The Role of Mitochondria in Sex-Dependent Differences in Hepatic Steatosis and Oxidative Stress in Response to Cafeteria Diet-Induced Obesity in Mice

 ,
,
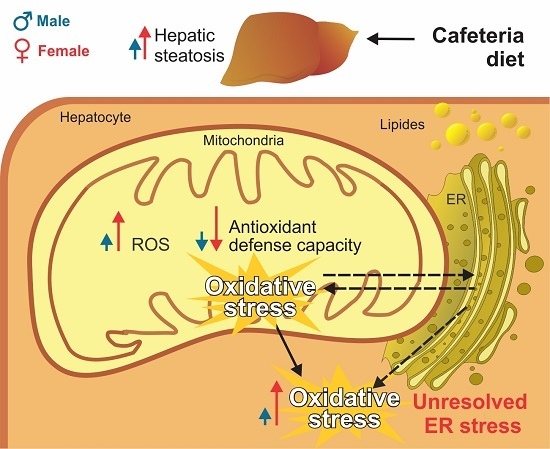
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Diet
2.4. Adiposity Index
2.5. Liver Histochemical Analysis and Determination of the Total Lipid Content
2.6. Liver Mitochondria Isolation
2.7. Determination of Oxidative Injury Parameters
2.8. Determination of Redox State Parameters
2.9. Determination of Antioxidant Enzyme Activities
2.10. Determination of the Respiratory Activity of Isolated Mitochondria
2.11. ATPase Activity
2.12. Peroxisomal Fatty Acyl-CoA Oxidase Activity
2.13. Liver Perfusion Experiments
2.14. Analytical Assays of Liver Perfusion Experiments
2.15. Statistical Analysis
3. Results
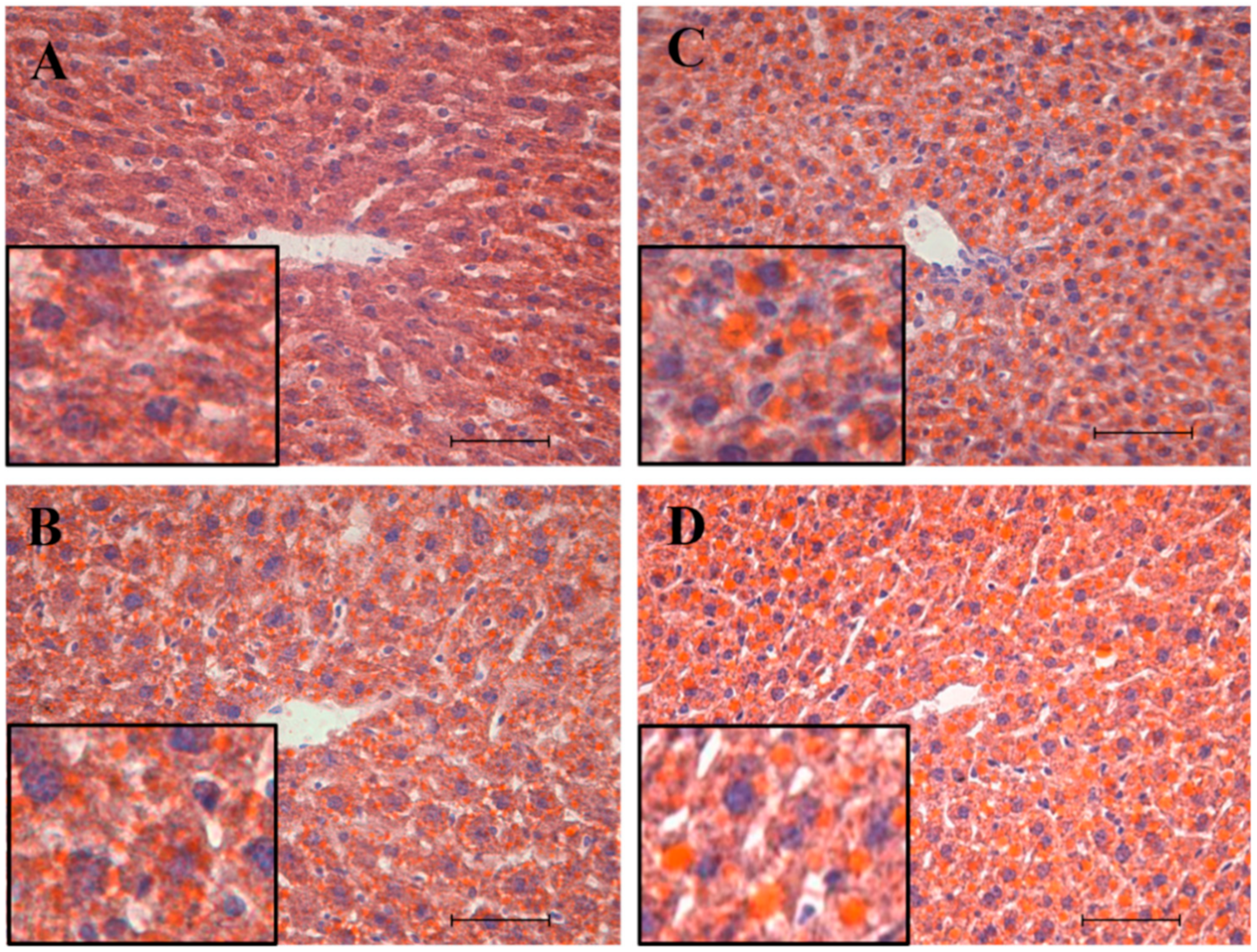
3.1. Biometric Parameters and Liver Histochemical Analysis
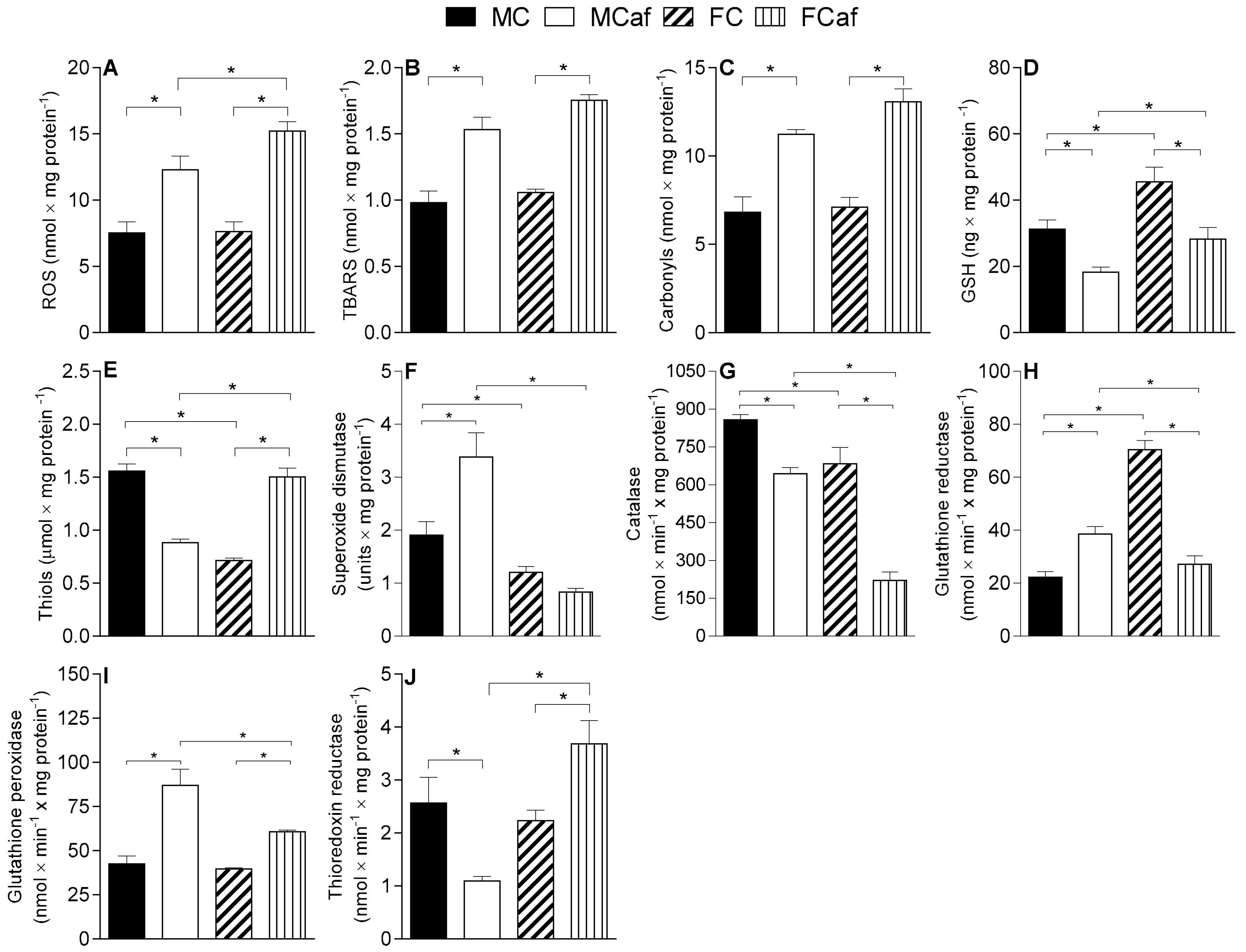
3.2. Parameters of Oxidative Stress and Antioxidant Enzyme Activities in Liver-Isolated Mitochondria
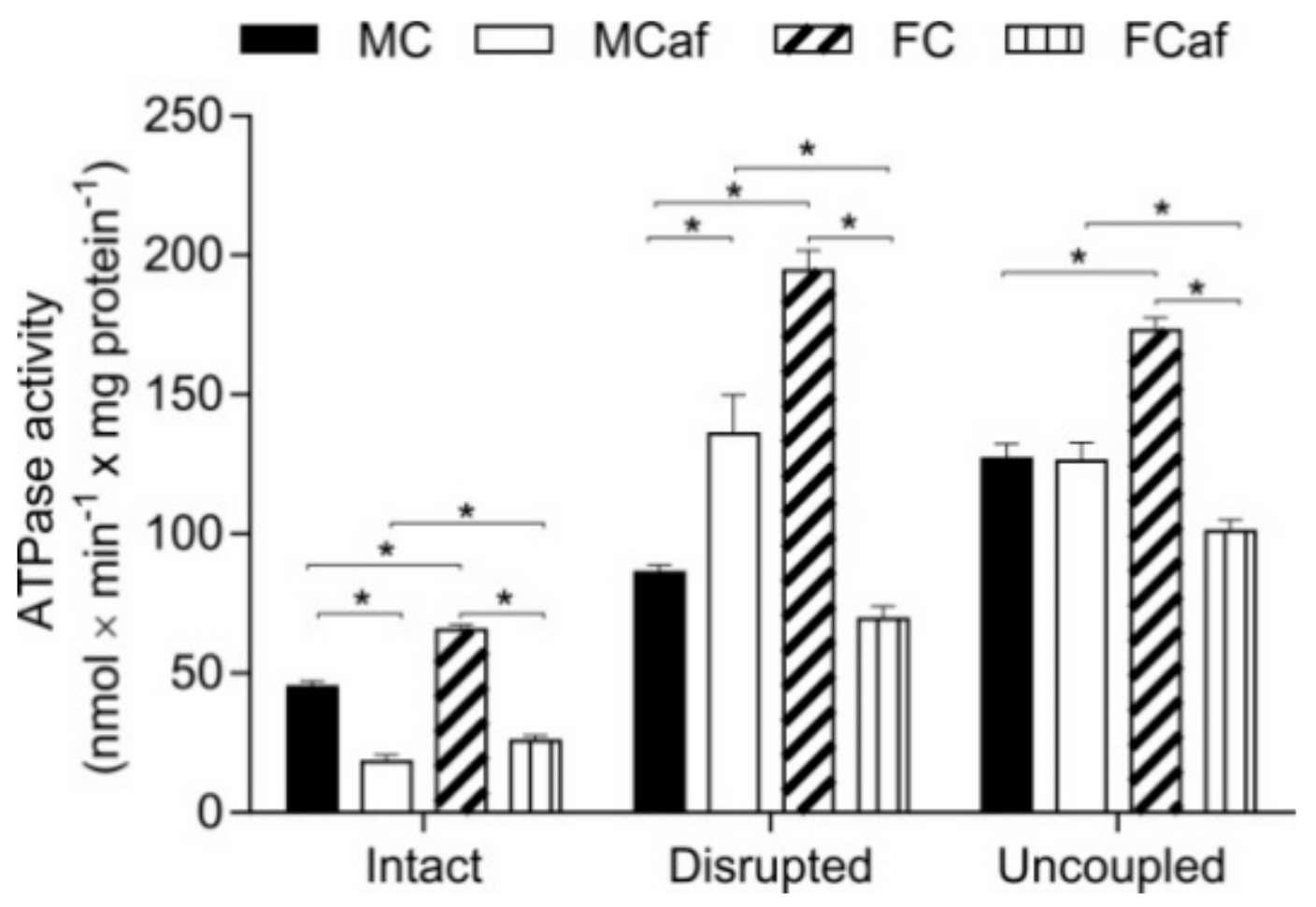
3.3. Parameters of Energy Transduction in Isolated Mitochondria
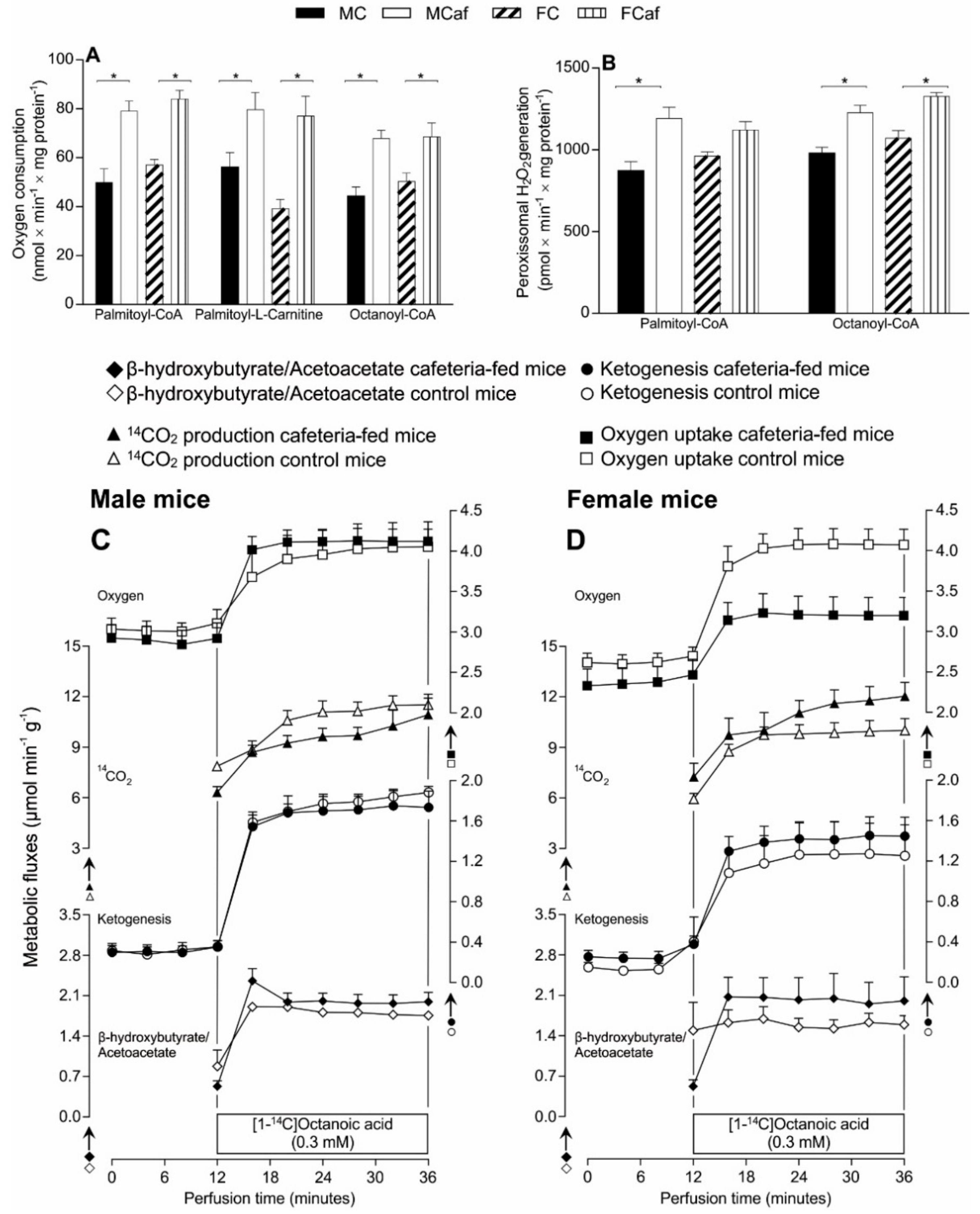
3.4. Fatty Acid Oxidation in Isolated Mitochondria and Peroxisomes
3.5. Oxygen Consumption, 14CO2 Production and Ketogenesis in Intact Livers
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gasparin, F.R.S.; Carreño, F.O.; Mewes, J.M.; Gilglioni, E.H.; Pagadigorria, C.L.S.; Natali, M.R.M.; Utsunomiya, K.S.; Constantin, R.P.; Ouchida, A.T.; Curti, C.; et al. Sex differences in the development of hepatic steatosis in cafeteria diet-induced obesity in young mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, M.; Godinho, L.F.; Costello, J.L.; Islinger, M. The different facets of organelle interplay—An overview