
Construction of Orthogonal Modular Proteinaceous Nanovaccine Delivery Vectors Based on mSA-Biotin Binding




{kind=link}
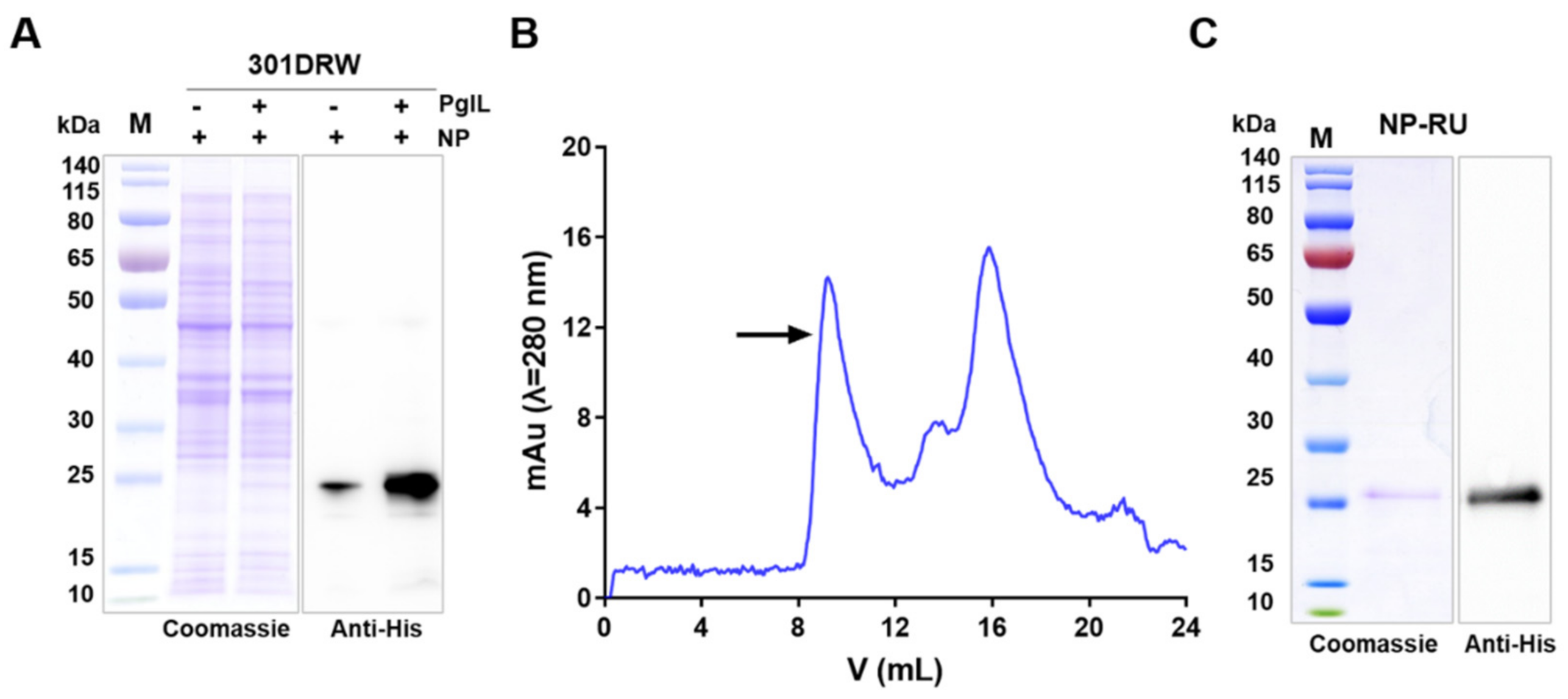{kind=link}
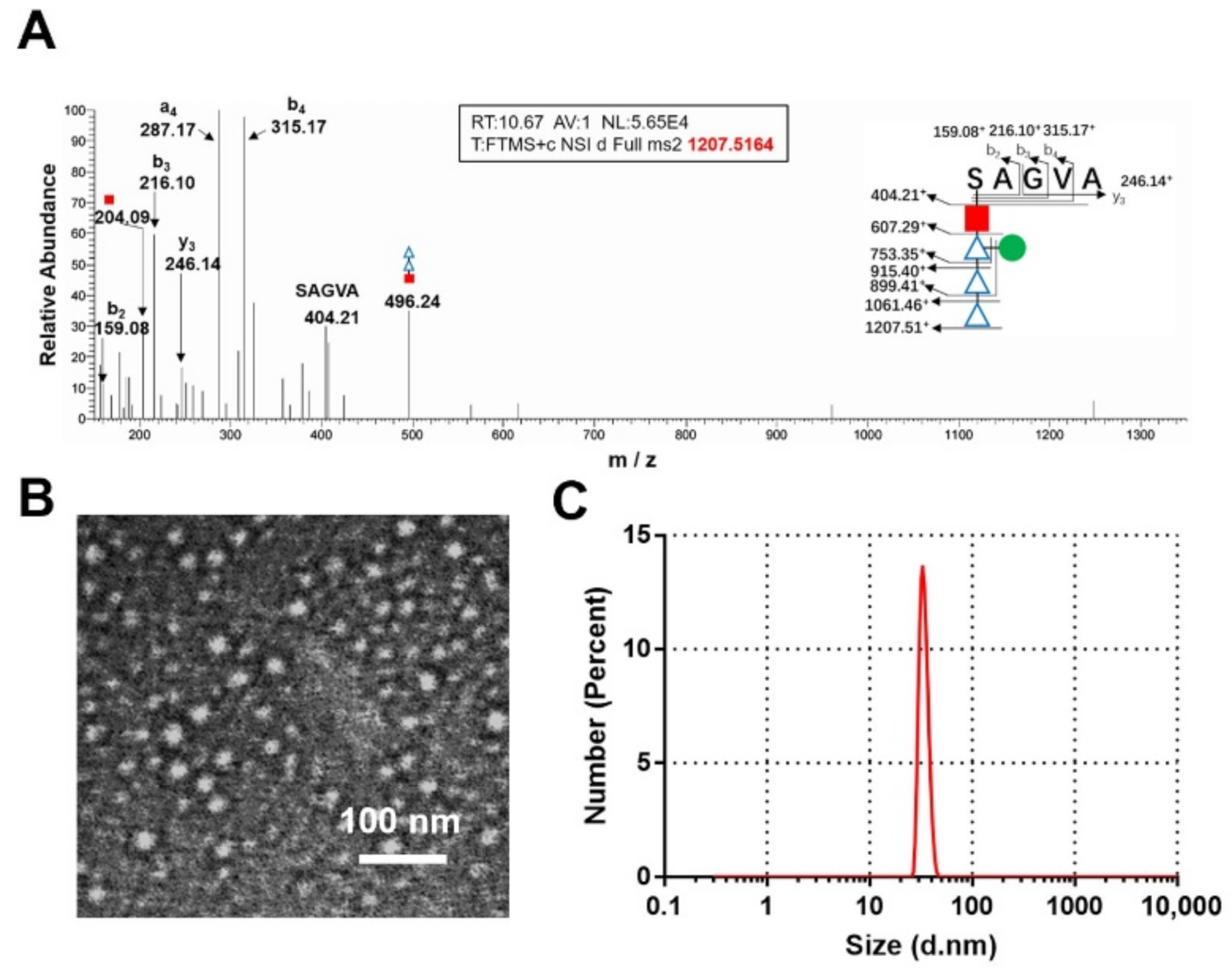{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
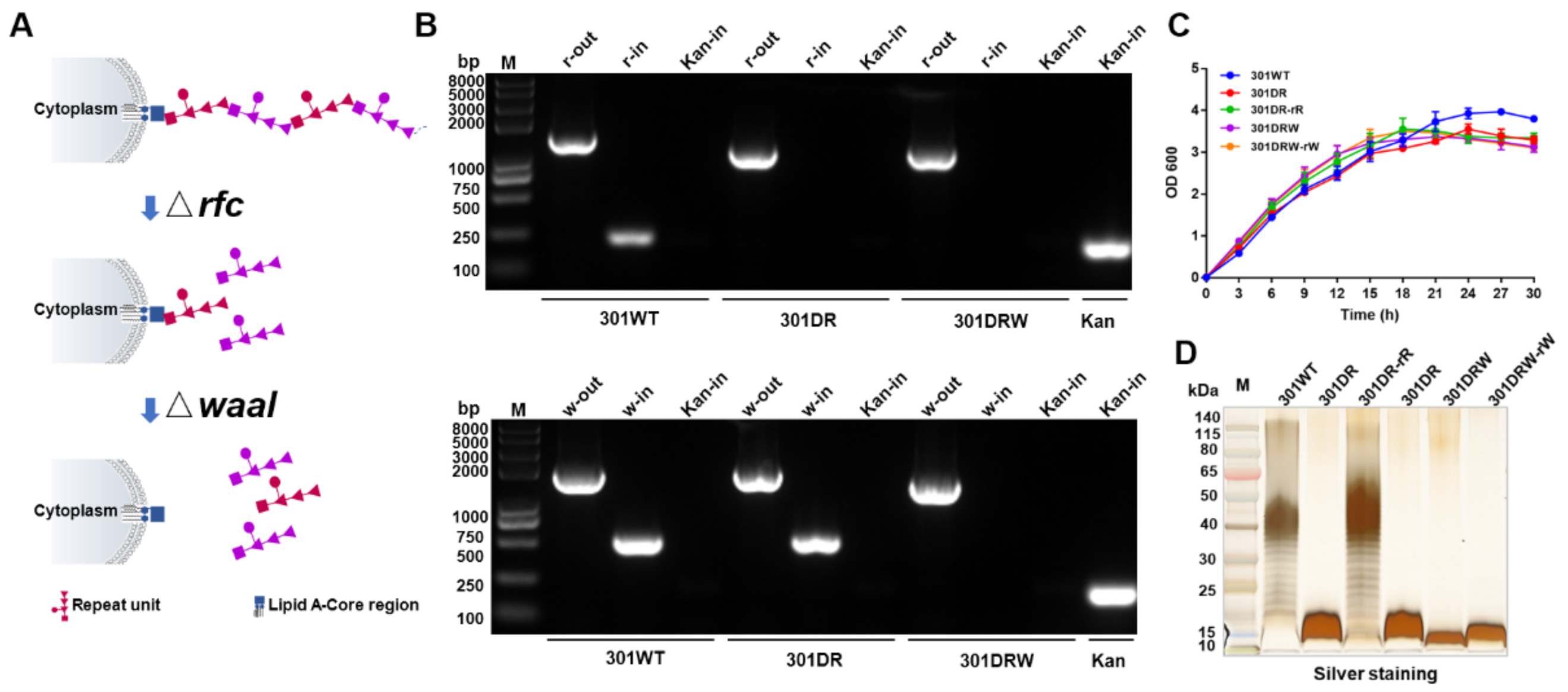
2.1. Construction of rfc and waaI Double Mutant S. flexneri 2a 301
2.2. Lipopolysaccharide (LPS) Preparation
2.3. Silver Staining
2.4. Protein Expression and Purification
2.5. Western Blotting
2.6. Mouse Immunization
2.7. Biotinylation and Connection
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results and Discussion
3.1. Establishment of Host Cells for Nanoparticle Expression by Knocking Out rfc and waaI in Shigella flexneri 301
3.2. Expression and Purification of Nanoparticles Modified with a Short Sugar Chain
3.3. Characterization of Nanoparticles Modified with Short Sugar Chains
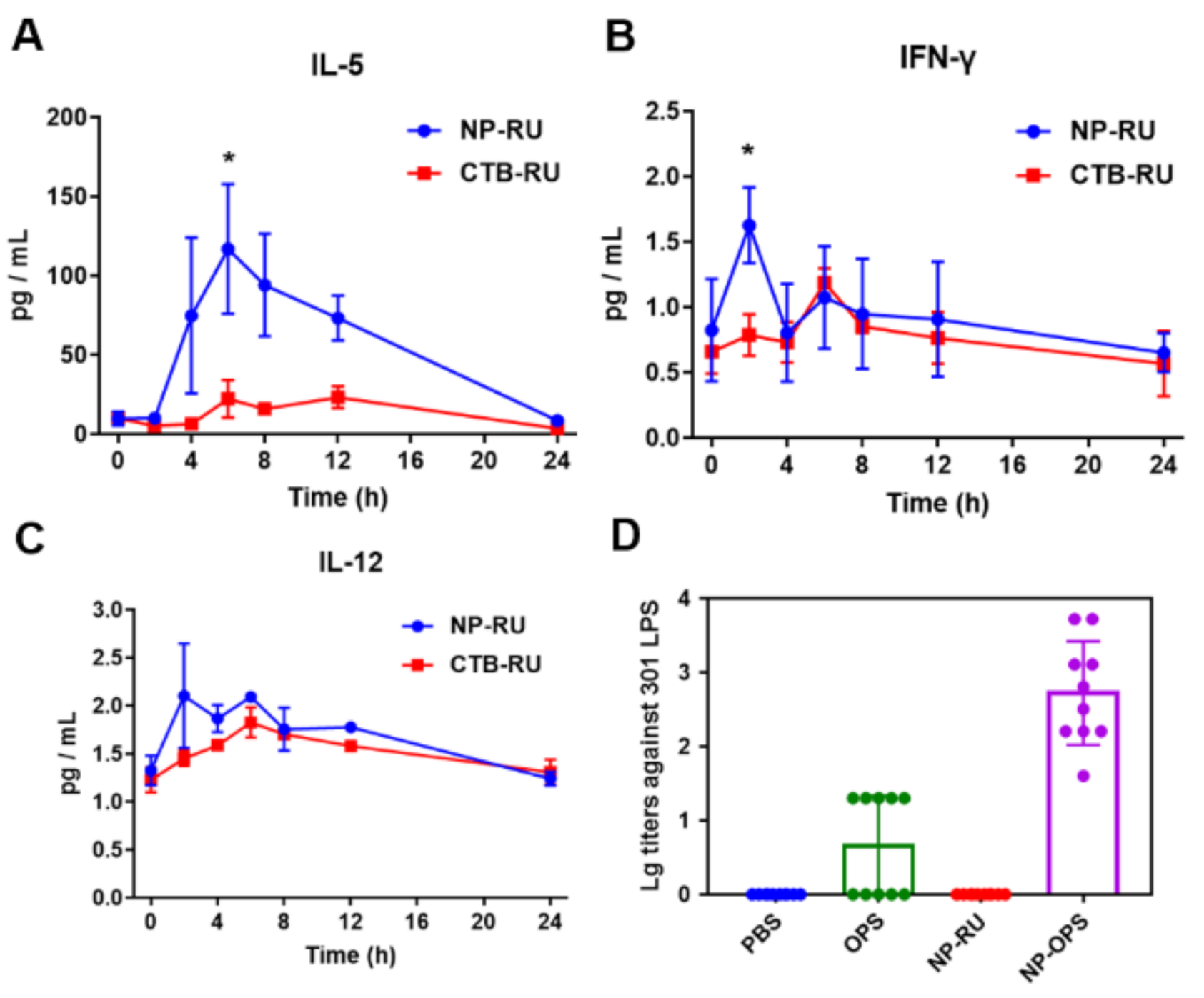
3.4. Activation of Immune Responses by Nanoparticles
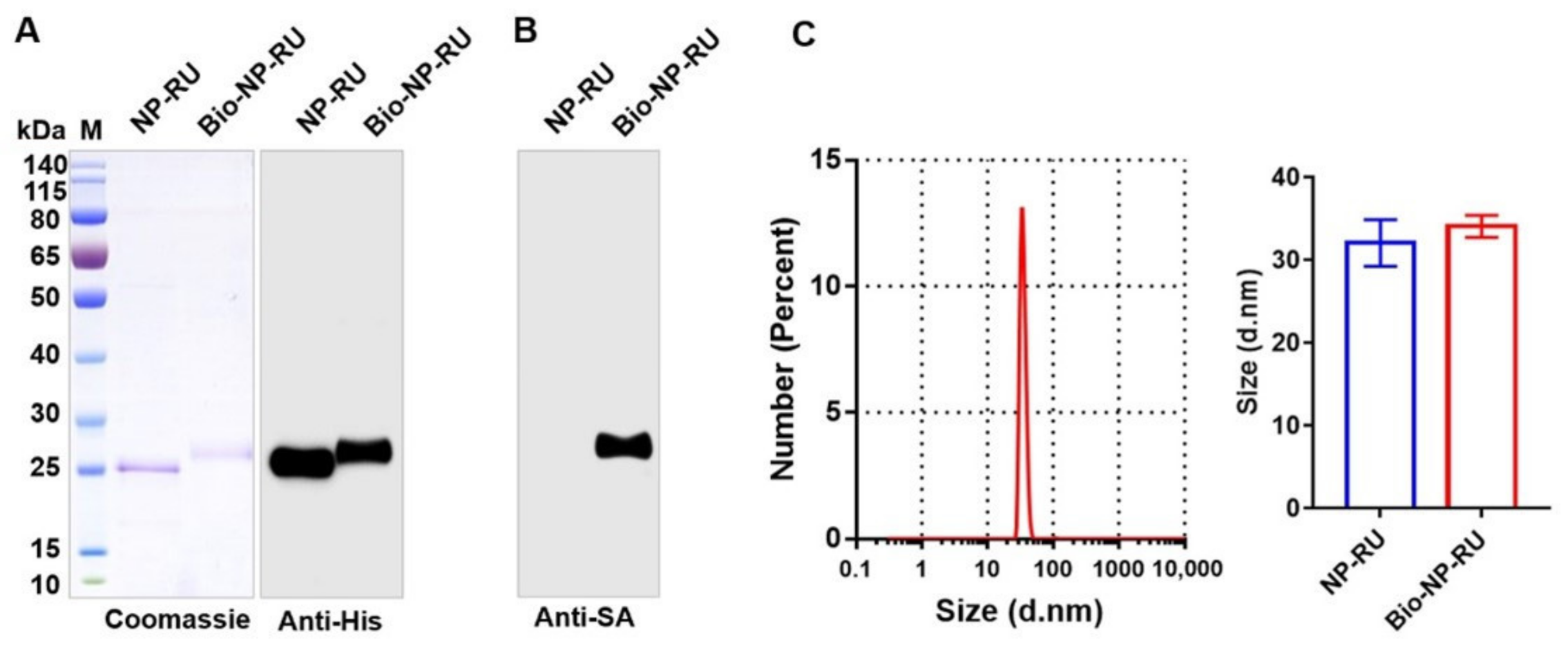
3.5. Construction of a Flexible Chassis Using Biotinylation of Nanoparticles
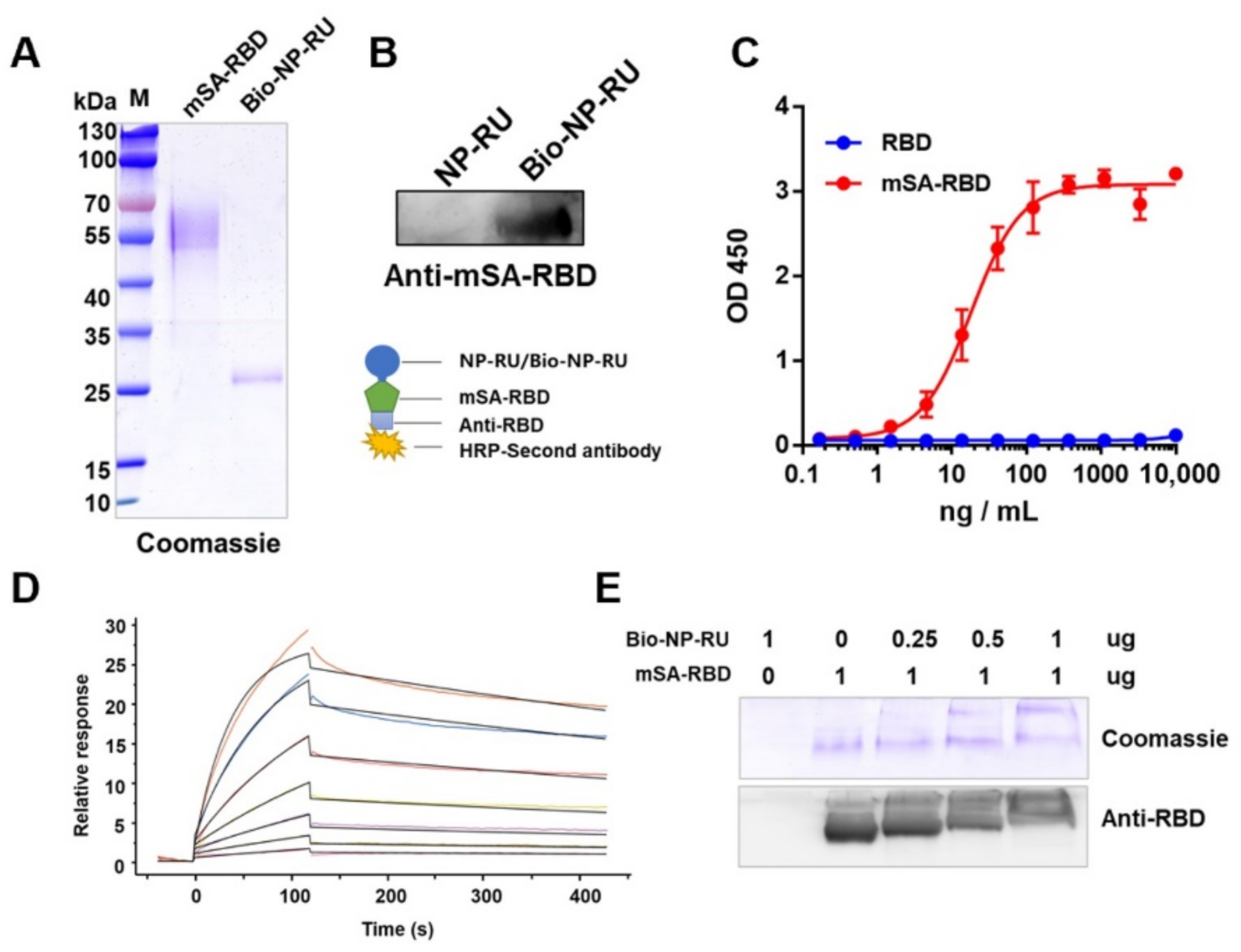
3.6. Display of Protein Antigen Types on NP-RU Chassis
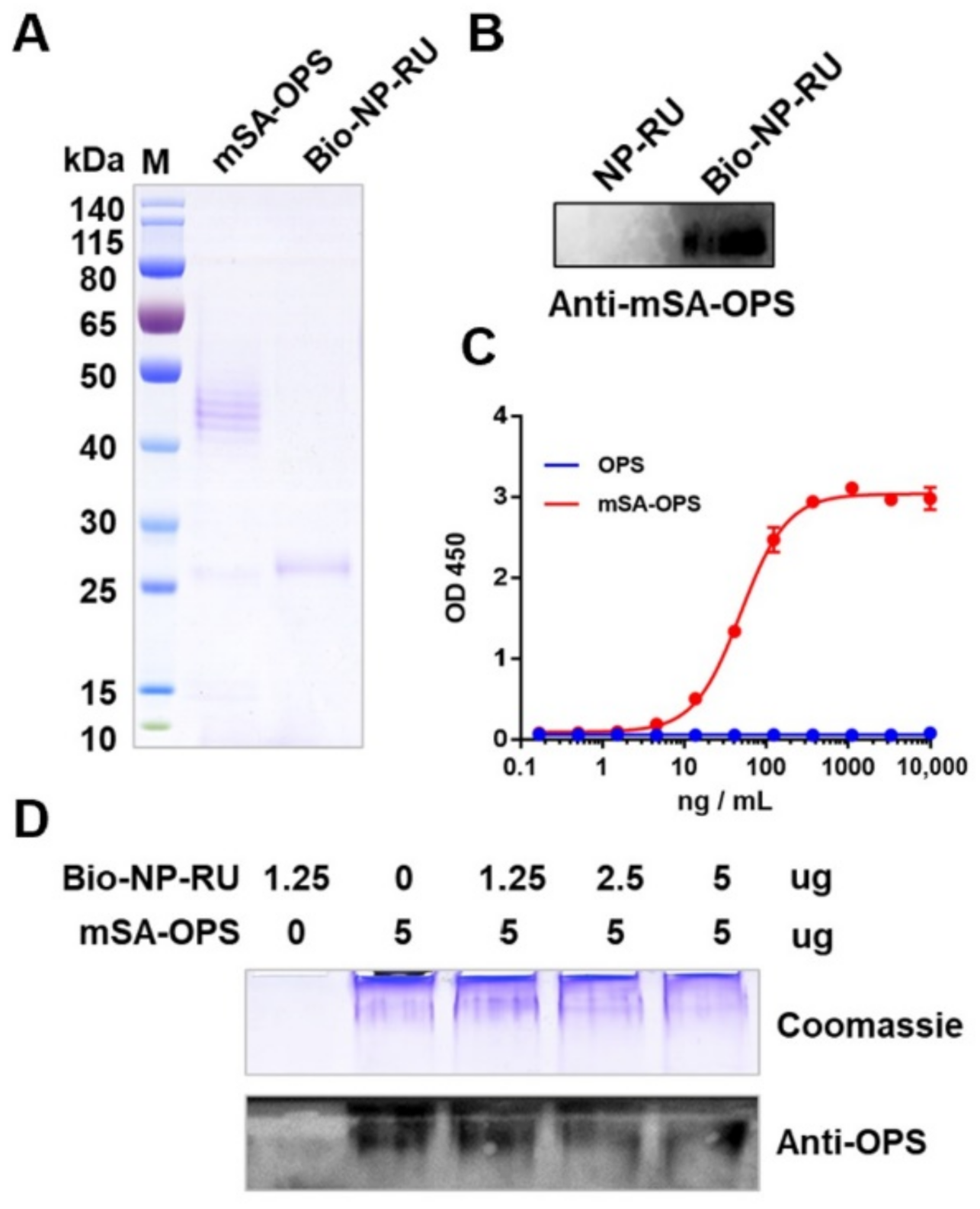
3.7. Display of Polysaccharide Antigen Types on the NP-RU Chassis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mamo, T.; Poland, G.A. Nanovaccinology: The next generation of vaccines meets 21st century materials science and engineering. Vaccine 2012, 30, 6609–6611. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Simon, J.K.; Baker, J.R., Jr. Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013, 13, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Yue, H.; Zhu, L.; Ma, G.-H.; Wang, H.-L. Prophylactic vaccine delivery systems against epidemic infectious diseases. Adv. Drug Deliv. Rev. 2021, 176, 113867. [Google Scholar] [CrossRef]
