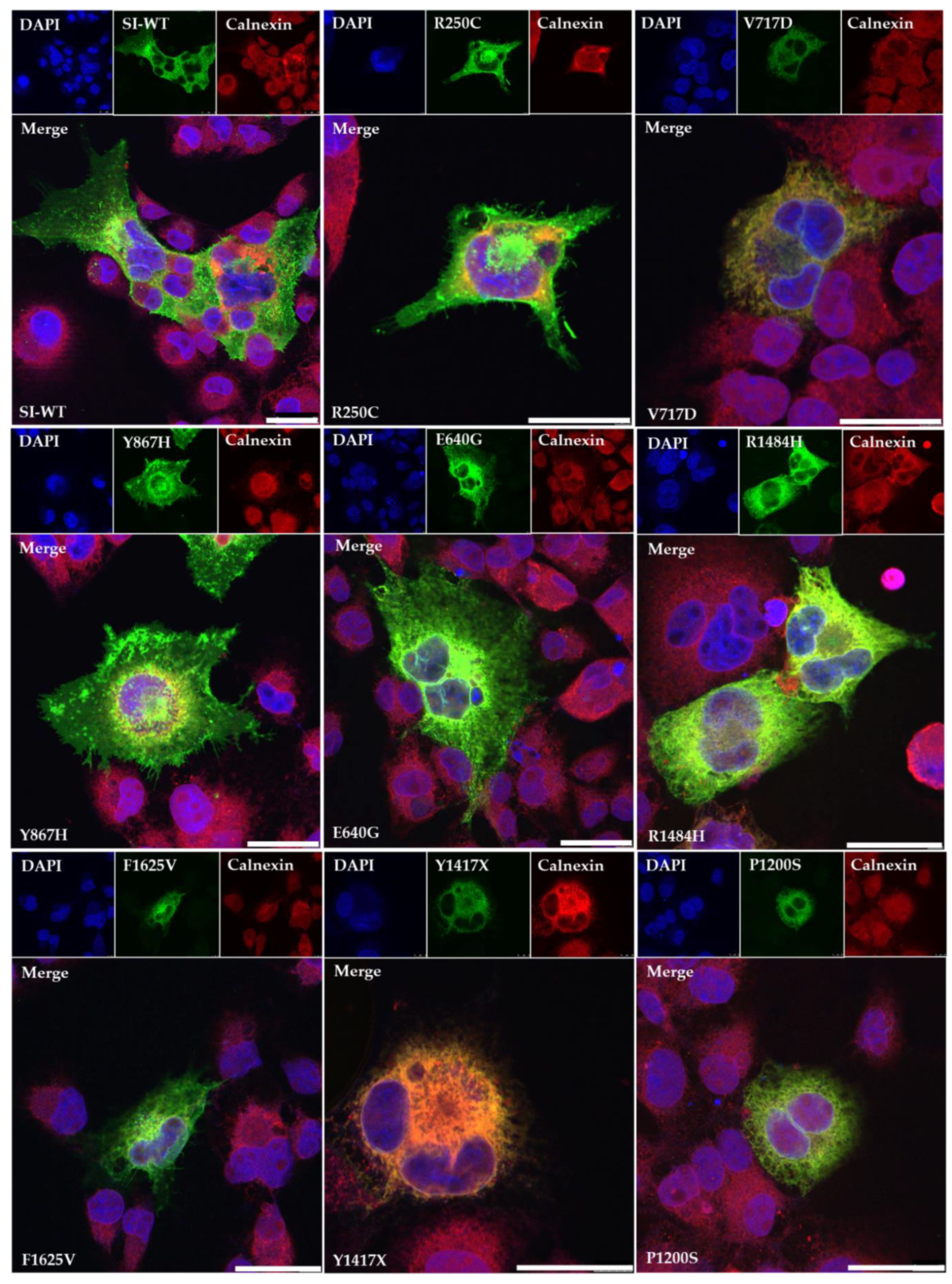
2.4. Confocal Fluorescence Microscopy
2.5. Statistical Analysis
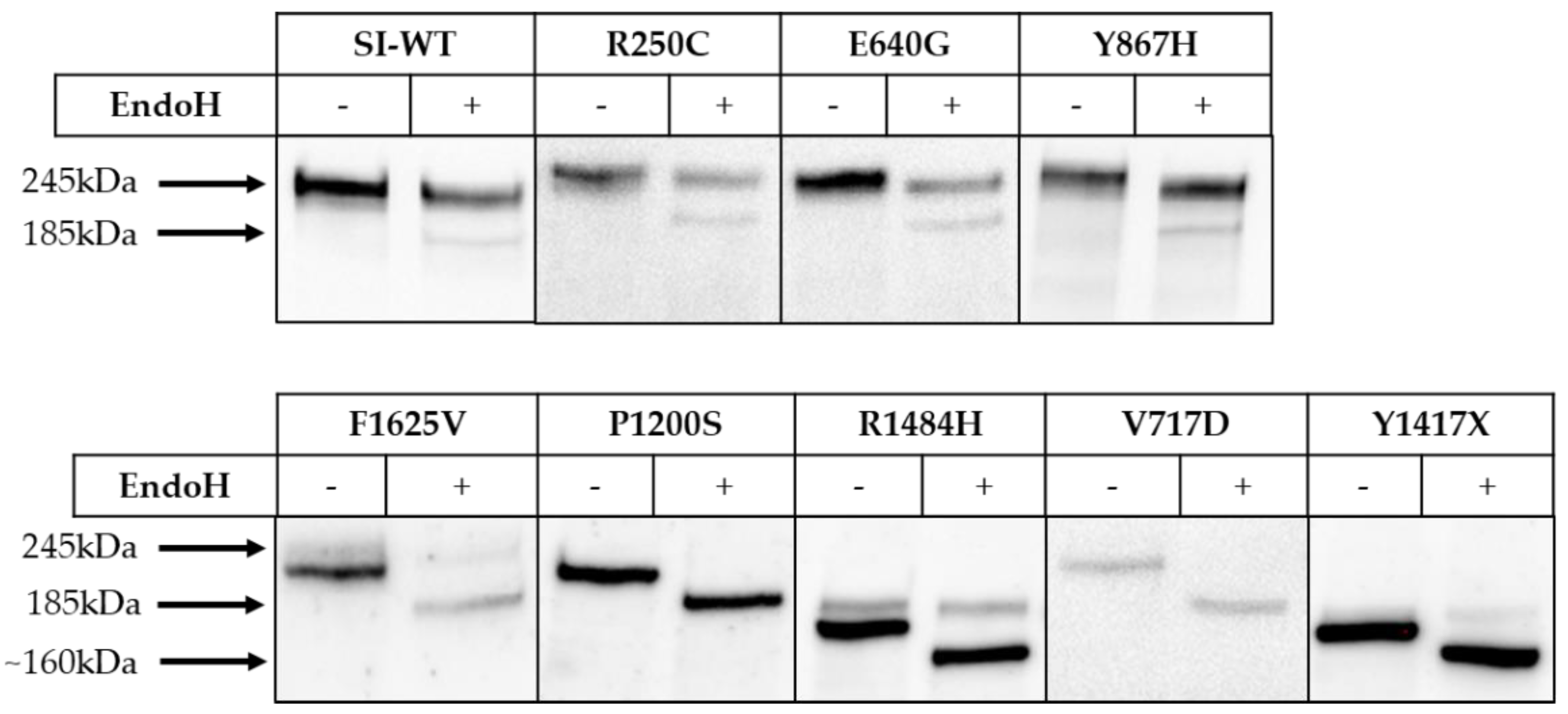
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conklin, K.A.; Yamashiro, K.M.; Gray, G.M. Human intestinal sucrase-isomaltase. Identification of free sucrase and isomaltase and cleavage of the hybrid into active distinct subunits. J. Biol. Chem. 1975, 250, 5735–5741. [Google Scholar] [PubMed]
- Treem, W.R. Congenital sucrase-isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 1995, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sander, P.; Alfalah, M.; Keiser, M.; Korponay-Szabo, I.; Kovacs, J.B.; Leeb, T.; Naim, H.Y. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum. Mutat. 2006, 27, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfalah, M.; Keiser, M.; Leeb, T.; Zimmer, K.-P.; Naim, H.Y. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology 2009, 136, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, C.E.; Ouwendijk, J.; Wittpoth, M.; Wisselaar, H.A.; Hauri, H.P.; Ginsel, L.A.; Naim, H.Y.; Fransen, J.A. A mutation in a highly conserved region in brush-border sucrase-isomaltase and lysosomal alpha-glucosidase results in Golgi retention. J. Cell Sci. 1997, 110 Pt 5, 557–567. [Google Scholar]
- Naim, H.Y.; Zimmer, K.-P. Genetically determined disaccharidase deficiency. In Walker’s Pediatric Gastrointestinal Disease; Kleinman, R., Goulet, O., Mieli-Vergani, G., Sanderson, I.S.P., Eds.; People Medical Publishing House-USA: Shelton, CT, USA, 2008; pp. 275–287. ISBN 978-1550093643. [Google Scholar]
- Naim, H.Y.; Heine, M.; Zimmer, K.-P. Congenital sucrase-isomaltase deficiency: Heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J. Pediatr. Gastroenterol. Nutr. 2012, 55 (Suppl. 2), S13–S20. [Google Scholar] [CrossRef] [Green Version]
- Robayo-Torres, C.C.; Quezada-Calvillo, R.; Nichols, B.L. Disaccharide digestion: Clinical and molecular aspects. Clin. Gastroenterol. Hepatol. 2006, 4, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Gericke, B.; Amiri, M.; Scott, C.R.; Naim, H.Y. Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Henstrom, M.; Diekmann, L.; Bonfiglio, F.; Hadizadeh, F.; Kuech, E.