of organelle interactions. Front. Cell Dev. Biol. 2015, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7–RILP–p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P.; Bultynck, G.; Haiech, J.; Heizmann, C.W.; Krebs, J.; Moreau, M. New functions of mitochondria associated membranes in cellular signaling ☆. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef]
- Lee, J.-S.; Zheng, Z.; Mendez, R.; Ha, S.-W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Rizzuto, R. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: More than just neighborhood? FEBS Lett. 2004, 567, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, H.J.; Ho Jung, M.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Eno, C.O.; Altman, B.J.; Zhu, Y.; Zhao, G.; Olberding, K.E.; Rathmell, J.C.; Li, C. ER stress modulates cellular metabolism. Biochem. J. 2011, 435, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoaris, C.R.; Rizo, G.V.; Roldi, L.P.; de Moraes, S.M.F.; de Proença, A.R.G.; Peralta, R.M.; Natali, M.R.M. Effects of cafeteria diet on the jejunum in sedentary and physically trained rats. Nutrition 2010, 26, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Bortolin, R.C.; Vargas, A.R.; Gasparotto, J.; Chaves, P.R.; Schnorr, C.E.; Martinello, K.B.; Silveira, A.K.; Rabelo, T.K.; Gelain, D.P.; Moreira, J.C.F. A new animal diet based on human Western diet is a robust diet-induced obesity model: Comparison to high-fat and cafeteria diets in term of metabolic and gut microbiota disruption. Int. J. Obes. 2018, 42, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Macedo, I.C.; Medeiros, L.F.; Oliveira, C.; Oliveira, C.M.; Rozynsky, J.R.; Scarabelot, V.L.; Souza, A.; Silva, F.R.; Santos, V.S.; Cioato, S.G.; et al. Cafeteria diet-induced obesity plus chronic stress alter serum leptin levels. Peptides 