
Advancement of Fluorescent and Structural Properties of Bovine Serum Albumin-Gold Bioconjugates in Normal and Heavy Water with pH Conditioning and Ageing

 , , ,
, , ,
Abstract
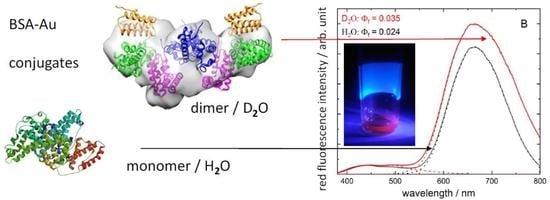
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Fluorescence Spectroscopy
2.4. Infrared Spectroscopy
2.5. Dynamic Light Scattering
2.6. Small-Angle X-ray Scattering
2.7. Small-Angle Neutron Scattering
3. Results and Discussion
3.1. Red Emission Characterized with Fluorescence Spectroscopy
3.2. Fine Structural Perturbations Observed by Infrared Spectroscopy
3.3. Conformational Changes of BSA–Au Conjugates Observed by Small-Angle X-ray and Neutron Scattering
3.4. Dynamic Light Scattering
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, H.-S.; Chiu, D.T. Soft fluorescent nanomaterials for biological and biomedical imaging. Chem. Soc. Rev. 2015, 44, 4699–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Zheng, Y.; Ying, J.Y. Protein-directed synthesis of highly fluorescent gold nanoclusters. J. Am. Chem. Soc. 2009, 131, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jin, R. On the ligand’s role in the fluorescence of gold nanoclusters. Nano Lett. 2010, 10, 2568–2573. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yu, P.; Toh, Y.-R.; Tang, J. Structure-correlated dual fluorescent bands in BSA-protected Au25 nanoclusters. J. Phys. Chem. C 2012, 116, 11830–11836. [Google Scholar] [CrossRef]
- Mohanty, J.S.; Chaudhari, K.; Sudhakar, C.; Pradeep, T. Metal-ion-induced luminescence enhancement in protein protected gold clusters. J. Phys. Chem. C 2019, 123, 28969–28976. [Google Scholar] [CrossRef]
- Le Guével, X.; Hötzer, B.; Jung, G.; Hollemeyer, K.; Trouillet, V.; Schneider, M. Formation of fluorescent metal (Au, Ag) nanoclusters capped in bovine serum albumin followed by fluorescence and spectroscopy. J. Phys. Chem. C 2011, 115, 10955–10963. [Google Scholar] [CrossRef]
- Chaudhari, K.; Xavier, P.L.; Pradeep, T. Understanding the evolution of luminescent gold quantum clusters in protein templates. ACS Nano 2011, 5, 8816–8827. [Google Scholar] [CrossRef]
- Sadler, P.J.; Tucker, A. PH-induced structural transitions of bovine serum albumin. Histidine PKa values and unfolding of the N-terminus during the N to F transition. Eur. J. Biochem. 1993, 212, 811–817. [Google Scholar] [CrossRef]
- Lin, H.; Imakita, K.; Fujii, M.; Sun, C.; Chen, B.; Kanno, T.; Sugimoto, H. New insights into the red luminescent bovine serum albumin conjugated gold nanospecies. J. Alloys Compd. 2017, 691, 860–865. [Google Scholar] [CrossRef]
- Dixon, J.M.; Egusa, S. Conformational change-induced fluorescence of bovine serum albumin–gold complexes. J. Am. Chem. Soc. 2018, 140, 2265–2271. [Google Scholar] [CrossRef]
- Dixon, J.M.; Tomida, J.; Egusa, S. Identifying the red-luminophore-forming domain in serum albumin–gold complexes. J. Phys. Chem. Lett. 2020, 11, 3345–3349. [Google Scholar] [CrossRef]
- Fehér, B.; Lyngsø, J.; Bartók, B.; Mihály, J.; Varga, Z.; Mészáros, R.; Pedersen, J.S.; Bóta, A.; Varga, I. Effect of PH on the conformation of bovine serume albumin–gold bioconjugates. J. Mol. Liq. 2020, 309, 113065. [Google Scholar] [CrossRef]
- Grdadolnik, J.; Maréchal, Y. Hydrogen–deuterium exchange in bovine serum albumin protein monitored by fourier transform infrared spectroscopy, Part I: Structural studies. Appl. Spectrosc. 2005, 59, 1347–1356. [Google Scholar] [CrossRef]
- Reslan, M.; Kayser, V. The effect of deuterium oxide on the conformational stability and aggregation of bovine serum albumin. Pharm. Dev. Technol. 2018, 23, 1030–1036. [Google Scholar] [CrossRef]
- Gardecki, J.A.; Maroncelli, M. Set of secondary emission standards for calibration of the spectral responsivity in emission spectroscopy. Appl. Spectrosc. 1998, 52, 1179–1189. [Google Scholar] [CrossRef]
- Crosby, G.A.; Demas, J.N. Measurement of photoluminescence quantum yields. Review. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar] [CrossRef]
- Varga, Z.; Mihály, J.; Berényi, S.; Bóta, A. Structural characterization of the poly(ethylene glycol) layer of sterically stabilized liposomes by means of FTIR spectroscopy. Eur. Polym. J. 2013, 49, 2415–2421. [Google Scholar] [CrossRef]
- CENIMAT. Dynamic Light Scattering (DLS)—AvidNano W130i. Available online: https://www.cenimat.fct.unl.pt/services/laboratory-electronic-and-optoelectronic-materials-and-devices/dynamic-light-scattering-dls-avidnano-w130i (accessed on 9 December 2021).
