
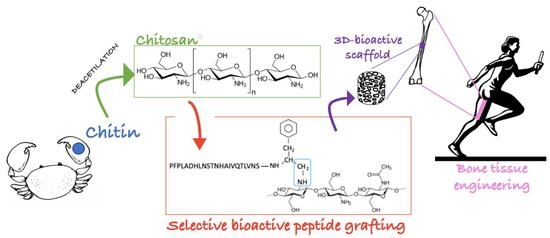
Chitosan Covalently Functionalized with Peptides Mapped on Vitronectin and BMP-2 for Bone Tissue Engineering

 ,
,  , , ,
, , ,  , ,
, ,  , , and
, , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis and Characterization
2.2.1. S-X-HVP
2.2.2. GBMP1a
2.3. S-X-HVP N-Terminal Conversion in an Alpha-oxo-Aldheyde
2.4. Scaffold Preparation
2.4.1. Chitosan Functionalization with ald-X-HVP and GBMP1a
2.4.2. Chitosan Spongy Scaffold Preparation
2.5. Scaffold Characterization
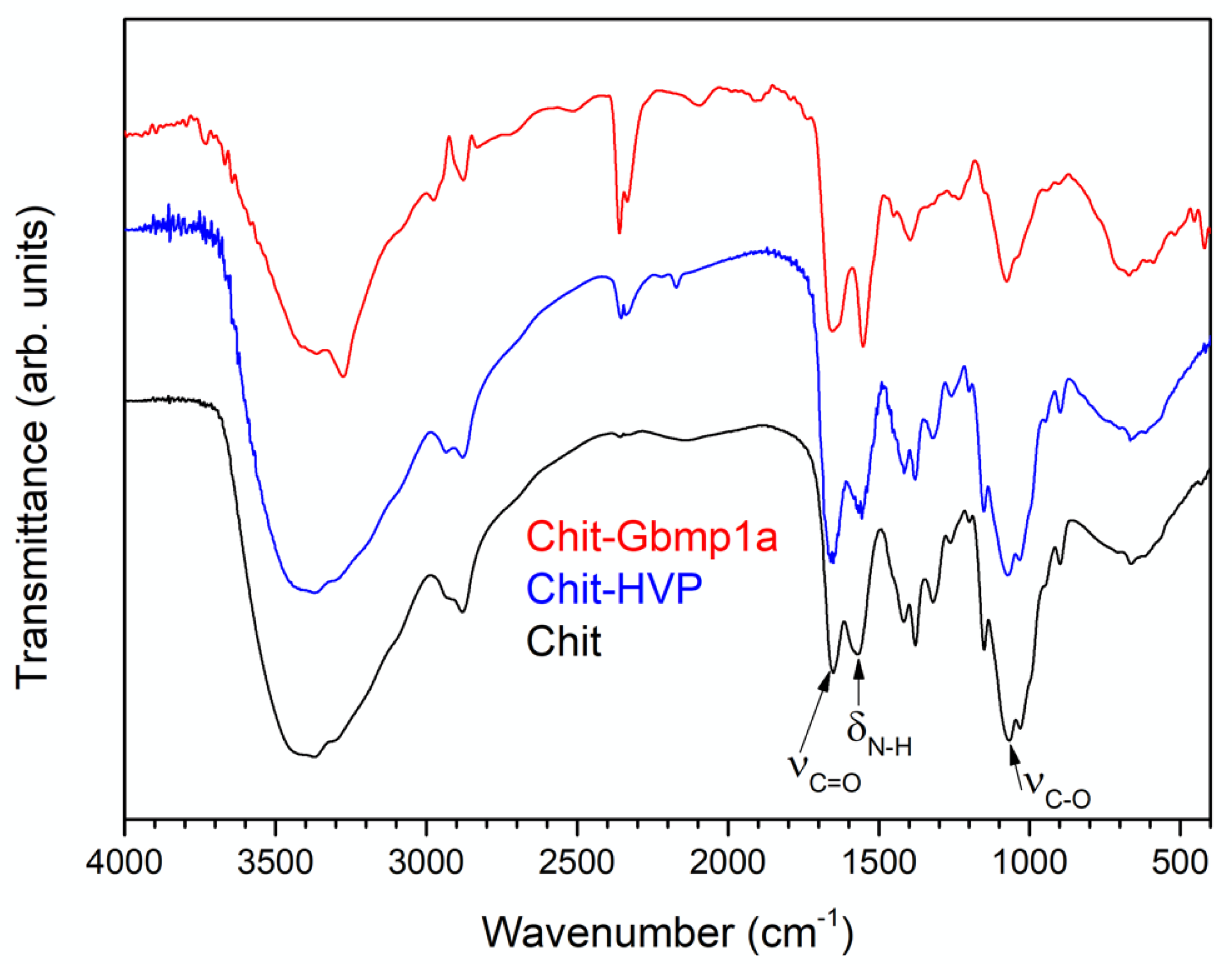
2.5.1. Fourier Transform Infrared Spectroscopy (FT-IR) Spectroscopy
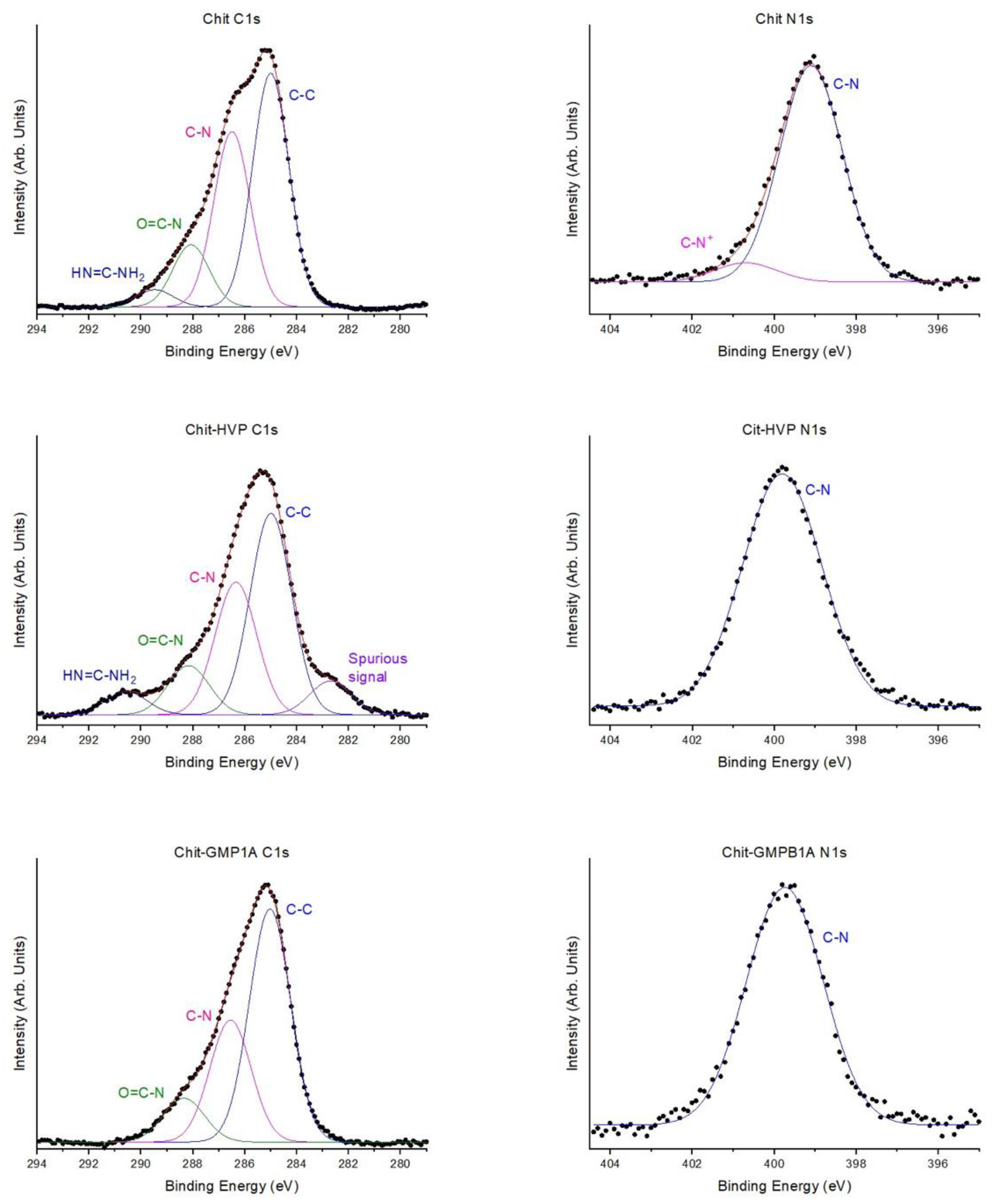
2.5.2. X-ray Photoelectron Spectroscopy (XPS)
2.5.3. Nuclear Magnetic Resonance analysis (NMR) of Chitosan, Chit-HVP, and Chit-GBMP1a in Solution
2.5.4. Scanning Electron Microscope Analysis (SEM)
2.6. Biological Assays
2.6.1. Cell Culture
2.6.2. Cell Viability Assay
2.6.3. Cell Proliferation Assay
2.6.4. Quantitative Real-Time Polymerase Chain Reaction
2.6.5. Calcium Assay
2.7. Statistical Analysis
3. Results
3.1. Scaffold Characterization
3.1.1. FT-IR
3.1.2. XPS Spectroscopy
3.1.3. NMR Analysis of Chitosan, Chit-HVP, and Chit-GBMP1a in Solution
3.1.4. SEM Analysis
3.2. Biological Assays
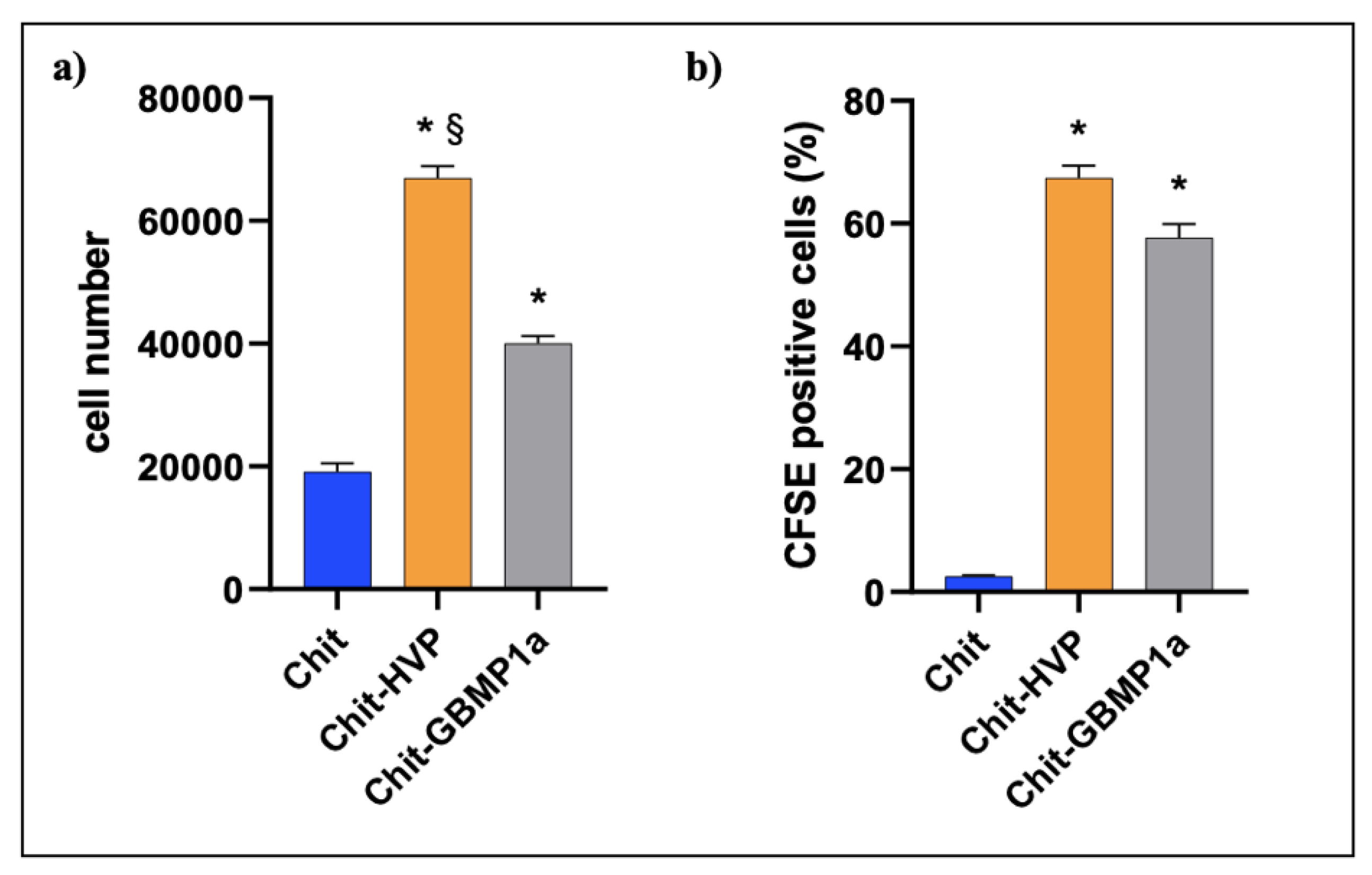
3.2.1. Functionalized Matrices Are Nontoxic for Osteoblast Cells and Support Cell Proliferation
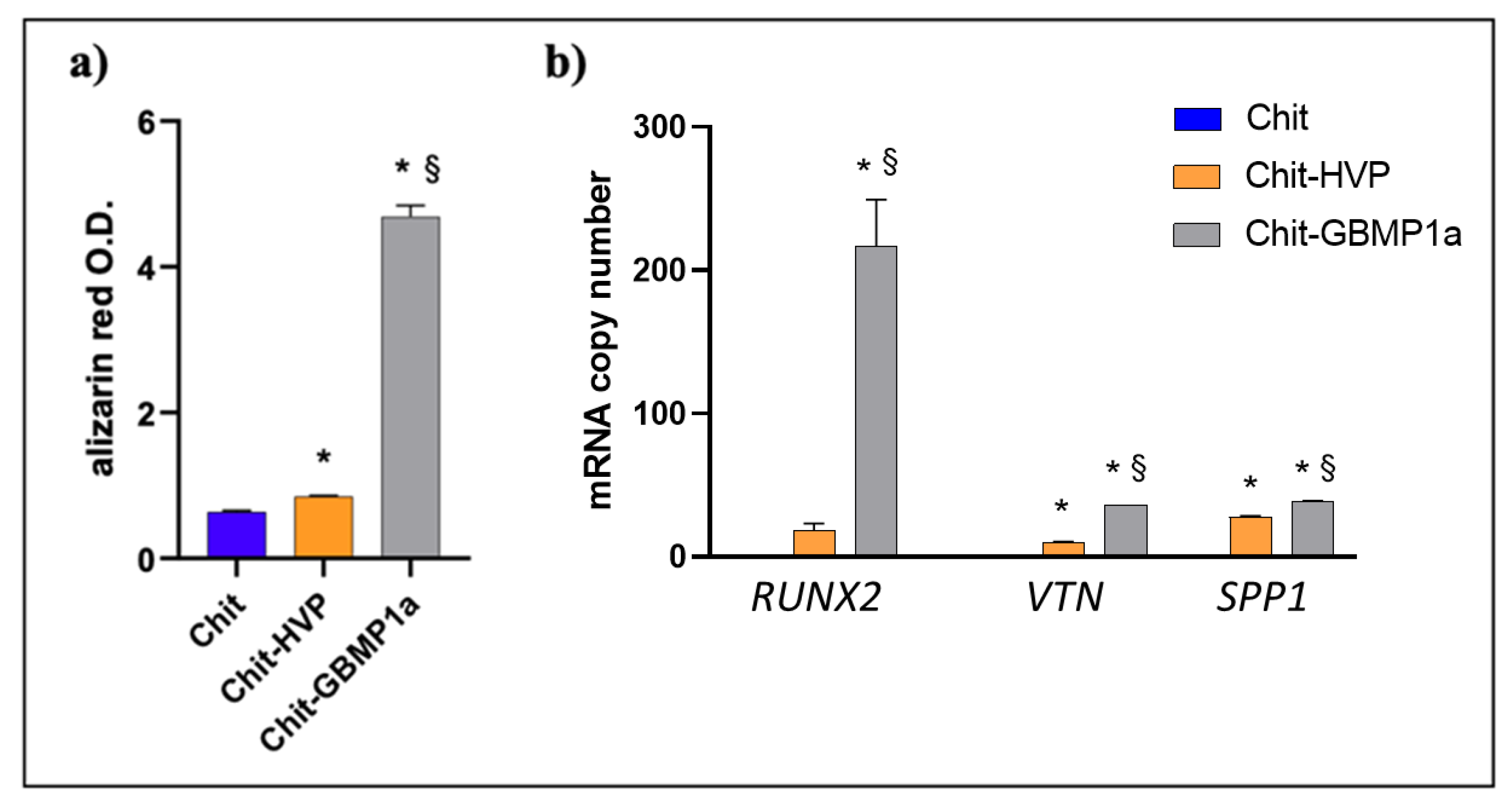
3.2.2. Functionalized Matrices Induce Differentiation in Cultured Osteoblast Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone Tissue Engineering: Recent Advances and Challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef] [Green Version]
- Oryan, A.; Alidadi, S.; Moshiri, A.; Maffulli, N. Bone regenerative medicine: Classic options, novel strategies, and future directions. J. Orthop. Surg. Res. 2014, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brydone, A.S.; Meek, D.; Maclaine, S. Bone grafting, orthopaedic biomaterials, and the clinical need for bone engineering. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 2010, 224, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Ehrler, D.M.; Vaccaro, A.R. The Use of Allograft Bone in Lumbar Spine Surgery. Clin. Orthop. Relat. Res. 2000, 371, 38–45. [Google Scholar] [CrossRef]
