Sinensetin Induces Autophagic Cell Death through p53-Related AMPK/mTOR Signaling in Hepatocellular Carcinoma HepG2 Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cell Viability and Morphology
2.4. Cell Cycle Progression
2.5. Annexin V-Propidium Iodide Apoptosis Detection
2.6. DNA Fragmentation Assay
2.7. Detection of Acidic Vesicular Organelles (AVO) with Acridine Orange (AO)
2.8. Immunoblotting Analysis
2.9. Transmission Electron Microscopy (TEM)
2.10. Preparation of the Protein and Ligands
2.11. Molecular Docking Studies
2.12. Statistical Analysis
3. Results
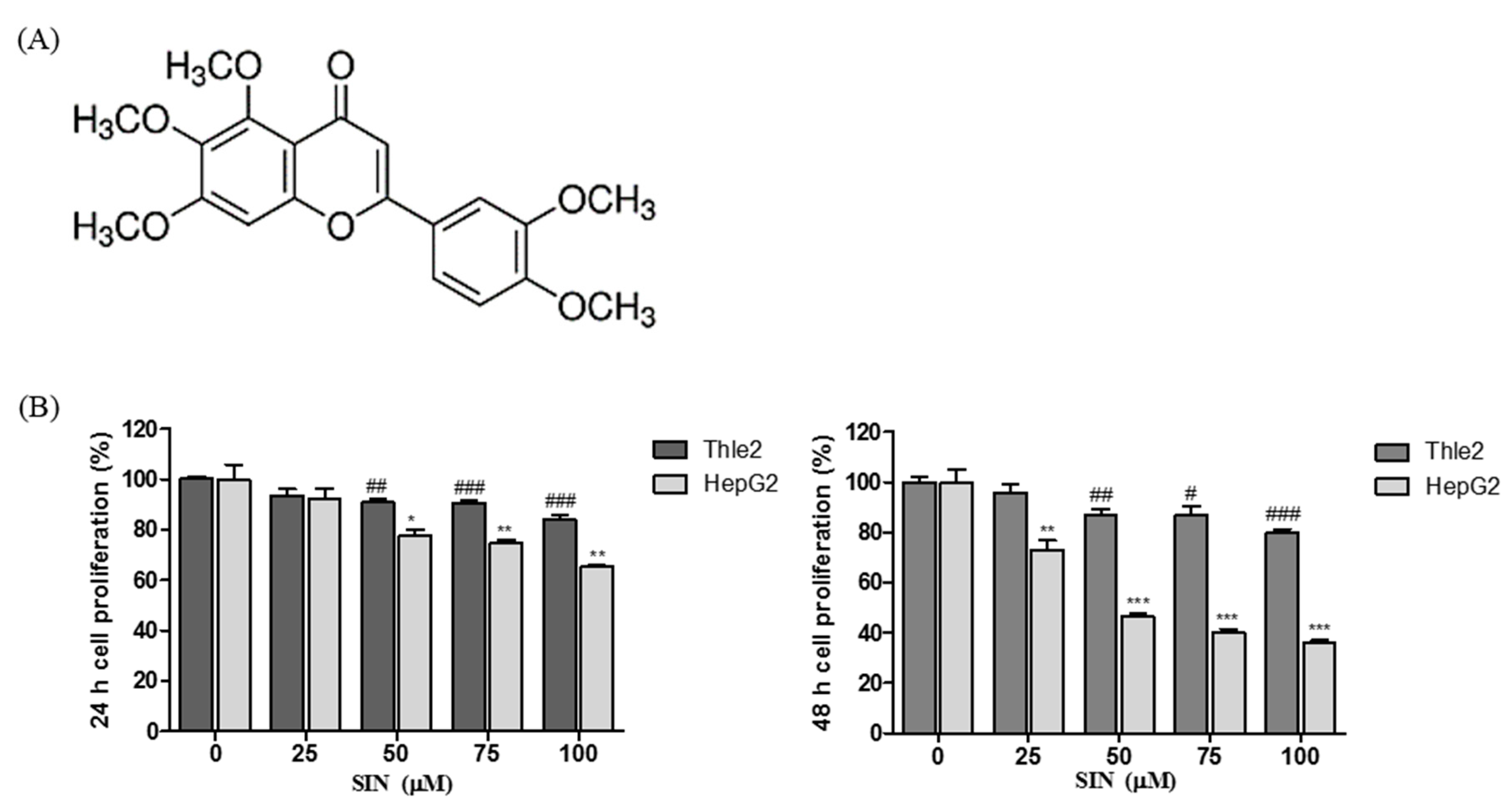
3.1. SIN Decreases Cell Viability in HepG2 Cells
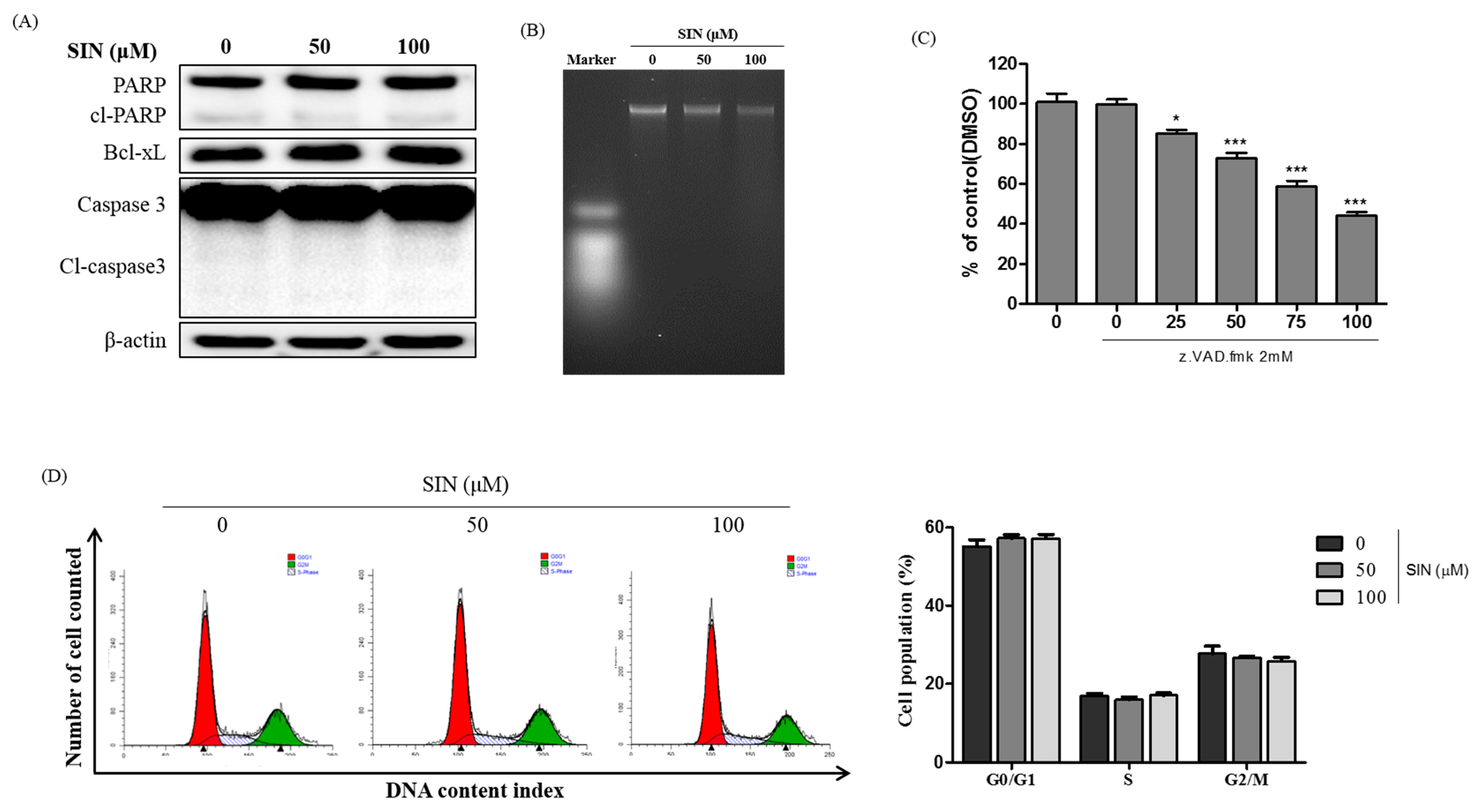
3.2. SIN Has No Apoptotic Effects and Cell Cycle Arrest in HepG2 Cells
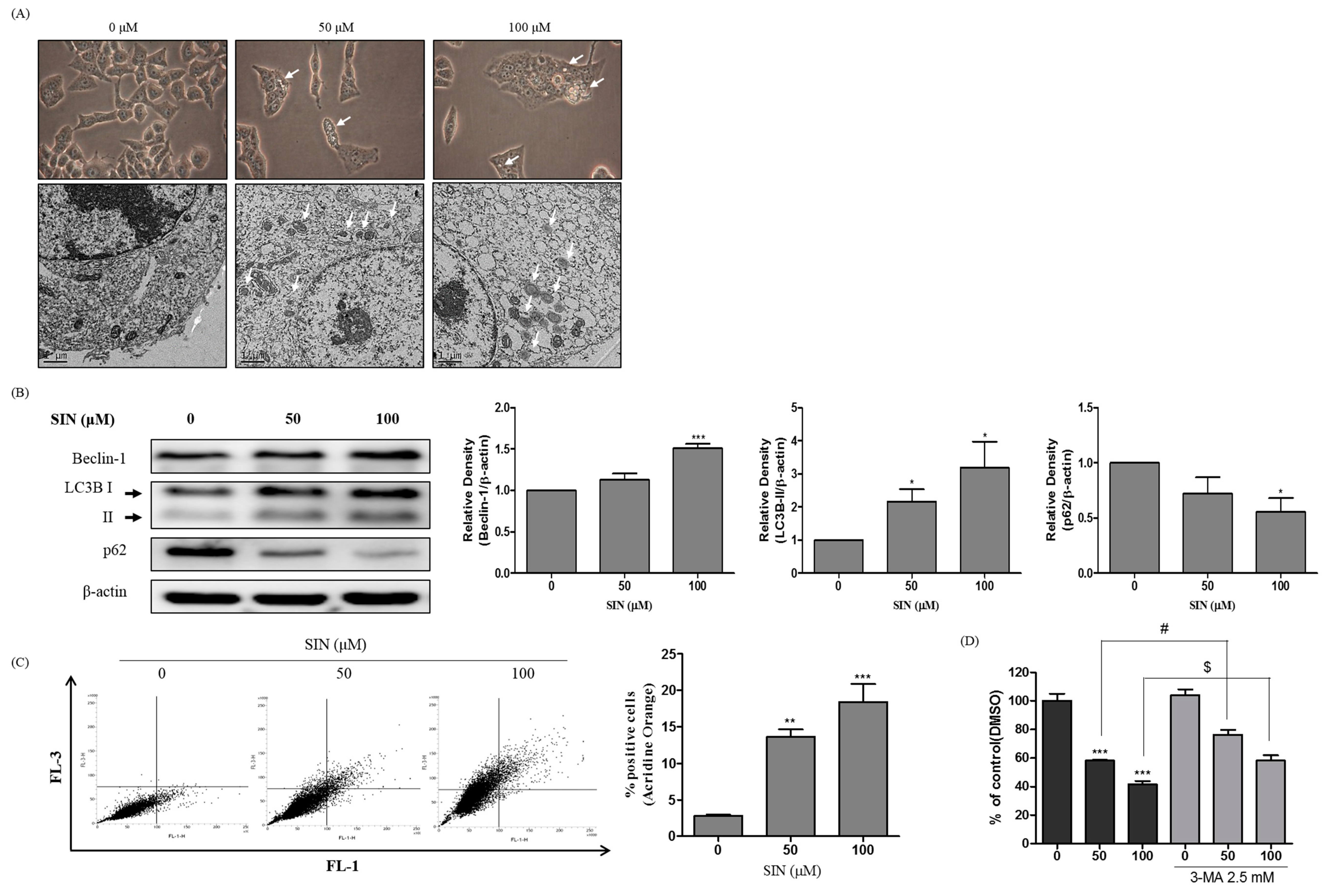
3.3. SIN Triggers Autophagic Cell Death in HepG2 Cells
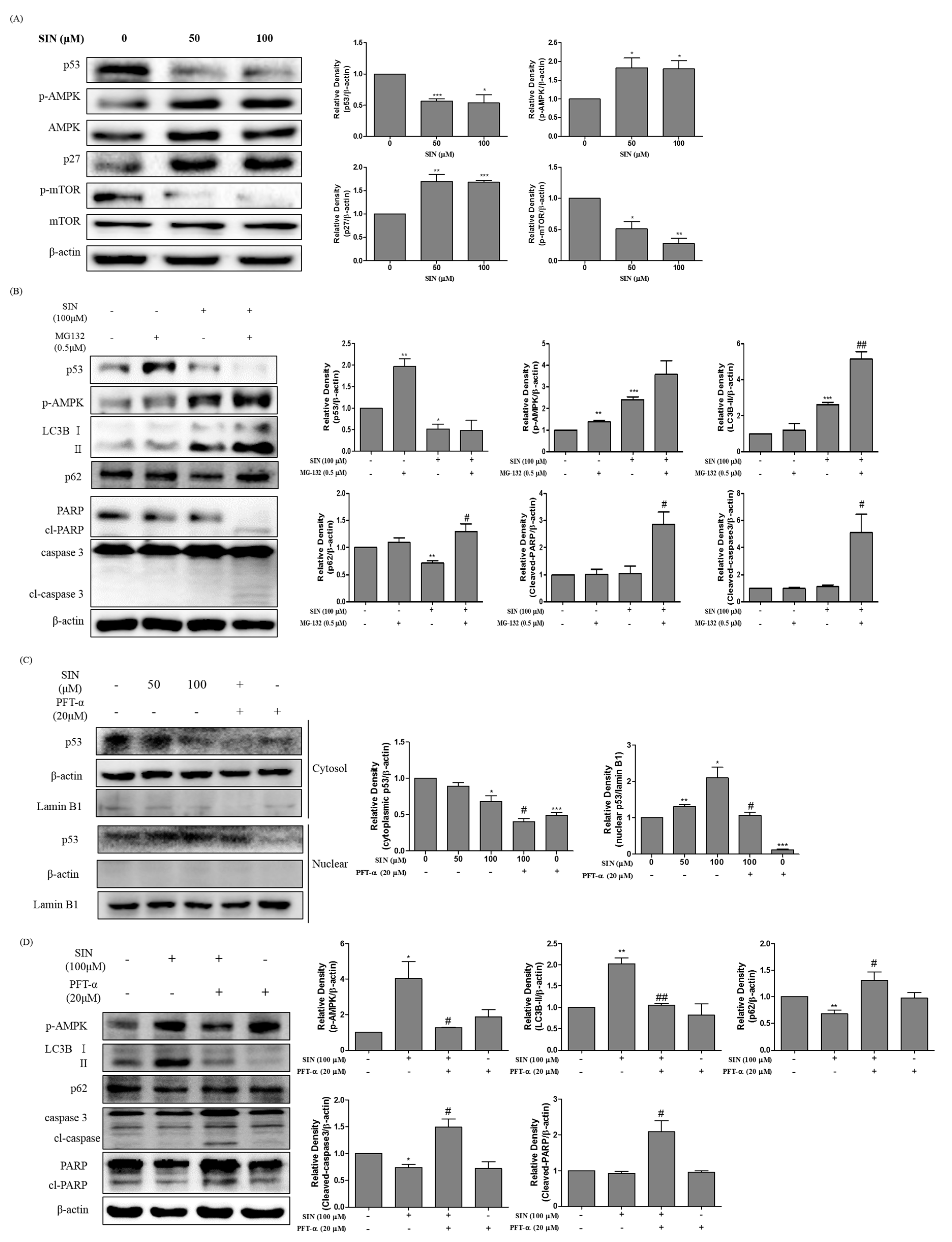
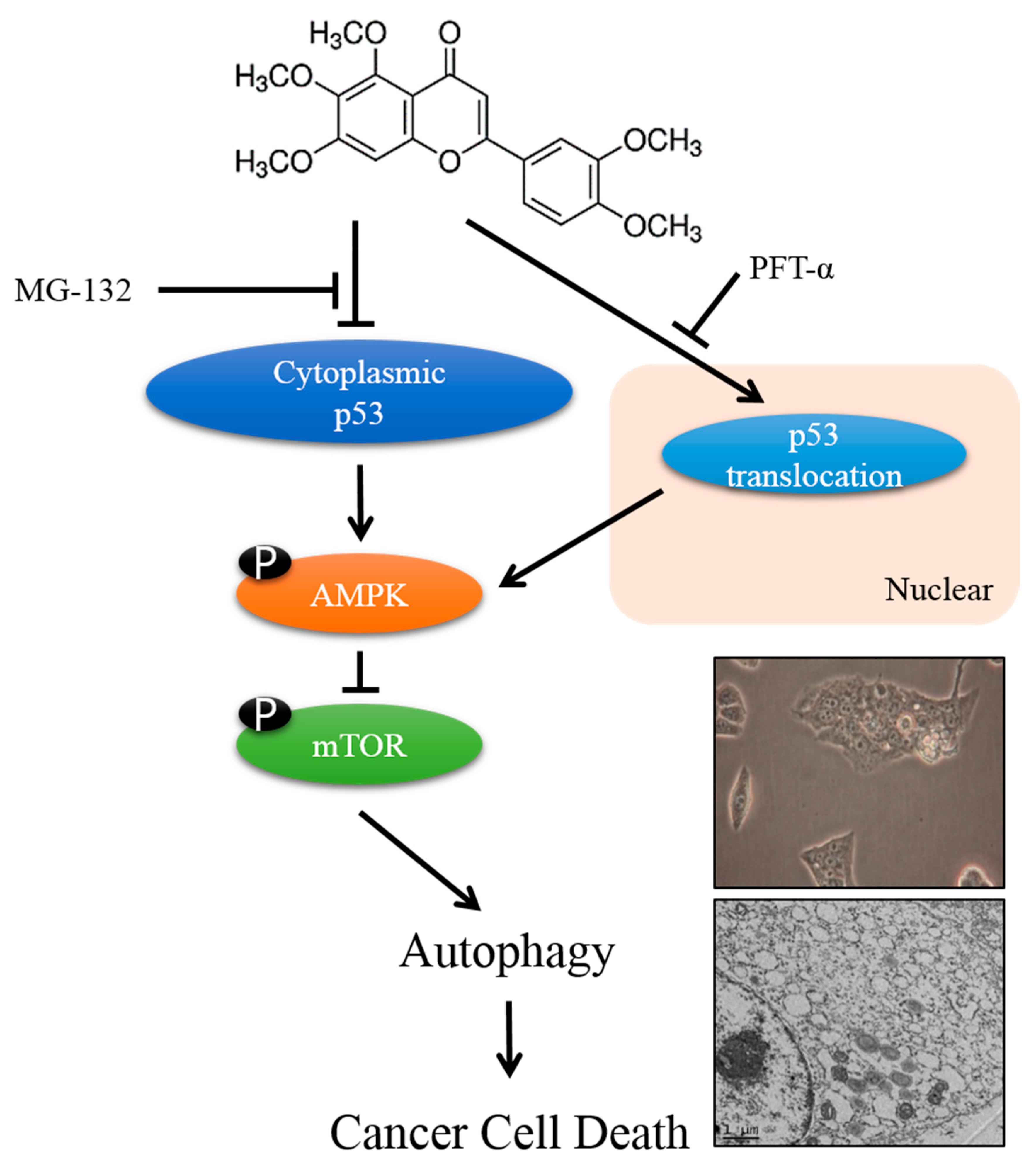
3.4. SIN Treatment Induces Autophagy through the p53-Mediated AMPK/mTOR Pathway in HepG2 Cells
3.5. SIN Increase Translocation of p53 in HepG2 Cells
3.6. SIN Induces Apoptosis in p53-Null Hep3B Cells
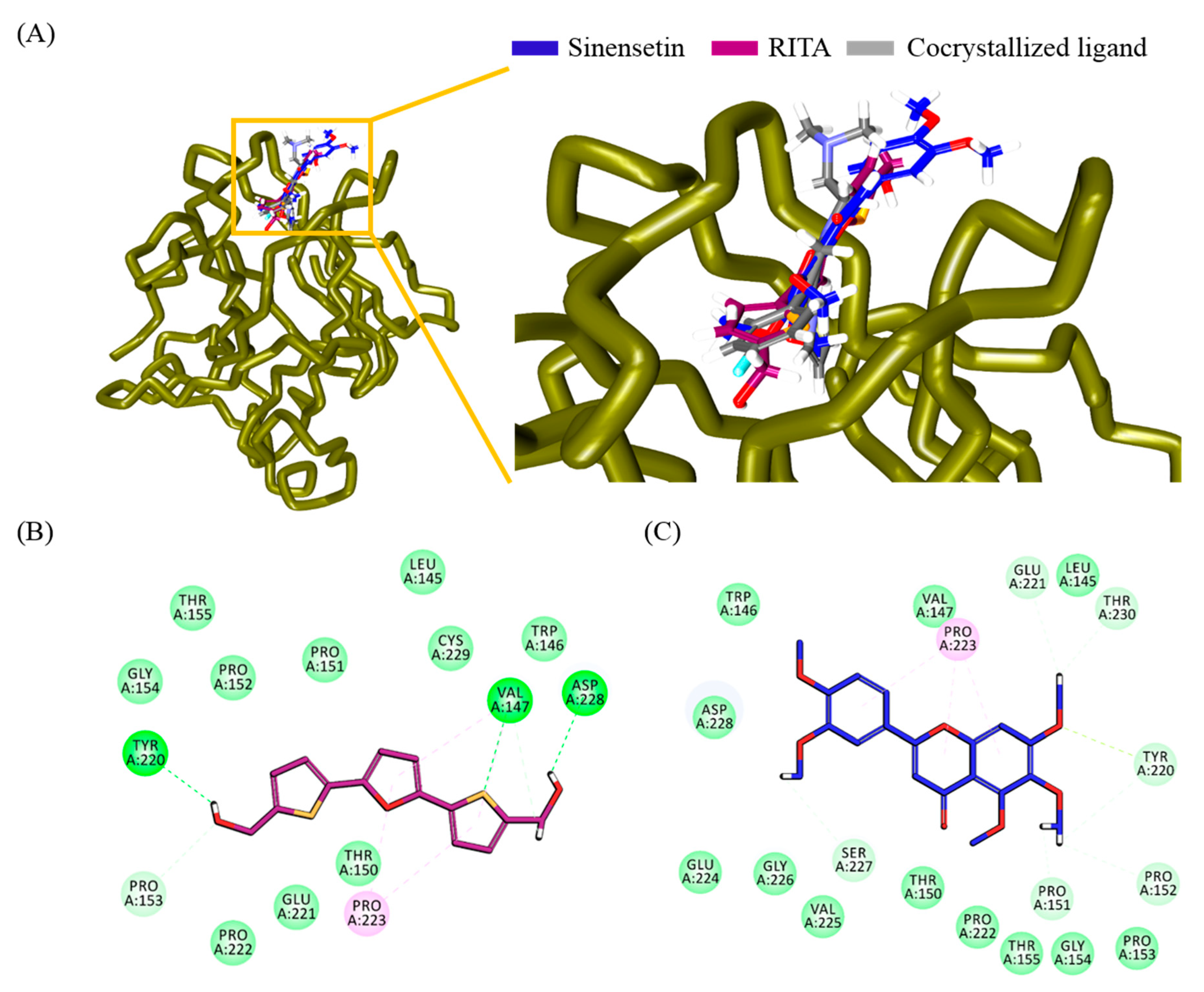
3.7. Evaluation and Validation by Molecular Docking
