
Preparation of Cu/Sn-Organic Nano-Composite Catalysts for Potential Use in Hydrogen Evolution Reaction and Electrochemical Characterization

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalysts Preparation
2.2. Catalysts Characterizations
2.3. Electrochemical Measurements
3. Results
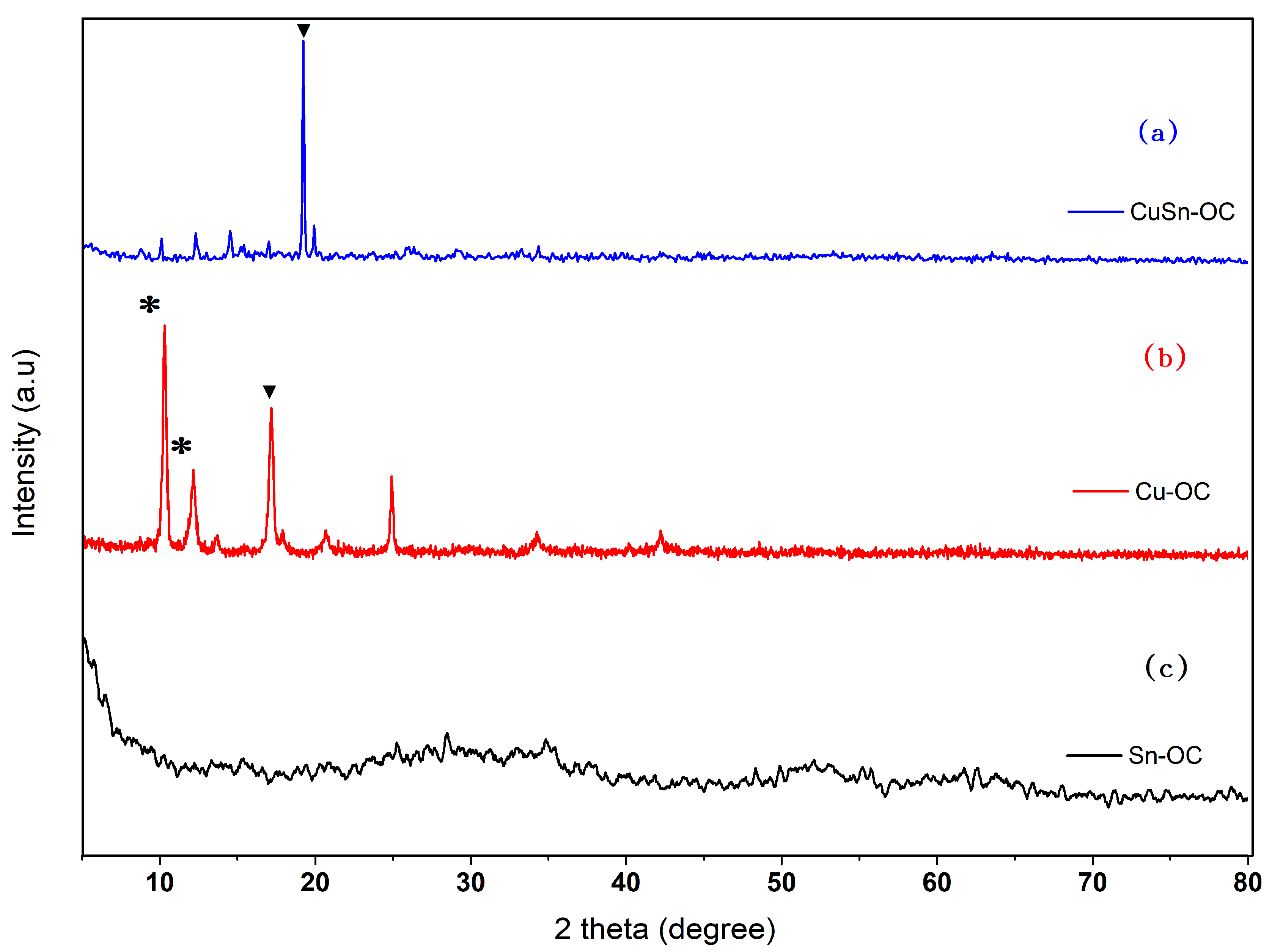
3.1. Synthesis and Microstructural Characterization
3.2. Electrochemical Properties
3.2.1. Cyclic Voltammetric Studies
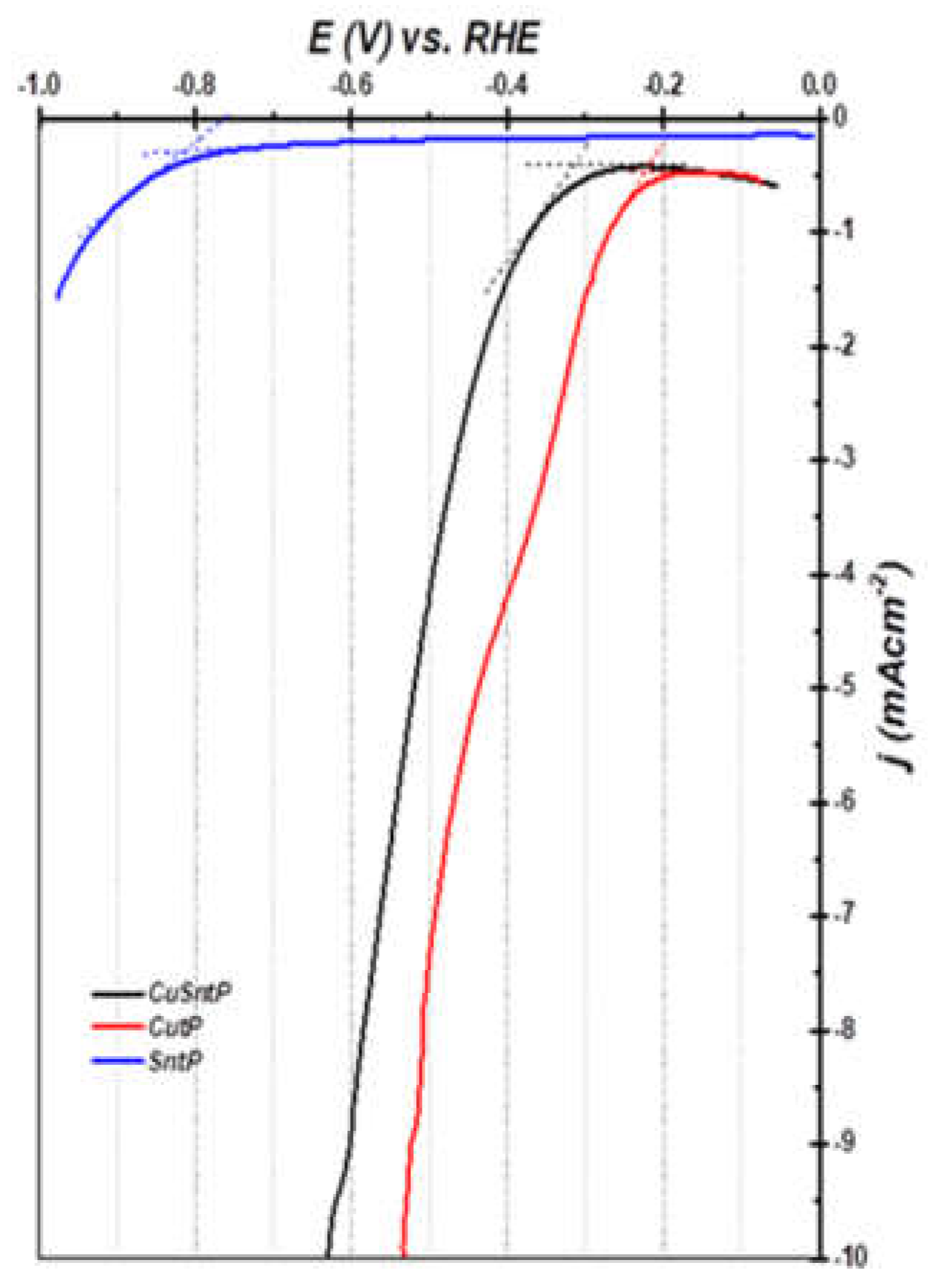
3.2.2. HER Performance Measurements
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, Y.; Xia, B.Y.; Zhao, B.; Wang, X. A review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting. J. Mater. Chem. A 2016, 4, 17587–17603. [Google Scholar] [CrossRef] [Green Version]
- Medrano-Banda, A.; Crespo-Yapur, A.; Velasco-Soto, M.; Videa, M. Galvanostatically Deposited PtNi Thin-Films as Electrocatalysts for the Hydrogen Evolution Reaction. Chem. Open 2022, 11, e202100241. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, C.; Qin, M.; Ben Li, B.; Lin, B.; Mao, Q.; Yang, H.; Bin Liu, B.; Wang, Y. Reversible hydrogen spillover in Ru-WO3-x enhances hydrogen evolution activity in neutral pH water splitting. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Thia, L.; Xia, B.Y.; Ge, X.; Liu, Z.; Fisher, A.; Wang, X. Construction of Efficient 3D Gas Evolution Electrocatalyst for Hydrogen Evolution: Porous FeP Nanowire Arrays on Graphene Sheets. Adv. Sci. 2015, 2, 1500120. [Google Scholar] [CrossRef]
- Vesborg, P.C.K.; Seger, B.; Chorkendorff, I. Recent development in hydrogen evolution reaction catalysts and their practical implementation. J. Phys. Chem. Lett. 2015, 6, 951–957. [Google Scholar] [CrossRef]
- Kumar, S.S.; Himabindu, V. Hydrogen production by PEM water electrolysis—A review. Mater. Sci. Energy Technol. 2019, 2, 442–454. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Velazquez Abad, A.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Yao, Y.; Zhang, J.-W.; Pan, L.; Zhang, X.; Zou, J.-J. Electrocatalysts for Hydrogen Evolution in Alkaline Electrolytes: Mechanisms, Challenges, and Prospective Solutions. Adv. Sci. 2017, 5, 1700464. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C.; Figueiredo, J.L. Hydrogen production by alkaline water electrolysis. Quim. Nova 2013, 36, 1176–1193. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Chen, W.; Wang, X. A Review of Phosphide-Based Materials for Electrocatalytic Hydrogen Evolution. Adv. Energy Mater. 2015, 5, 1500985. [Google Scholar] [CrossRef]