- Develioglu, H.; Saraydin, S.U.; Kartal, U. The bone-healing effect of a xenograft in a rat calvarial defect model. Dent. Mater. J. 2009, 28, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Moghaddam, A. Allograft bone matrix versus synthetic bone graft substitutes. Injury 2011, 42, S16–S21. [Google Scholar] [CrossRef]
- Barone, A.; Ricci, M.; Mangano, F.; Covani, U. Morbidity Associated With Iliac Crest Harvesting in the Treatment of Maxillary and Mandibular Atrophies: A 10-Year Analysis. J. Oral Maxillofac. Surg. 2011, 69, 2298–2304. [Google Scholar] [CrossRef]
- Nkenke, E.; Weisbach, V.; Winckler, E.; Kessler, P.; Schultze-Mosgau, S.; Wiltfang, J.; Neukam, F. Morbidity of harvesting of bone grafts from the iliac crest for preprosthetic augmentation procedures: A prospective study. Int. J. Oral Maxillofac. Surg. 2004, 33, 157–163. [Google Scholar] [CrossRef]
- Keating, J.F.; McQueen, M.M. Substitutes For Autologous Bone Graft In Orthopaedic Trauma. J. Bone Jt. Surgery. Br. Vol. 2001, 83-B, 3–8. [Google Scholar] [CrossRef]
- Moshiri, A.; Oryan, A. Role of tissue engineering in tendon reconstructive surgery and regenerative medicine: Current concepts, approaches and concerns. Hard Tissue 2012, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Janicki, P.; Schmidmaier, G. What should be the characteristics of the ideal bone graft substitute? Combining scaffolds with growth factors and/or stem cells. Injury 2011, 42, S77–S81. [Google Scholar] [CrossRef] [PubMed]
- Calori, G.; Mazza, E.; Colombo, M.; Ripamonti, C. The use of bone-graft substitutes in large bone defects: Any specific needs? Injury 2011, 42, S56–S63. [Google Scholar] [CrossRef]
- Reddy, M.; Ponnamma, D.; Choudhary, R.; Sadasivuni, K. A Comparative Review of Natural and Synthetic Biopolymer Composite Scaffolds. Polymers 2021, 13, 1105. [Google Scholar] [CrossRef] [PubMed]
- Oryan, A.; Alidadi, S.; Bigham-Sadegh, A.; Moshiri, A.; Kamali, A. Effectiveness of tissue engineered chitosan-gelatin composite scaffold loaded with human platelet gel in regeneration of critical sized radial bone defect in rat. J. Control. Release 2017, 254, 65–74. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Kundu, B.; Naskar, D.; Kim, H.-W.; Maiti, T.K.; Bhattacharya, D.; Kundu, S.C. Silk scaffolds in bone tissue engineering: An overview. Acta Biomater. 2017, 63, 1–17. [Google Scholar] [CrossRef]