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Banjerdpongchai, R.; Wudtiwai, B.; Khaw-On, P.; Rachakhom, W.; Duangnil, N.; Kongtawelert, P. Hesperidin from citrus seed induces human hepatocellular carcinoma hepg2 cell apoptosis via both mitochondrial and death receptor pathways. Tumor Biol. 2016, 37, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Zhou, Y.; Li, Y.; Zhou, T.; Zheng, J.; Li, S.; Li, H.B. Dietary natural products for prevention and treatment of liver cancer. Nutrients 2016, 8, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. CR 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Klionsky, D.J. Autophagy and human disease. Cell Cycle 2007, 6, 1837–1849. [Google Scholar] [CrossRef] [Green Version]
- Moretti, L.; Yang, E.S.; Kim, K.W.; Lu, B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist. Updat. 2007, 10, 135–143. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. Amp-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef] [Green Version]
- Budanov, A.V.; Karin, M. P53 target genes sestrin1 and sestrin2 connect genotoxic stress and mtor signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis An. Int. J. Program. Cell Death 2008, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrakovcic, M.; Frohlich, L.F. P53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Jiang, Z.; Ma, H.; Ning, L.; Chen, H.; Li, L.; Qi, H. Volatile oil of acori graminei rhizoma-induced apoptosis and autophagy are dependent on p53 status in human glioma cells. Sci. Rep. 2016, 6, 21148. [Google Scholar] [CrossRef]
- Park, H.S.; Park, K.I.; Lee, D.H.; Kang, S.R.; Nagappan, A.; Kim, J.A.; Kim, E.H.; Lee, W.S.; Shin, S.C.; Hah, Y.S.; et al. Polyphenolic extract isolated from korean lonicera japonica thunb. Induce g2/m cell cycle arrest and apoptosis in hepg2 cells: Involvements of pi3k/akt and mapks. Food Chem. Toxicol. 2012, 50, 2407–2416. [Google Scholar] [CrossRef]
- Deng, G.L.; Zeng, S.; Shen, H. Chemotherapy and target therapy for hepatocellular carcinoma: New advances and challenges. World J. Hepatol. 2015, 7, 787–798. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, J.O.; Han, S.W.; Oh, D.Y.; Im, S.A.; Kim, T.Y.; Bang, Y.J. Clinical outcomes of sorafenib treatment in patients with metastatic hepatocellular carcinoma who had been previously treated with fluoropyrimidine plus platinum-based chemotherapy. Am. J. Clin. Oncol. 2011, 34, 125–129. [Google Scholar] [CrossRef]