2012, 38, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampey, B.P.; Vanhoose, A.M.; Winfield, H.M.; Freemerman, A.J.; Muehlbauer, M.J.; Fueger, P.T.; Newgard, C.B.; Makowski, L. Cafeteria diet is a robust model of human metabolic syndrome with liver and adipose inflammation: Comparison to high-fat diet. Obesity 2011, 19, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Higa, T.S.; Spinola, A.V.; Fonseca-Alaniz, M.H.; Evangelista, F.S.A. Comparison between cafeteria and high-fat diets in the induction of metabolic dysfunction in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 47–54. [Google Scholar] [PubMed]
- Voss, D.O.; Campello, A.P.; Bacila, M. The respiratory chain and the oxidative phosphorylation of rat brain mitochondria. Biochem. Biophys. Res. Commun. 1961, 4, 48–51. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Guarnier, F.A.; Cecchini, A.L.; Suzukawa, A.A.; Maragno, A.L.G.C.; Simão, A.N.C.; Gomes, M.D.; Cecchini, R. Time course of skeletal muscle loss and oxidative stress in rats with Walker 256 solid tumor. Muscle Nerve 2010, 42, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Gagliano, N.; Lusini, L.; Milzani, A.; Di Simplicio, P.; Colombo, R. Actin carbonylation: From a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic. Biol. Med. 2001, 31, 1075–1083. [Google Scholar] [CrossRef]
- Harishekar, M.B.; Kiran, B. Effect of lead, alcohol and vitamin e on protein carbonyl content in rats. J. Appl. Pharm. Sci. 2011, 63, 50–57. [Google Scholar]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Siqueira, I.R.; Fochesatto, C.; da Silva Torres, I.L.; Dalmaz, C.; Alexandre Netto, C. Aging affects oxidative state in hippocampus, hypothalamus and adrenal glands of Wistar rats. Life Sci. 2005, 78, 271–278. [Google Scholar] [CrossRef]