- Pati, R.; Shevtsov, M.; Sonawane, A. Nanoparticle Vaccines Against Infectious Diseases. Front. Immunol. 2018, 9, 2224. [Google Scholar] [CrossRef] [Green Version]
- Lynn, G.M.; Laga, R.; Darrah, P.A.; Ishizuka, A.S.; Balaci, A.J.; Dulcey, A.E.; Pechar, M.; Pola, R.; Gerner, M.Y.; Yamamoto, A.; et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 2015, 33, 1201–1210. [Google Scholar] [CrossRef]
- Lizotte, P.; Wen, A.M.; Sheen, M.R.; Fields, J.; Rojanasopondist, P.; Steinmetz, N.F.; Fiering, S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol. 2016, 11, 295–303. [Google Scholar] [CrossRef]
- Grgacic, E.V.; Anderson, D. Virus-like particles: Passport to immune recognition. Methods 2006, 40, 60–65. [Google Scholar] [CrossRef]
- Jardine, J.; Julien, J.-P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.-S.; MacPherson, S.; Jones, M.; et al. Rational HIV Immunogen Design to Target Specific Germline B Cell Receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef]
- Choi, B.; Moon, H.; Hong, S.J.; Shin, C.; Do, Y.; Ryu, S.; Kang, S. Effective Delivery of Antigen–Encapsulin Nanoparticle Fusions to Dendritic Cells Leads to Antigen-Specific Cytotoxic T Cell Activation and Tumor Rejection. ACS Nano 2016, 10, 7339–7350. [Google Scholar] [CrossRef]
- He, L.; De Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.-P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; Van Montfort, T.; Stunnenberg, M.; Labranche, C.C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, M.; Wei, C.-J.; Yassine, H.M.; McTamney, P.M.; Boyington, J.C.; Whittle, J.R.R.; Rao, S.S.; Kong, W.-P.; Wang, L.; Nabel, G.J. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 2013, 499, 102–106. [Google Scholar] [CrossRef]