- Zou, L.; Zhang, Y.; Liu, X.; Chen, J.; Zhang, Q. Biomimetic mineralization on natural and synthetic polymers to prepare hybrid scaffolds for bone tissue engineering. Colloids Surfaces B Biointerfaces 2019, 178, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vázquez, M.; Vega-Ruiz, B.; Ramos-Zúñiga, R.; Saldaña-Koppel, D.A.; Quiñones-Olvera, L.F. Chitosan and Its Potential Use as a Scaffold for Tissue Engineering in Regenerative Medicine. BioMed. Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, A.; Zein, N.; Harmouch, E.; Hafdi, B.; Bornert, F.; Offner, D.; Clauss, F.; Fioretti, F.; Huck, O.; Benkirane-Jessel, N.; et al. Application of Chitosan in Bone and Dental Engineering. Molecules 2019, 24, 3009. [Google Scholar] [CrossRef] [Green Version]
- Yadav, L.R.; Chandran, S.V.; Lavanya, K.; Selvamurugan, N. Chitosan-based 3D-printed scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2021, 183, 1925–1938. [Google Scholar] [CrossRef]
- Kumar, M.N.V.R. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Matica, M.A.; Aachmann, F.L.; Tøndervik, A.; Sletta, H.; Ostafe, V. Chitosan as a Wound Dressing Starting Material: Antimicrobial Properties and Mode of Action. Int. J. Mol. Sci. 2019, 20, 5889. [Google Scholar] [CrossRef] [Green Version]
- Oryan, A.; Sahvieh, S. Effectiveness of chitosan scaffold in skin, bone and cartilage healing. Int. J. Biol. Macromol. 2017, 104, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Leena, R.S.; Selvamurugan, N. Chitosan based biocomposite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ren, Q.; Li, Z.; Zhang, L. Chitosan-Based Biomimetically Mineralized Composite Materials in Human Hard Tissue Repair. Molecules 2020, 25, 4785. [Google Scholar] [CrossRef]
- Deepthi, S.; Venkatesan, J.; Kim, S.-K.; Bumgardner, J.D.; Jayakumar, R. An overview of chitin or chitosan/nano ceramic composite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1338–1353. [Google Scholar] [CrossRef]
- Stępniewski, M.; Martynkiewicz, J.; Gosk, J. Chitosan and its composites: Properties for use in bone substitution. Polym. Med. 2017, 47, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pighinelli, L.; Kucharska, M. Chitosan–hydroxyapatite composites. Carbohydr. Polym. 2013, 93, 256–262. [Google Scholar] [CrossRef]