- Cao, D.; Xu, H.; Cheng, D. Construction of Defect-Rich RhCu Nanotubes with Highly Active Rh 3 Cu 1 Alloy Phase for Overall Water Splitting in All pH Values. Adv. Energy Mater. 2020, 10, 1903038. [Google Scholar] [CrossRef]
- Otani, M.; Hamada, I.; Sugino, O.; Morikawa, Y.; Okamoto, Y.; Ikeshoji, T. Electrode Dynamics from First Principles. J. Phys. Soc. Jpn. 2008, 77, 024802. [Google Scholar] [CrossRef] [Green Version]
- Dubouis, N.; Grimaud, A. The hydrogen evolution reaction: From material to interfacial descriptors. Chem. Sci. 2019, 10, 9165–9181. [Google Scholar] [CrossRef] [Green Version]
- Marković, N.M.; Grgur, B.N.; Ross, P.N. Temperature-Dependent Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Acid Solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Y.; Chu, P.K.; Liu, Q.; Liu, X.; Zhang, S.; Luo, J.; Wang, X.; Hu, G. Recent advances in non-noble metal-based bifunctional electrocatalysts for overall seawater splitting. J. Alloys Compd. 2022, 922, 153851. [Google Scholar] [CrossRef]
- Sheng, W.; Myint, M.; Chen, J.G.; Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 2013, 6, 1509–1512. [Google Scholar] [CrossRef]
- Sheng, W. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef] [Green Version]
- Tymoczko, J.; Calle-Vallejo, F.; Schuhmann, W.; Bandarenka, A.S. Making the hydrogen evolution reaction in polymer electrolyte membrane electrolysers even faster. Nat. Commun. 2016, 7, 10990. [Google Scholar] [CrossRef]
- Marković, N.M.; Ross, P.N. Surface science studies of model fuel cell electrocatalysts. In Fuel Cells Compendium; Elsevier: Amsterdam, The Netherlands, 2005; pp. 275–374. [Google Scholar] [CrossRef]
- Mugheri, A.Q.; Daudpoto, M.R.; Sangah, A.A.; Mugheri, N.; El-Bahy, Z.M.; El-Badry, Y.A. A new hyperbranched water-splitting technique based on Co3O4/MoS2 nano composite catalyst for High-Performance of hydrogen evolution reaction. Int. J. Hydrogen Energy 2022, 47, 2124–2133. [Google Scholar] [CrossRef]