- El Bissati, K.; Zhou, Y.; Paulillo, S.M.; Raman, S.K.; Karch, C.P.; Roberts, C.W.; Lanar, D.E.; Reed, S.; Fox, C.; Carter, D.; et al. Protein nanovaccine confers robust immunity against Toxoplasma. NPJ Vaccines 2017, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Wu, J.; Qing, S.; Zhang, X.; Zhang, L.; Yue, H.; Zeng, M.; Wang, B.; Yuan, Z.; Qiu, Y.; et al. Biosynthesis of Self-Assembled Proteinaceous Nanoparticles for Vaccination. Adv. Mater. 2020, 32, 2002940. [Google Scholar] [CrossRef]
- Pan, C.; Sun, P.; Liu, B.; Liang, H.; Peng, Z.; Dong, Y.; Wang, D.; Liu, X.; Wang, B.; Zeng, M.; et al. Biosynthesis of Conjugate Vaccines Using an O-Linked Glycosylation System. mBio 2016, 7, e00443-16. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Pan, C.; Zeng, M.; Liu, B.; Liang, H.; Wang, D.; Liu, X.; Wang, B.; Lyu, Y.; Wu, J.; et al. Design and production of conjugate vaccines against S. Paratyphi A using an O-linked glycosylation system in vivo. NPJ Vaccines 2018, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Pan, C.; Sun, P.; Feng, E.; Wu, J.; Zhu, L.; Wang, H. Application of an O-Linked Glycosylation System in Yersinia enterocolitica Serotype O:9 to Generate a New Candidate Vaccine against Brucella abortus. Microorganisms 2020, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Hug, I.; Feldman, M.F. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 2010, 21, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Faridmoayer, A.; Fentabil, M.A.; Mills, D.C.; Klassen, J.S.; Feldman, M.F. Functional Characterization of Bacterial Oligosaccharyltransferases Involved in O-Linked Protein Glycosylation. J. Bacteriol. 2007, 189, 8088–8098. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Pan, C.; Sun, P.; Peng, Z.; Feng, E.; Wu, J.; Wang, H.; Zhu, L. Orthogonal modular biosynthesis of nanoscale conjugate vaccines for vaccination against infection. Nano Res. 2021, 15, 1645–1653. [Google Scholar] [CrossRef]