- Lall, R.K.; Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Dietary polyphenols in prevention and treatment of prostate cancer. Int. J. Mol. Sci. 2015, 16, 3350–3376. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer. Res. 2015, 35, 645–651. [Google Scholar]
- Kang, S.I.; Shin, H.S.; Kim, S.J. Sinensetin enhances adipogenesis and lipolysis by increasing cyclic adenosine monophosphate levels in 3t3-l1 adipocytes. Biol. Pharm. Bull. 2015, 38, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Ji, G.; Cao, A.; Shi, J.; Shi, H.; Xie, J.; Wu, D. Effects of sinensetin on proliferation and apoptosis of human gastric cancer ags cells. Zhongguo Zhong Yao Za Zhi 2011, 36, 790–794. [Google Scholar] [PubMed]
- Lee, S.H.; Yumnam, S.; Hong, G.E.; Raha, S.; Saralamma, V.V.; Lee, H.J.; Heo, J.D.; Lee, S.J.; Lee, W.S.; Kim, E.H.; et al. Flavonoids of korean citrus aurantium l. Induce apoptosis via intrinsic pathway in human hepatoblastoma hepg2 cells. Phytother. Res. 2015, 29, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.T.; Lin, M.X.; Lin, S.C.; Tung, Y.T.; Lin, S.H.; Lin, C.C. Sinensetin induces apoptosis and autophagy in the treatment of human t-cell lymphoma. Anticancer. Drugs 