- Zan, Q.; Wang, C.; Dong, L.; Cheng, P.; Tian, J. Design and preparation of chitosan/HA composite scaffolds for tissue engineering with long-bone-like structure. Int. J. Mater. Prod. Technol. 2010, 37, 271. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Park, Y.-J.; Lee, S.-J.; Ku, Y.; Han, S.-B.; Klokkevold, P.R.; Chung, C.-P. The Bone Regenerative Effect of Platelet-Derived Growth Factor-BB Delivered With a Chitosan/Tricalcium Phosphate Sponge Carrier. J. Periodontol. 2000, 71, 418–424. [Google Scholar] [CrossRef]
- Peter, M.; Kumar, P.T.S.; Binulal, N.S.; Nair, S.V.; Tamura, H.; Jayakumar, R. Development of novel α-chitin/nanobioactive glass ceramic composite scaffolds for tissue engineering applications. Carbohydr. Polym. 2009, 78, 926–931. [Google Scholar] [CrossRef]
- Pourhaghgouy, M.; Zamanian, A.; Shahrezaee, M.; Masouleh, M.P. Physicochemical properties and bioactivity of freeze-cast chitosan nanocomposite scaffolds reinforced with bioactive glass. Mater. Sci. Eng. C 2016, 58, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ramay, H.R.; Hauch, K.D.; Xiao, D.; Zhang, M. Chitosan–alginate hybrid scaffolds for bone tissue engineering. Biomaterials 2005, 26, 3919–3928. [Google Scholar] [CrossRef]
- Venkatesan, J.; Bhatnagar, I.; Manivasagan, P.; Kang, K.-H.; Kim, S.-K. Alginate composites for bone tissue engineering: A review. Int. J. Biol. Macromol. 2015, 72, 269–281. [Google Scholar] [CrossRef]
- Sivashankari, P.; Prabaharan, M. Prospects of chitosan-based scaffolds for growth factor release in tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1382–1389. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Fan, Y.; Li, X. The use of bioactive peptides to modify materials for bone tissue repair. Regen. Biomater. 2017, 4, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Bullock, G.; Atkinson, J.; Gentile, P.; Hatton, P.; Miller, C. Osteogenic Peptides and Attachment Methods Determine Tissue Regeneration in Modified Bone Graft Substitutes. J. Funct. Biomater. 2021, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, A.A.; Hennessy, K.M.; Bellis, S.L. The effect of adsorbed serum proteins, RGD and proteoglycan-binding peptides on the adhesion of mesenchymal stem cells to hydroxyapatite. Biomaterials 2007, 28, 383–392. [Google Scholar] [CrossRef]