- Peng, Z.; Wu, J.; Wang, K.; Li, X.; Sun, P.; Zhang, L.; Huang, J.; Liu, Y.; Hua, X.; Yu, Y.; et al. Production of a Promising Biosynthetic Self-Assembled Nanoconjugate Vaccine against Klebsiella pneumoniae Serotype O2 in a General Escherichia coli Host. Adv. Sci. 2021, 8, 2100549. [Google Scholar] [CrossRef]
- Lim, K.H.; Huang, H.; Pralle, A.; Park, S. Engineered Streptavidin Monomer and Dimer with Improved Stability and Function. Biochem. 2011, 50, 8682–8691. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Huang, H.; Pralle, A.; Park, S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol. Bioeng. 2013, 110, 57–67. [Google Scholar] [CrossRef]
- Kroetsch, A.; Chin, B.; Nguyen, V.; Gao, J.; Park, S. Functional expression of monomeric streptavidin and fusion proteins in Escherichia coli: Applications in flow cytometry and ELISA. Appl. Microbiol. Biotechnol. 2018, 102, 10079–10089. [Google Scholar] [CrossRef]
- Dixon, J.E.; Osman, G.; Morris, G.E.; Markides, H.; Rotherham, M.; Bayoussef, Z.; El Haj, A.J.; Denning, C.; Shakesheff, K.M. Highly efficient delivery of functional cargoes by the synergistic effect of GAG binding motifs and cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2016, 113, E291–E299. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.S.; Milles, L.F.; Sedlak, S.M.; Gaub, H.E. Monomeric streptavidin: A versatile regenerative handle for force spectroscopy. bioRxiv 2018, 276444. [Google Scholar] [CrossRef]
- Csizmar, C.; Petersburg, J.R.; Hendricks, A.; Stern, L.; Hackel, B.J.; Wagner, C.R. Engineering Reversible Cell–Cell Interactions with Lipid Anchored Prosthetic Receptors. Bioconjugate Chem. 2018, 29, 1291–1301. [Google Scholar] [CrossRef]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Narendra, V.; Drury, W.J., 