2019, 30, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rosengarth, A.; Luecke, H. Structure of the human p53 core domain in the absence of DNA. Acta Cryst. D Biol. Cryst. 2007, 63, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhao, J.; Chen, Z. Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: A case of oligopeptide binding protein. Comput. Math. Methods Med. 2018, 2018, 3502514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.X.; Ouyang, L.; Cheng, Y.; Liu, B. Plant natural compounds: Targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif. 2012, 45, 466–476. [Google Scholar] [CrossRef]
- Qiu, G.H.; Xie, X.; Xu, F.; Shi, X.; Wang, Y.; Deng, L. Distinctive pharmacological differences between liver cancer cell lines hepg2 and hep3b. Cytotechnology 2015, 67, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.W.; Leung, Y.K.; Chan, C.P. Herbal medicine in the treatment of cancer. Curr. Med. Chem. Anticancer. Agents 2002, 2, 209–214. [Google Scholar] [CrossRef]
- Lee, H.J.; Nagappan, A.; Park, H.S.; Hong, G.E.; Yumnam, S.; Raha, S.; Saralamma, V.V.; Lee, W.S.; Kim, E.H.; Kim, G.S. Flavonoids isolated from citrus platymamma induce mitochondrial-dependent apoptosis in ags cells by modulation of the pi3k/akt and mapk pathways. Oncol. Rep. 2015, 34, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Lam, I.K.; Alex, D.; Wang, Y.H.; Liu, P.; Liu, A.L.; Du, G.H.; Lee, S.M. In vitro and in vivo structure and activity relationship analysis of polymethoxylated flavonoids: Identifying sinensetin as a novel antiangiogenesis agent. Mol. Nutr. Food Res. 2012, 56, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.A.; Siddiqui, M.J.; Ang, L.F.; Sadikun, A.; Chan, S.H.; Tan, S.C.; Asmawi, M.Z.; Yam, M.F. Potent alpha-glucosidase and alpha-amylase inhibitory activities of standardized 50% ethanolic extracts and sinensetin from orthosiphon stamineus benth as anti-diabetic mechanism. BMC Complement. Altern. Med. 2012, 12, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.S.; Kang, S.I.; Yoon, S.A.; Ko, H.C.; Kim, S.J. Sinensetin attenuates lps-induced inflammation by regulating the protein level of ikappab-alpha. Biosci. Biotechnol. Biochem. 2012, 76, 847–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Venkatarame Gowda Saralamma, V.; Kim, S.M.; Ha, S.E.; Raha, S.; Lee, W.S.; Kim, E.H.; Lee, S.J.; Heo, J.D.; Kim, G.S. Pectolinarigenin induced cell cycle arrest, autophagy, and