- Battista, E.; Causa, F.; Lettera, V.; Panzetta, V.; Guarnieri, D.; Fusco, S.; Gentile, F.; Netti, P.A. Ligand engagement on material surfaces is discriminated by cell mechanosensoring. Biomaterials 2015, 45, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.-H.; Wang, D.-M.; Hsieh, H.-J.; Liu, H.-C.; Hsien, T.-Y.; Lai, J.-Y.; Hou, L.-T. Preparation and characterization of RGD-immobilized chitosan scaffolds. Biomaterials 2005, 26, 3197–3206. [Google Scholar] [CrossRef]
- Tiğli, R.S.; Gümüşderelioğlu, M. Evaluation of RGD- or EGF-immobilized chitosan scaffolds for chondrogenic activity. Int. J. Biol. Macromol. 2008, 43, 121–128. [Google Scholar] [CrossRef]
- Masuko, T.; Minami, A.; Iwasaki, N.; Majima, T.; Nishimura, A.S.-I.; Lee, Y.C. Thiolation of Chitosan. Attachment of Proteins via Thioether Formation. Biomacromolecules 2005, 6, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, K.; Nomizu, M. Mixed Peptide-Conjugated Chitosan Matrices as Multi-Receptor Targeted Cell-Adhesive Scaffolds. Int. J. Mol. Sci. 2018, 19, 2713. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-B.; Chen, Y.-R.; Li, W.-T.; Lai, J.-Y.; Liu, H.-L. RGD-conjugated UV-crosslinked chitosan scaffolds inoculated with mesenchymal stem cells for bone tissue engineering. Carbohydr. Polym. 2012, 89, 379–387. [Google Scholar] [CrossRef]
- Hansson, A.; Hashom, N.; Falson, F.; Rousselle, P.; Jordan, O.; Borchard, G. In vitro evaluation of an RGD-functionalized chitosan derivative for enhanced cell adhesion. Carbohydr. Polym. 2012, 90, 1494–1500. [Google Scholar] [CrossRef]
- Brun, P.; Zamuner, A.; Battocchio, C.; Cassari, L.; Todesco, M.; Graziani, V.; Iucci, G.; Marsotto, M.; Tortora, L.; Secchi, V.; et al. Bio-Functionalized Chitosan for Bone Tissue Engineering. Int. J. Mol. Sci. 2021, 22, 5916. [Google Scholar] [CrossRef]
- Secchi, V.; Franchi, S.; Ciccarelli, D.; Dettin, M.; Zamuner, A.; Serio, A.; Iucci, G.; Battocchio, C. Biofunctionalization of TiO2 Surfaces with Self-Assembling Layers of Oligopeptides Covalently Grafted to Chitosan. ACS Biomater. Sci. Eng. 2019, 5, 2190–2199. [Google Scholar] [CrossRef]
- Spears, R.J.; Fascione, M.A. Site-selective incorporation and ligation of protein aldehydes. Org. Biomol. Chem. 2016, 14, 7622–7638. [Google Scholar] [CrossRef] [Green Version]