3rd; Casey, R.; Jansen, P.W.; Yuan, Z.F.; Garcia, B.A.; Vermeulen, M.; Bonasio, R. In vivo proximity labeling for the detection of protein-protein and protein-RNA interactions. J. Proteome Res. 2014, 13, 6135–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.K.; Demonte, D.; Dundas, C.M.; Park, S. Cell labeling and proximity dependent biotinylation with engineered monomeric streptavidin. Technology 2016, 4, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Liu, X.; Peng, Z.; Sun, H.; Zhang, M.; Zhang, J.; Liu, S.; Hao, L.; Lu, G.; Zheng, K.; et al. Retargeted human avidin-CAR T cells for adoptive immunotherapy of EGFRvIII expressing gliomas and their evaluation via optical imaging. Oncotarget 2015, 6, 23735–23747. [Google Scholar] [CrossRef] [Green Version]
- Lohmueller, J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. OncoImmunology 2018, 7, e1368604. [Google Scholar] [CrossRef]
- Thrane, S.; Janitzek, C.M.; Agerbæk, M.Ø.; Ditlev, S.B.; Resende, M.; Nielsen, M.A.; Theander, T.G.; Salanti, A.; Sander, A.F. A Novel Virus-Like Particle Based Vaccine Platform Displaying the Placental Malaria Antigen VAR2CSA. PLoS ONE 2015, 10, e0143071. [Google Scholar] [CrossRef] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [Green Version]
- Morona, R.; Mavris, M.; Fallarino, A.; Manning, P.A. Characterization of the rfc region of Shigella flexneri. J. Bacteriol. 1994, 176, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.A.; McKenzie, A.N.J. TH2 cell development and function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef]
- Athie-Morales, V.; Smits, H.; Cantrell, D.; Hilkens, C. Sustained IL-12 Signaling Is Required for Th1 Development. J. Immunol. 2003, 172, 61–69. [Google Scholar] [CrossRef]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 Increases the Innate Immunity of Tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef]