- Mahale, N.K.; Ingle, S.T. Electrocatalytic hydrogen evolution reaction on nano-nickel decorated graphene electrode. Energy 2017, 119, 872–878. [Google Scholar] [CrossRef]
- Zhou, K.L.; Wang, Z.; Han, C.B.; Ke, X.; Wang, C.; Jin, Y.; Zhang, Q.; Liu, J.; Wang, H.; Yan, H. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 2021, 12, 3783. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.; Findeis, B.; Haukka, M.; Graff, R.; Pakkanen, T.A.; Gade, L.H. Metal ± Metal Bonds between Group 12 Metals and Tin: Structural Characterization of the Complete Series of Sn-M-Sn (M ˆ Zn, Cd, Hg) Heterodimetallic Complexes. Chem. Eur. J. 2002, 8, 14. [Google Scholar] [CrossRef]
- Solla-Gullón, J.; Rodríguez, P.; Herrero, E.; Aldaz, A.; Feliu, J.M. Surface characterization of platinum electrodes. Phys. Chem. Chem. Phys. 2007, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Faid, A.Y.; Barnett, A.O.; Seland, F.; Sunde, S. Ni/NiO nanosheets for alkaline hydrogen evolution reaction: In situ electrochemical-Raman study. Electrochim. Acta 2020, 361, 137040. [Google Scholar] [CrossRef]
- Cariou, J.; Guern, Y.; Le Jeune, B.; Le Brun, G.; Lortrian, J. Scattering through fluids: Speckle size measurement and Monte Carlo simulations close to and into the multiple scattering. Opt. Express 2004, 12, 176–188. [Google Scholar] [CrossRef]
- Room Temperature Synthesis and Characterizations of Cu-MOF using Natural Polysaccharide as Potential Organic Linker. Int. J. Recent Technol. Eng. 2019, 8, 6490–6498. [CrossRef]
- Kumar, R.S.; Kumar, S.S.; Kulandainathan, M.A. Efficient electrosynthesis of highly active Cu3(BTC)2-MOF and its catalytic application to chemical reduction. Micropor. Mesoporous Mater. 2013, 168, 57–64. [Google Scholar] [CrossRef]
- Dhumal, N.R.; Singh, M.P.; Anderson, J.A.; Kiefer, J.; Kim, H.J. Molecular Interactions of a Cu-Based Metal–Organic Framework with a Confined Imidazolium-Based Ionic Liquid: A Combined Density Functional Theory and Experimental Vibrational Spectroscopy Study. J. Phys. Chem. C 2016, 120, 3295–3304. [Google Scholar] [CrossRef] [Green Version]
- Téllez, C.A.; Hollauer, E.; Mondragon, M.A.; Castañ, V.M. Fourier transform infrared and Raman spectra, vibrational assignment and ab initio calculations of terephthalic acid and related compounds. Spectrochim. Acta Part A 2001, 57, 993–1007. [Google Scholar] [CrossRef]