- Wacha, A.; Varga, Z.; Bóta, A. CREDO: A new general-purpose laboratory instrument for small-angle X-ray scattering. J. Appl. Cryst. 2014, 47, 1749–1754. [Google Scholar] [CrossRef] [Green Version]
- Wacha, A. Optimized pinhole geometry for small-angle scattering. J. Appl. Cryst. 2015, 48, 1843–1848. [Google Scholar] [CrossRef] [Green Version]
- SANS. Budapest Neutron Centre. Available online: https://www.bnc.hu/?q=ys-sans (accessed on 19 December 2021).
- Almásy, L. New measurement control software on the yellow submarine SANS instrument at the Budapest Neutron Centre. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2021, 15, 527–531. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta-Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Li, W.-W.; Katzir, A.; Raichlin, Y.; Yu, H.-Q.; Mizaikoff, B. Probing the secondary structure of bovine serum albumin during heat-induced denaturation using mid-infrared fiberoptic sensors. Analyst 2015, 140, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Cryst. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Jeffries, C.M.; Graewert, M.A.; Blanchet, C.E.; Langley, D.B.; Whitten, A.E.; Svergun, D.I. Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. Nat. Protoc. 2016, 11, 2122–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| β-Turns 1687 cm−1 | α-Helix 1656 m−1 | Loose α-Helix 1651 cm−1 | Random Coil 1648 cm−1 | β-Sheet 1638 cm−1 | Inter. β-Sheet 1611 cm−1 | |
|---|---|---|---|---|---|---|
| pH12 | 17 | - | 74 | 9 | - | |
| pH12 (2 d) | 35 | 23 | - | 42 | - | |
| pH7 (2 h) | 32 | 33 | - | 33 | 2 | |
| pH7 (2 d) | 27 | - | 41 | - | 29 | 3 |
| β-Turns 1687 cm−1 | α-Helix 1656 cm−1 | Loose α-Helix 1651 cm−1 | Random Coil 1648 cm−1 | β-Sheet 1638 cm−1 | Inter. β-Sheet 1611 cm−1 | |
|---|---|---|---|---|---|---|
| pD12 | 21 | - | - | 70 | - | 9 |
| pD12 (2 d) | 35 | - | - | - | 62 | 3 |
| pD7 (2 h) | 29 | 1 | - | 70 | - | |
| pD7 (2 d) | 34 | 15 | - | 51 | - |
| BSA pD7 SANS | BSA–Au pD12 SANS | BSA–Au pD7 SANS | BSA–Au pD12 SAXS | BSA–Au pD7 SAXS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I(0) (cm−1) | 0.037 ± 1 × 10−3 | 0.076 ± 1 × 10−3 | 0.200 ± 4 × 10−3 | 0.948 ± 1 × 10−3 | 1.970 ± 0.026 | |||||
| RG (Å) Guinier fit | 28.4 ± 0.9 | 27.1 ± 0.1 | 38.3 ± 0.4 | 31.60 ± 0.06 | 43.0 ± 0.390 | |||||
| RG (Å) IFT | 29.75 ± 1.12 | 28.47 ± 0.40 | 39.37 ± 0.46 | 33.71 ± 0.04 | 44.98 ± 0.30 | |||||
| Fitting method | DAMMIF | SASREF | DAMMIF | SASREF | DAMMIF | SASREF | DAMMIF | SASREF | DAMMIF | SASREF |
| χ2 | 1.11 | 1.09 | 1.09 | 1.95 | 1.63 | 1.04 | 1.95 | 6.56 | 0.73 | 1.46 |
| BSA pH7 | BSA pH12 | BSA–Au pH12 | BSA–Au pH7 | BSA pD7 | BSA pD12 | BSA–Au pD12 | BSA–Au pD7 | |
|---|---|---|---|---|---|---|---|---|
| Diameter, Dh (Å) | 37 | 99 | 131 | 115 | 38 | 81 | 126 | 233/119 * |
| Polydisp. (%) | 57.7 | 16.7 | 23.8 | 27.5 | 41.0 | 19.9 | 21.5 | 15.9/70.7 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, B.; Mihály, J.; Demeter, A.; Almásy, L.; Wacha, A.; Varga, Z.; Varga, I.; Pedersen, J.S.; Bóta, A. Advancement of Fluorescent and Structural Properties of Bovine Serum Albumin-Gold Bioconjugates in Normal and Heavy Water with pH Conditioning and Ageing. Nanomaterials 2022, 12, 390. https://doi.org/10.3390/nano12030390
Fehér B, Mihály J, Demeter A, Almásy L, Wacha A, Varga Z, Varga I, Pedersen JS, Bóta A. Advancement of Fluorescent and Structural Properties of Bovine Serum Albumin-Gold Bioconjugates in Normal and Heavy Water with pH Conditioning and Ageing. Nanomaterials. 2022; 12(3):390. https://doi.org/10.3390/nano12030390
Chicago/Turabian StyleFehér, Bence, Judith Mihály, Attila Demeter, László Almásy, András Wacha, Zoltán Varga, Imre Varga, Jan Skov Pedersen, and Attila Bóta. 2022. "Advancement of Fluorescent and Structural Properties of Bovine Serum Albumin-Gold Bioconjugates in Normal and Heavy Water with pH Conditioning and Ageing" Nanomaterials 12, no. 3: 390. https://doi.org/10.3390/nano12030390