- Sedlak, J.; Lindsay, R.H. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar] [CrossRef]
- Faure, P.; Lafond, J.-L. Measurement of Plasma Sulfhydryl and Carbonyl Groups as a Possible Indicator of Protein Oxidation. In Analysis of Free Radicals in Biological Systems; Birkhäuser Basel: Basel, Switzerland, 1995; pp. 237–248. [Google Scholar]
- Aebi, H. Catalase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Elsevier: Weinheim, Germany, 1974; pp. 673–684. [Google Scholar]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar] [PubMed]
- Mize, C.E.; Langdon, R.G. Hepatic glutathione reductase. I. Purification and general kinetic properties. J. Biol. Chem. 1962, 237, 1589–1595. [Google Scholar] [PubMed]
- Arnér, E.S.J.; Zhong, L.; Holmgren, A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1998, 300, 226–239. [Google Scholar]
- Clark, L.J.; Kaplan, S.; Mattews, E.; Edwards, F.; Helmsworth, J. Monitor and control of blood oxygen tension and pH during total body perfusion. Trans. Am. Soc. Artif. Intern. Organs 1956, 2, 41–48. [Google Scholar]
- Chance, B.; Williams, G. Respiratory enzymes in oxidative phosphorylation. J. Biol. Chem. 1955, 271, 383–393. [Google Scholar]
- Garland, P.B.; Shepherd, D.; Nicholls, D.G.; Yates, D.W.; Light, P.A. Interactions Between Fatty Acid Oxidation and the Tricarboxylic Acid Cycle. In Citric Acid Cycle; Lowenstein, J., Ed.; Dekker: New York, NY, USA, 1969; pp. 163–212. ISBN 9780080881287. [Google Scholar]
- Pullman, M.E.; Penefsky, H.S.; Datta, A.; Racker, E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J. Biol. Chem. 1960, 235, 3322–3329. [Google Scholar] [PubMed]
- Fiske, C.H.; Subbarow, Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar]
- Small, G.M.; Burdett, K.; Connock, M.J. A sensitive spectrophotometric assay for peroxisomal acyl-CoA oxidase. Biochem. J. 1985, 227, 205–210. [Google Scholar] [CrossRef]