-M.; von Kockritz-Blickwede, M.; Thingholm, L.B.; Zheng, T.; Assadi, G.; Dierks, C.; et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut 2018, 67, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Etxebarria, K.; Zheng, T.; Bonfiglio, F.; Bujanda, L.; Dlugosz, A.; Lindberg, G.; Schmidt, P.T.; Karling, P.; Ohlsson, B.; Simren, M.; et al. Increased Prevalence of Rare Sucrase-isomaltase Pathogenic Variants in Irritable Bowel Syndrome Patients. Clin. Gastroenterol. Hepatol. 2018, 16, 1673–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husein, D.M.; Naim, H. Impaired cell surface expression and digestive function of sucrase-isomaltase gene variants are associated with reduced efficacy of low FODMAPs diet in patients with IBS-D. Gut 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chey, W.D.; Kurlander, J.; Eswaran, S. Irritable bowel syndrome: A clinical review. JAMA 2015, 313, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Self, M.M.; Czyzewski, D.I.; Chumpitazi, B.P.; Weidler, E.M.; Shulman, R.J. Subtypes of irritable bowel syndrome in children and adolescents. Clin. Gastroenterol. Hepatol. 2014, 12, 1468–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Eswaran, S.; Photenhauer, A.L.; Merchant, J.L.; Chey, W.D.; D’Amato, M. Reduced efficacy of low FODMAPs diet in patients with IBS-D carrying sucrase-isomaltase (SI) hypomorphic variants. Gut 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwendijk, J.; Moolenaar, C.E.; Peters, W.J.; Hollenberg, C.P.; Ginsel, L.A.; Fransen, J.A.; Naim, H.Y. Congenital sucrase-isomaltase deficiency. Identification of a glutamine to proline substitution that leads to a transport block of sucrase-isomaltase in a pre-Golgi compartment. J. Clin. Investig. 1996, 97, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Naim, H.Y.; Lacey, S.W.; Sambrook, J.F.; Gething, M.J. Expression of a full-length cDNA coding for human intestinal lactase-phlorizin hydrolase reveals an uncleaved, enzymatically active, and transport-competent protein. J. Biol. Chem. 1991, 266, 12313–12320. [Google Scholar] [PubMed]