- Holmgren, J.; Harandi, A.M.; Czerkinsky, C. Mucosal adjuvants and anti-infection and anti-immunopathology vaccines based on cholera toxin, cholera toxin B subunit and CpG DNA. Expert Rev. Vaccines 2003, 2, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.; Fredriksson, M.; Nordström, I.; Holmgren, J. Cholera Toxin and Its B Subunit Promote Dendritic Cell Vaccination with Different Influences on Th1 and Th2 Development. Infect. Immun. 2003, 71, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Pan, C.; Liu, Z.; Sun, P.; Hua, X.; Feng, E.; Yu, Y.; Wu, J.; Zhu, L.; Wang, H. Safety and immunogenicity of a new glycoengineered vaccine against Acinetobacter baumannii in mice. Microb. Biotechnol. 2021. [Google Scholar] [CrossRef]
- Xu, H.; Huang, J.; Liu, Z.; Li, X.; Wang, K.; Feng, E.; Wu, J.; Zhu, L.; Yao, K.; Pan, C.; et al. Expression of Bordetella pertussis Antigens Fused to Different Vectors and Their Effectiveness as Vaccines. Vaccines 2021, 9, 542. [Google Scholar] [CrossRef]
- Anjuère, F.; George-Chandy, A.; Audant, F.; Rousseau, D.; Holmgren, J.; Czerkinsky, C. Transcutaneous Immunization with Cholera Toxin B Subunit Adjuvant Suppresses IgE Antibody Responses Via Selective Induction of Th1 Immune Responses. J. Immunol. 2003, 170, 1586–1592. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Yoo, J.-K.; Sohn, H.-J.; Kang, H.-K.; Kim, D.; Shin, H.-J.; Kim, J.-H. Protective immunity against Naegleria fowleri infection on mice immunized with the rNfa1 protein using mucosal adjuvants. Parasitol. Res. 2015, 114, 1377–1385. [Google Scholar] [CrossRef]