apoptosis in gastric cancer cell via pi3k/akt/mtor signaling pathway. Nutrients 2018, 10, 1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raha, S.; Yumnam, S.; Hong, G.E.; Lee, H.J.; Saralamma, V.V.; Park, H.S.; Heo, J.D.; Lee, S.J.; Kim, E.H.; Kim, J.A.; et al. Naringin induces autophagy-mediated growth inhibition by downregulating the pi3k/akt/mtor cascade via activation of mapk pathways in ags cancer cells. Int. J. Oncol. 2015, 47, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19. [Google Scholar]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. Lc3, a mammalian homologue of yeast apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Mariggio, M.A.; Racanelli, V.; Vacca, A.; Frassanito, M.A. Autophagy: A new mechanism of prosurvival and drug resistance in multiple myeloma. Transl. Oncol. 2018, 11, 1350–1357. [Google Scholar] [CrossRef]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/ampk/mtor signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Abrams, J. P53: The janus of autophagy? Nat. Cell Biol. 2008, 10, 637–639. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Galluzzi, L.; Morselli, E.; Kepp, O.; Malik, S.A.; Kroemer, G. Autophagy regulation by p53. Curr. Opin. Cell Biol. 2010, 22, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline induces autophagic cell death via p53/sestrin2/ampk signalling in human hepatocellular carcinoma cells. Br. J. Pharm. 2011, 164, 731–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.M.; Ha, S.E.; Lee, H.J.; Rampogu, S.; Vetrivel, P.; Kim, H.H.; Venkatarame Gowda Saralamma, V.; Lee, K.W.; Kim, G.S. Sinensetin Induces Autophagic Cell Death through p53-Related AMPK/mTOR Signaling in Hepatocellular Carcinoma HepG2 Cells. Nutrients 2020, 12, 2462. https://doi.org/10.3390/nu12082462
Kim SM, Ha SE, Lee HJ, Rampogu S, Vetrivel P, Kim HH, Venkatarame Gowda Saralamma V, Lee KW, Kim GS. Sinensetin Induces Autophagic Cell Death through p53-Related AMPK/mTOR Signaling in Hepatocellular Carcinoma HepG2 Cells. Nutrients. 2020; 12(8):2462. https://doi.org/10.3390/nu12082462
Chicago/Turabian StyleKim, Seong Min, Sang Eun Ha, Ho Jeong Lee, Shailima Rampogu, Preethi Vetrivel, Hun Hwan Kim, Venu Venkatarame Gowda Saralamma, Keun Woo Lee, and Gon Sup Kim. 2020. "Sinensetin Induces Autophagic Cell Death through p53-Related AMPK/mTOR Signaling in Hepatocellular Carcinoma HepG2 Cells" Nutrients 12, no. 8: 2462. https://doi.org/10.3390/nu12082462