- Ruoslahti, E. RGD and Other Recognition Sequences for Integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Massia, S.P.; Hubbell, J.A. Covalently Attached GRGD on Polymer Surfaces Promotes Biospecific Adhesion of Mammalian Cells. Ann. N. Y. Acad. Sci. 1990, 589, 261–270. [Google Scholar] [CrossRef]
- Dettin, M.; Zamuner, A.; Iucci, G.; Messina, G.M.L.; Battocchio, C.; Picariello, G.; Gallina, G.; Marletta, G.; Castagliuolo, I.; Brun, P. Driving h-osteoblast adhesion and proliferation on titania: Peptide hydrogels decorated with growth factors and adhesive conjugates. J. Pept. Sci. 2014, 20, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomateials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]
- Dettin, M.; Zamuner, A.; Roso, M.; Iucci, G.; Samouillan, V.; Danesin, R.; Modesti, M.; Conconi, M.T. Facile and selective covalent grafting of an RGD-peptide to electrospun scaffolds improves HUVEC adhesion. J. Pept. Sci. 2015, 21, 786–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Scorzeto, M.; Vassanelli, S.; Castagliuolo, I.; Palù, G.; Ghezzo, F.; Messina, G.M.; Iucci, G.; Battaglia, V.; Sivolella, S.; et al. Mechanisms underlying the attachment and spreading of human osteoblasts: From transient interactions to focal adhesions on vitronectin-grafted bioactive surfaces. Acta Biomater. 2013, 9, 6105–6115. [Google Scholar] [CrossRef]
- Zamuner, A.; Brun, P.; Ciccimarra, R.; Ravanetti, F.; Veschini, L.; Elsayed, H.; Sivolella, S.; Iucci, G.; Porzionato, A.; Di Silvio, L.; et al. Biofunctionalization of bioactive ceramic scaffolds to increase the cell response for bone regeneration. Biomed. Mater. 2021, 16, 055007. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Dettin, M.; Maule, F.; Scabello, A.; Calvanese, L.; D’Auria, G.; Falcigno, L.; Porcù, E.; Zamuner, A.; Della Puppa, A.; et al. A synthetic BMP-2 mimicking peptide induces glioblastoma stem cell differentiation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2282–2292. [Google Scholar] [CrossRef]
- Watts, J.F. High resolution XPS of organic polymers: The Scienta ESCA 300 database. G. Beamson and D. Briggs. 280pp., £65. John Wiley & Sons, Chichester, ISBN 0471 935921, (1992). Book Review. Surf. Interface Anal. 1993, 20, 267. [Google Scholar] [CrossRef]