- Lv, X.; Yang, J.; Song, H.; Li, T.; Guo, L.; Xing, Y.; Xi, T. Therapeutic efficacy of the multi-epitope vaccine CTB-UE against Helicobacter pylori infection in a Mongolian gerbil model and its microRNA-155-associated immuno-protective mechanism. Vaccine 2014, 32, 5343–5352. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, K.; Sun, J.-B.; Nordström, I.; Fredriksson, M.; Lindblad, M.; Li, B.-L.; Holmgren, J. Coupling of antigen to cholera toxin for dendritic cell vaccination promotes the induction of MHC class I-restricted cytotoxic T cells and the rejection of a cognate antigen-expressing model tumor. Eur. J. Immunol. 2004, 34, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- De Haan, L.; Hearn, A.R.; Rivett, A.J.; Hirst, T.R. Enhanced Delivery of Exogenous Peptides into the Class I Antigen Processing and Presentation Pathway. Infect. Immun. 2002, 70, 3249–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isomura, I.; Yasuda, Y.; Tsujimura, K.; Takahashi, T.; Tochikubo, K.; Morita, A. Recombinant Cholera Toxin B Subunit Activates Dendritic Cells and Enhances Antitumor Immunity. Microbiol. Immunol. 2005, 49, 79–87. [Google Scholar] [CrossRef]
- Yamamoto, T.; Aoki, K.; Sugiyama, A.; Doi, H.; Kodama, T.; Shimizu, Y.; Kanai, M. Design and Synthesis of Biotin Analogues Reversibly Binding with Streptavidin. Chem.–Asian J. 2015, 10, 1071–1078. [Google Scholar] [CrossRef]
- Le, Q.; Nguyen, V.; Park, S. Recent advances in the engineering and application of streptavidin-like molecules. Appl. Microbiol. Biotechnol. 2019, 103, 7355–7365. [Google Scholar] [CrossRef]
- Spiller, K.L.; Nassiri, S.; Witherel, C.E.; Anfang, R.R.; Ng, J.; Nakazawa, K.R.; Yu, T.; Vunjak-Novakovic, G. Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials 2015, 37, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Cheng, K. The principles and applications of avidin-based nanoparticles in drug delivery and diagnosis. J. Control Release 2017, 245, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Jones, C. Vaccines based on the cell surface carbohydrates of pathogenic bacteria. Anais Acad. Bras. Ciências 2005, 77, 293–324. [Google Scholar] [CrossRef] [Green Version]