- Taguchi, H.; Ogura, Y.; Takanashi, T.; Hashizoe, M.; Honda, Y. In vivo Quantitation of Peroxides in the Vitreous Humor by Fluorophotometry. Invest. Ophthalmol. Vis. Sci. 1996, 37, 1444–1450. [Google Scholar]
- Scholz, R.; Bücher, T. Hemoglobin-Free Perfusion of Rat Liver. In Control of Energy Metabolism; Chance, B., Estabrook, R.W., Williamson, J.R., Eds.; Elsevier: New York, NY, USA, 1965; pp. 393–414. ISBN 978-1-4832-3161-7. [Google Scholar]
- Soboll, S.; Heldt, H.-W.; Scholz, R. Changes in the Subcellular Distribution of Metabolites Due to Ethanol Oxidation in the Perfused Rat Liver. Hoppe-Seyler´s Zeitschrift für Physiol. Chemie 1981, 362, 247–260. [Google Scholar] [CrossRef]
- Williamson, D.H.; Mellanby, J. D-(-)- /3-Hydroxybutyrate and Acetoacetate. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic Press: Cambridge, MA, USA, 1974; pp. 1836–1839. ISBN 9780120913022. [Google Scholar]
- Scholz, R.; Olson, M.S.; Schwab, A.J.; Schwabe, U.; Noell, C.; Braun, W. The effect of fatty acids on the regulation of pyruvate dehydrogenase in perfused rat liver. Eur. J. Biochem. 1978, 86, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracht, A.; Ishii-Iwamoto, E.L.; Kelmer-Bracht, A.M. O Estudo do Metabolismo no Fígado em Perfusão. In Métodos de Laboratório em Bioquímica; Bracht, A., Ishii-Iwamoto, E.L., Eds.; Malone: São Paulo, Brasil, 2003; pp. 275–288. [Google Scholar]
- Castro, J.P.; Grune, T.; Speckmann, B. The two faces of reactive oxygen species (ROS) in adipocyte function and dysfunction. Biol. Chem. 2016, 397, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Jung, T.; Grune, T. Pathophysiological importance of aggregated damaged proteins. Free Radic. Biol. Med. 2014, 71, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Jarukamjorn, K.; Jearapong, N.; Pimson, C.; Chatuphonprasert, W. A High-fat, high-fructose diet induces antioxidant imbalance and increases the risk and progression of nonalcoholic fatty liver disease in mice. Scientifica (Cairo) 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Oxidative stress and antioxidants balance in fatty liver disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Holmgren, A.; Arnér, E.S.J.; Schmidt, E.E. NADPH-dependent and -independent disulfide reductase systems. Free Radic. Biol. Med. 2018, 127, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Schmidt, E.E. Disulfide reductase systems in liver. Br. J. Pharmacol. 2019, 176, 532–543. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, F.; Diotel, N.; Girard, D.; Rondeau, P.; Faadiel Essop, M.; Bourdon, E.; Faadiel, M.; Ee Girard, D. Enhanced oxidative stress in adipose tissue from diabetic mice, possible contribution of glycated albumin. Biochem. Biophys. Res. Commun. 2016, 473, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhang, X.; Ye, F.; Zhong, L. High-fat diet-induced changes in liver thioredoxin and thioredoxin reductase as a novel feature of insulin resistance. FEBS Open Bio 2014, 4, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, H.P.; Ogata, F.T.; Stern, A. Thioredoxin promotes survival signaling events under nitrosative/oxidative stress associated with cancer development. Biomed. J. 2017, 40, 189–199. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic reticulum stress in metabolic disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics; Academic Press: Cambridge, MA, USA, 2013; ISBN 9780123884251. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Constantin, R.P.; do Nascimento, G.S.; Constantin, R.P.; Salgueiro, C.L.; Bracht, A.; Ishii-Iwamoto, E.L.; Yamamoto, N.S.; Constantin, J. Citrus flavanones affect hepatic fatty acid oxidation in rats by acting as prooxidant agents. Biomed. Res. Int. 2013, 2013, 342973. [Google Scholar] [CrossRef] [PubMed]