- Yeh, J.; Lindau, I. Atomic subshell photoionization cross sections and asymmetry parameters: 1 ≤ Z ≤ 103. At. Data Nucl. Data Tables 1985, 32, 1–155. [Google Scholar] [CrossRef]
- Ortiz, J.; Chou, L.L. Calcium upregulated survivin expression and associated osteogenesis of normal human osteoblasts. J. Biomed. Mater. Res. Part A 2012, 100A, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Ghezzo, F.; Roso, M.; Danesin, R.; Palù, G.; Bagno, A.; Modesti, M.; Castagliuolo, I.; Dettin, M. Electrospun scaffolds of self-assembling peptides with poly(ethylene oxide) for bone tissue engineering. Acta Biomater. 2011, 7, 2526–2532. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, Y.; Maye, P.; Rowe, D.W. Examination of Mineralized Nodule Formation in Living Osteoblastic Cultures Using Fluorescent Dyes. Biotechnol. Prog. 2006, 22, 1697–1701. [Google Scholar] [CrossRef]
- Kumirska, J.; Czerwicka, M.; Kaczyński, Z.; Bychowska, A.; Brzozowski, K.; Thöming, J.; Stepnowski, P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drugs 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [Green Version]
- Lebugle, A.; Subirade, M.; Gueguen, J. Structural characteristics of a globular protein investigated by X-ray photoelectron spectroscopy: Comparison between a legumin film and a powdered legumin. Biochim. Biophys. Acta 1995, 1248, 107–114. [Google Scholar] [CrossRef]
- Deligianni, D.; Katsala, N.; Ladas, S.; Sotiropoulou, D.; Amedee, J.; Missirlis, Y. Effect of surface roughness of the titanium alloy Ti–6Al–4V on human bone marrow cell response and on protein adsorption. Biomaterials 2001, 22, 1241–1251. [Google Scholar] [CrossRef]
- Franchi, S.; Secchi, V.; Santi, M.; Dettin, M.; Zamuner, A.; Battocchio, C.; Iucci, G. Biofunctionalization of TiO2 surfaces with self-assembling oligopeptides in different pH and Ionic Strength conditions: Charge effects and molecular organization. Mater. Sci. Eng. C 2018, 90, 651–656. [Google Scholar] [CrossRef]
- Naumkin, A.V.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. X-ray Photoelectron Spectroscopy Database XPS, Version 4.1, NIST Standard Reference Database 20; National Institute of Standards and Technology: Gaithersburg, MD, USA, 1989. [CrossRef]
- Ding, X.-X.; Zhou, Y.-M.; Xiang, X.-C.; Meng, L.; Qin, Q.; Ye, S. Research progress on chitosan composite scaffolds in bone tissue engineering. Hua Xi Kou Qiang Yi Xue Za Zhi Huaxi Kouqiang Yixue Zazhi West China J. Stomatol. 2018, 36, 441–446. [Google Scholar] [CrossRef]