- Giri, S.D.; Sarkar, A. Electrochemical Study of Bulk and Monolayer Copper in Alkaline Solution. J. Electrochem. Soc. 2016, 163, H252–H259. [Google Scholar] [CrossRef]
- Ambrose, J.; Barradas, R.G.; Shoesmith, D.W. Investigations of copper in aqueous alkaline solutions by cyclic Voltammetry. Electroanal. Chem. Interfacial Electrochem. 1973, 47, 44–47. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Ingredient | Formula | mmol | V (mL), DMF | Catalyst Code |
|---|---|---|---|---|---|
| Cu-OC | Cupric acetate | Cu2(CH3COO)4 | 2.75 | 50 | Cu-OC |
| Sn-OC | Tin (IV) chloride | SnCl4 | 1.923 | 50 | Sn-OC |
| CuSn-OC | Cupric (II) acetate monohydrate | Cu(CH3COO)2.H2O | 2.75 | 100 | CuSn-OC |
| Tin (IV) chloride | SnCl4 | 1.923 |
| Electrocatalyst | Onset Potential (mV) vs. RHE | Cdl (μF cm−2) | ECSA (m2 g−1) | Tafel Slope mVdec−1 | jo mA/cm2 | α | V (mV) vs. RHE at −10 mA/cm2 |
|---|---|---|---|---|---|---|---|
| Cu-OC | −320 | 50 | 0.41 | 335 | 2.1 × 10−1 | 0.18 | −630 |
| Sn-OC | −760 | 40 | 0.33 | 250 | 3.1 × 10−5 | 0.24 | -- |
| CuSn-OC | −250 | 60 | 0.50 | 195 | 6.3557 × 10−3 | 0.31 | −530 |
| Element, Orbit | Weight % | Atomic % |
|---|---|---|
| C, K | 23.58 | 41.50 |
| O, K | 31.24 | 41.27 |
| S, K | 9.74 | 6.42 |
| Cu, K | 24.95 | 8.30 |
| Sn, L | 8.28 | 1.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khdary, N.H.; El Enany, G.; Almalki, A.S.; Alhassan, A.M.; Altamimi, A.; Alshihri, S. Preparation of Cu/Sn-Organic Nano-Composite Catalysts for Potential Use in Hydrogen Evolution Reaction and Electrochemical Characterization. Nanomaterials 2023, 13, 911. https://doi.org/10.3390/nano13050911
Khdary NH, El Enany G, Almalki AS, Alhassan AM, Altamimi A, Alshihri S. Preparation of Cu/Sn-Organic Nano-Composite Catalysts for Potential Use in Hydrogen Evolution Reaction and Electrochemical Characterization. Nanomaterials. 2023; 13(5):911. https://doi.org/10.3390/nano13050911
Chicago/Turabian StyleKhdary, Nezar H., Gaber El Enany, Amani S. Almalki, Ahmed M. Alhassan, Abdullah Altamimi, and Saeed Alshihri. 2023. "Preparation of Cu/Sn-Organic Nano-Composite Catalysts for Potential Use in Hydrogen Evolution Reaction and Electrochemical Characterization" Nanomaterials 13, no. 5: 911. https://doi.org/10.3390/nano13050911