- Ciapaite, J.; van den Broek, N.M.; te Brinke, H.; Nicolay, K.; Jeneson, J.A.; Houten, S.M.; Prompers, J.J. Differential effects of short- and long-term high-fat diet feeding on hepatic fatty acid metabolism in rats. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Bucci, M.; Roivainen, A.; Någren, K.; Järvisalo, M.J.; Kiss, J.; Guiducci, L.; Fielding, B.; Naum, A.G.; Borra, R.; et al. Fatty Acid Metabolism in the Liver, Measured by Positron Emission Tomography, Is Increased in Obese Individuals. Gastroenterology 2010, 139, 846–856.e6. [Google Scholar] [CrossRef] [PubMed]
- De Lazarin, M.O.; Ishii-Iwamoto, E.L.; Yamamoto, N.S.; Constantin, R.P.; Garcia, R.F.; da Costa, C.E.M.; de Vitoriano, A.S.; de Oliveira, M.C.; Salgueiro-Pagadigorria, C.L. Liver mitochondrial function and redox status in an experimental model of non-alcoholic fatty liver disease induced by monosodium l-glutamate in rats. Exp. Mol. Pathol. 2011, 91, 687–694. [Google Scholar]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chami, M.; Oulès, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Bréchot, P. Role of SERCA1 Truncated Isoform in the Proapoptotic Calcium Transfer from ER to Mitochondria during ER Stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Parameter | Male Mice | Female Mice | ||
|---|---|---|---|---|---|
| Control | Cafeteria | Control | Cafeteria | ||
| Succinate | state III | 261.11 ± 7.50 | 129.86 ± 5.65 D | 256.92 ± 7.36 | 168.56 ± 8.77 DS |
| state IV | 55.12 ± 3.39 | 31.60 ± 2.25 D | 55.61 ± 3.13 | 42.02 ± 2.17 DS | |
| RC | 4.66 ± 0.40 | 5.23 ± 0.52 | 3.43 ± 0.34 | 3.93 ± 0.25 | |