- Helppolainen, S.H.; Nurminen, K.P.; Määttä, J.A.E.; Halling, K.K.; Slotte, J.P.; Huhtala, T.; Liimatainen, T.; Ylä-Herttuala, S.; Airenne, K.J.; Närvänen, A.; et al. Rhizavidin from Rhizobium etli: The first natural dimer in the avidin protein family. Biochem. J. 2007, 405, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Lu, Y.-J.; Malley, R. Multiple antigen-presenting system (MAPS) to induce comprehensive B- and T-cell immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 13564–13569. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Li, F.; Wang, S.; Lu, G.; Bao, W.; Wang, Y.; Tian, Z.; Wei, W.; Ma, G. Near-infrared light–triggered platelet arsenal for combined photothermal-immunotherapy against cancer. Sci. Adv. 2021, 7, eabd7614. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Pan, C.; Wang, K.; Liu, Y.; Sun, Y.; Guo, Y.; Sun, P.; Wu, J.; Lu, Y.; Zhu, L.; et al. Construction of Orthogonal Modular Proteinaceous Nanovaccine Delivery Vectors Based on mSA-Biotin Binding. Nanomaterials 2022, 12, 734. https://doi.org/10.3390/nano12050734
Shi Y, Pan C, Wang K, Liu Y, Sun Y, Guo Y, Sun P, Wu J, Lu Y, Zhu L, et al. Construction of Orthogonal Modular Proteinaceous Nanovaccine Delivery Vectors Based on mSA-Biotin Binding. Nanomaterials. 2022; 12(5):734. https://doi.org/10.3390/nano12050734
Chicago/Turabian StyleShi, Yixin, Chao Pan, Kangfeng Wang, Yan Liu, Yange Sun, Yan Guo, Peng Sun, Jun Wu, Ying Lu, Li Zhu, and et al. 2022. "Construction of Orthogonal Modular Proteinaceous Nanovaccine Delivery Vectors Based on mSA-Biotin Binding" Nanomaterials 12, no. 5: 734. https://doi.org/10.3390/nano12050734