- Batista, M.; Pinto, L.; Gomes, C.; Gomes, P. Novel highly-soluble peptide–chitosan polymers: Chemical synthesis and spectral characterization. Carbohydr. Polym. 2006, 64, 299–305. [Google Scholar] [CrossRef] [Green Version]
- El-Amin, S.; Lu, H.; Khan, Y.; Burems, J.; Mitchell, J.; Tuan, R.; Laurencin, C. Extracellular matrix production by human osteoblasts cultured on biodegradable polymers applicable for tissue engineering. Biomaterials 2003, 24, 1213–1221. [Google Scholar] [CrossRef]
- Hanna, H.; Mir, L.M.; Andre, F.M. In vitro osteoblastic differentiation of mesenchymal stem cells generates cell layers with distinct properties. Stem Cell Res. Ther. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.D.S.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence |
|---|---|
| GAPDH | Fw: 5′-agtgccagcctcgtcccgta-3′ Rv: 5′-caggcgcccaatacggccaa-3′ |
| RUNX2 | Fw: 5′-cagtgacaccatgtcagcaa-3′ Rv: 5′-gctcacgtcgctcattttg-3′ |
| VTN | Fw: 5′- ggaggacatcttcgagcttct-3′ Rv: 5′- gctaatgaactggggctgtc-3′ |
| SPP1 | Fw: 5′-aagtttcgcagacctgacatc-3′ Rv: 5′-ggctgtcccaatcagaagg-3′ |
| Sample | N/C Ratio |
|---|---|
| Chit | 0.07 |
| Chit-HVP | 0.15 |
| Chit-GBMP1a | 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, P.; Zamuner, A.; Cassari, L.; D’Auria, G.; Falcigno, L.; Franchi, S.; Contini, G.; Marsotto, M.; Battocchio, C.; Iucci, G.; et al. Chitosan Covalently Functionalized with Peptides Mapped on Vitronectin and BMP-2 for Bone Tissue Engineering. Nanomaterials 2021, 11, 2784. https://doi.org/10.3390/nano11112784
Brun P, Zamuner A, Cassari L, D’Auria G, Falcigno L, Franchi S, Contini G, Marsotto M, Battocchio C, Iucci G, et al. Chitosan Covalently Functionalized with Peptides Mapped on Vitronectin and BMP-2 for Bone Tissue Engineering. Nanomaterials. 2021; 11(11):2784. https://doi.org/10.3390/nano11112784
Chicago/Turabian StyleBrun, Paola, Annj Zamuner, Leonardo Cassari, Gabriella D’Auria, Lucia Falcigno, Stefano Franchi, Giorgio Contini, Martina Marsotto, Chiara Battocchio, Giovanna Iucci, and et al. 2021. "Chitosan Covalently Functionalized with Peptides Mapped on Vitronectin and BMP-2 for Bone Tissue Engineering" Nanomaterials 11, no. 11: 2784. https://doi.org/10.3390/nano11112784