| ADP/O | 2.14 ± 0.18 | 2.15 ± 0.22 | 2.19 ± 0.23 | 2.05 ± 0.24 | |
| β-Hydroxybutyrate | state III | 69.29 ± 2.83 | 73.54 ± 4.52 | 162.02 ± 4.64 S | 172.78 ± 11.46 S |
| state IV | 25.51 ± 2.65 | 21.73 ± 2.85 | 53.71 ± 1.52 S | 45.0 ± 3.16 DS | |
| RC | 3.52 ± 0.16 | 5.12 ± 0.39 D | 3.06 ± 0.18 | 3.46 ± 0.17 S | |
| ADP/O | 2.72 ± 0.20 | 2.72 ± 0.13 | 3.33 ± 0.29 | 3.50 ± 0.38 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morais Mewes, J.; Rodrigues Silva Gasparin, F.; Yoshida, T.; Amâncio Daniel da Silva, M.; Raquel Marçal Natali, M.; Francisco Veiga Bizerra, P.; Sayuri Utsunomiya, K.; Hideo Gilglioni, E.; Shigueaki Mito, M.; Cristiane Mantovanelli, G.; et al. The Role of Mitochondria in Sex-Dependent Differences in Hepatic Steatosis and Oxidative Stress in Response to Cafeteria Diet-Induced Obesity in Mice. Nutrients 2019, 11, 1618. https://doi.org/10.3390/nu11071618
Morais Mewes J, Rodrigues Silva Gasparin F, Yoshida T, Amâncio Daniel da Silva M, Raquel Marçal Natali M, Francisco Veiga Bizerra P, Sayuri Utsunomiya K, Hideo Gilglioni E, Shigueaki Mito M, Cristiane Mantovanelli G, et al. The Role of Mitochondria in Sex-Dependent Differences in Hepatic Steatosis and Oxidative Stress in Response to Cafeteria Diet-Induced Obesity in Mice. Nutrients. 2019; 11(7):1618. https://doi.org/10.3390/nu11071618
Chicago/Turabian StyleMorais Mewes, Juliana, Fabiana Rodrigues Silva Gasparin, Tiago Yoshida, Mariana Amâncio Daniel da Silva, Maria Raquel Marçal Natali, Paulo Francisco Veiga Bizerra, Karina Sayuri Utsunomiya, Eduardo Hideo Gilglioni, Marcio Shigueaki Mito, Gislaine Cristiane Mantovanelli, and et al. 2019. "The Role of Mitochondria in Sex-Dependent Differences in Hepatic Steatosis and Oxidative Stress in Response to Cafeteria Diet-Induced Obesity in Mice" Nutrients 11, no. 7: 1618. https://doi.org/10.3390/nu11071618