- Hauri, H.P.; Sterchi, E.E.; Bienz, D.; Fransen, J.A.; Marxer, A. Expression and intracellular transport of microvillus membrane hydrolases in human intestinal epithelial cells. J. Cell Biol. 1985, 101, 838–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spodsberg, N.; Jacob, R.; Alfalah, M.; Zimmer, K.P.; Naim, H.Y. Molecular basis of aberrant apical protein transport in an intestinal enzyme disorder. J. Biol. Chem. 2001, 276, 23506–23510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.H.-M.; Hamaker, B.R.; Nichols, B.L.J. Direct starch digestion by sucrase-isomaltase and maltase-glucoamylase. J. Pediatr. Gastroenterol. Nutr. 2012, 55 (Suppl. 2), S43–S45. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-H.; Eskandari, R.; Jones, K.; Reddy, K.R.; Quezada-Calvillo, R.; Nichols, B.L.; Rose, D.R.; Hamaker, B.R.; Pinto, B.M. Modulation of starch digestion for slow glucose release through “toggling” of activities of mucosal alpha-glucosidases. J. Biol. Chem. 2012, 287, 31929–31938. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Mutation | Primer | Amino Acid Exchange |
|---|---|---|
| c.748C>T | gagattttgtcatgatttatcctggaaaacat | Arg→Cyst (p.R250C) |
| c.1919A>G | tgtggctggtaccacagaagaactttg | Glu→Gly (p.E640G) |
| 2150T>A | aagaccagatcttcatgagttttatgagg | Val→Asp (p.V717D) |
| c.2599T>C | attcatcacatcaggaaggaactaccttag | Tyr→His (p.Y867H) |
| c.3598C>T | tttgggctcaactccacaagttgcaa | Pro→Ser (p.P1200S) |
| c.4251T>A | acgaacttaattaaccaccttatttcccagaac | Tyr→Stop (p.Y1417X) |
| c.4451G>A | aatttctcattccacgtatcctactagtgg | Tyr→Hist (p.Y1484H) |
| c.4873T>G | gggatatagtcaagcagttcttatgggg | Phe→Val (p.F1625V) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Husein, D.M.; Rizk, S.; Naim, H.Y. Differential Effects of Sucrase-Isomaltase Mutants on Its Trafficking and Function in Irritable Bowel Syndrome: Similarities to Congenital Sucrase-Isomaltase Deficiency. Nutrients 2021, 13, 9. https://doi.org/10.3390/nu13010009
Husein DM, Rizk S, Naim HY. Differential Effects of Sucrase-Isomaltase Mutants on Its Trafficking and Function in Irritable Bowel Syndrome: Similarities to Congenital Sucrase-Isomaltase Deficiency. Nutrients. 2021; 13(1):9. https://doi.org/10.3390/nu13010009
Chicago/Turabian StyleHusein, Diab M., Sandra Rizk, and Hassan Y. Naim. 2021. "Differential Effects of Sucrase-Isomaltase Mutants on Its Trafficking and Function in Irritable Bowel Syndrome: Similarities to Congenital Sucrase-Isomaltase Deficiency" Nutrients 13, no. 1: 9. https://doi.org/10.3390/nu13010009