Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery

 , ,
, ,
Abstract
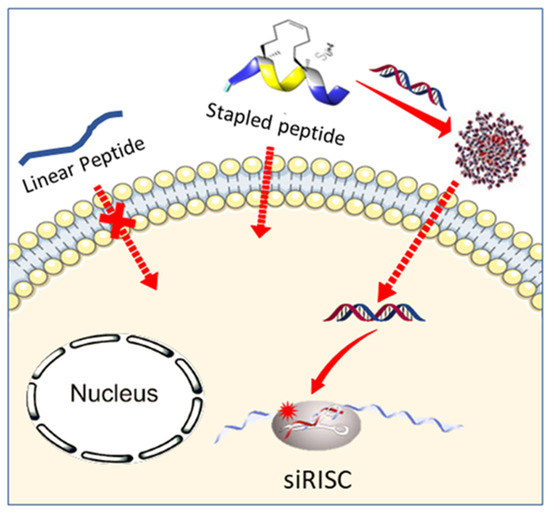
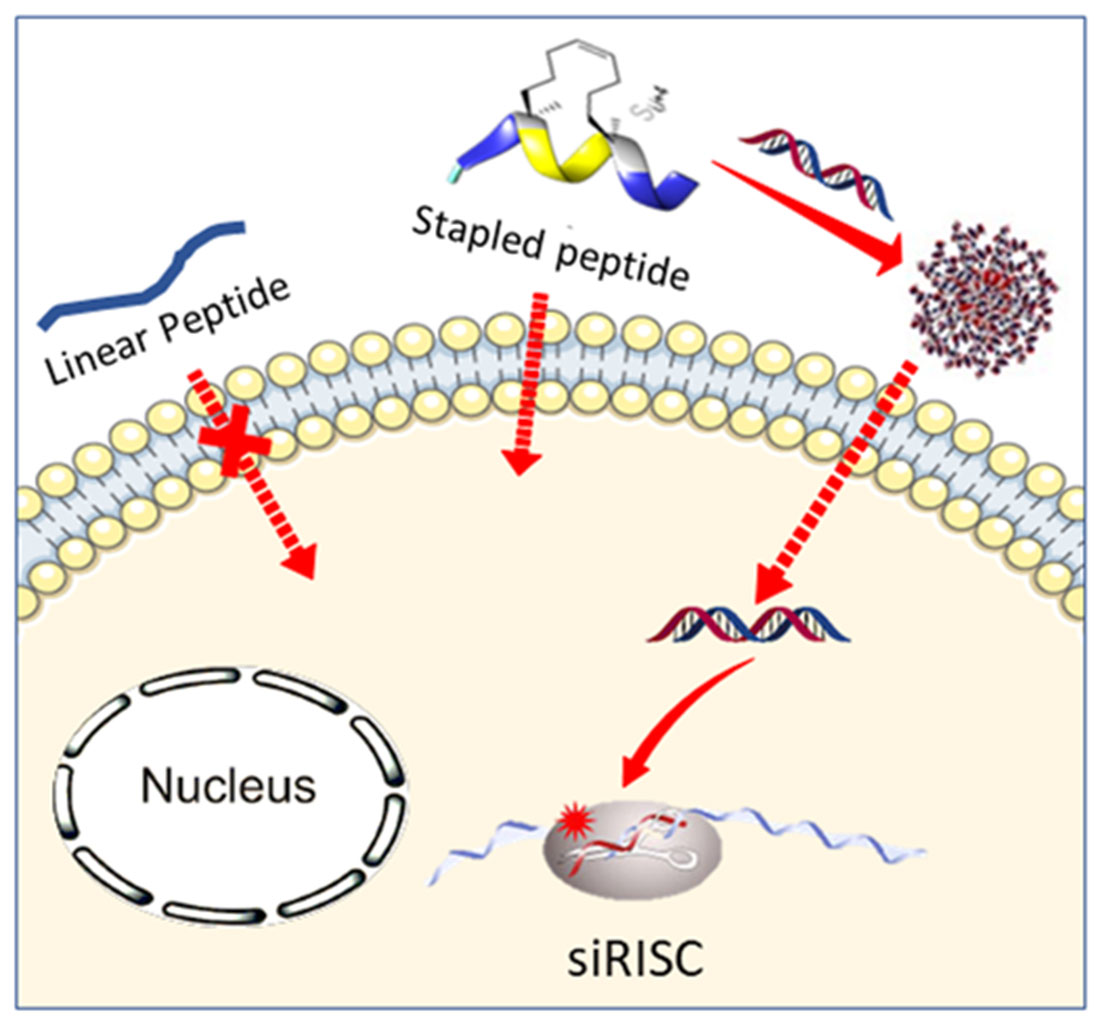
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Synthesis and Purification of Peptides and Stapled Peptides
2.3. Circular Dichroism (CD) and Determination of Helical Content
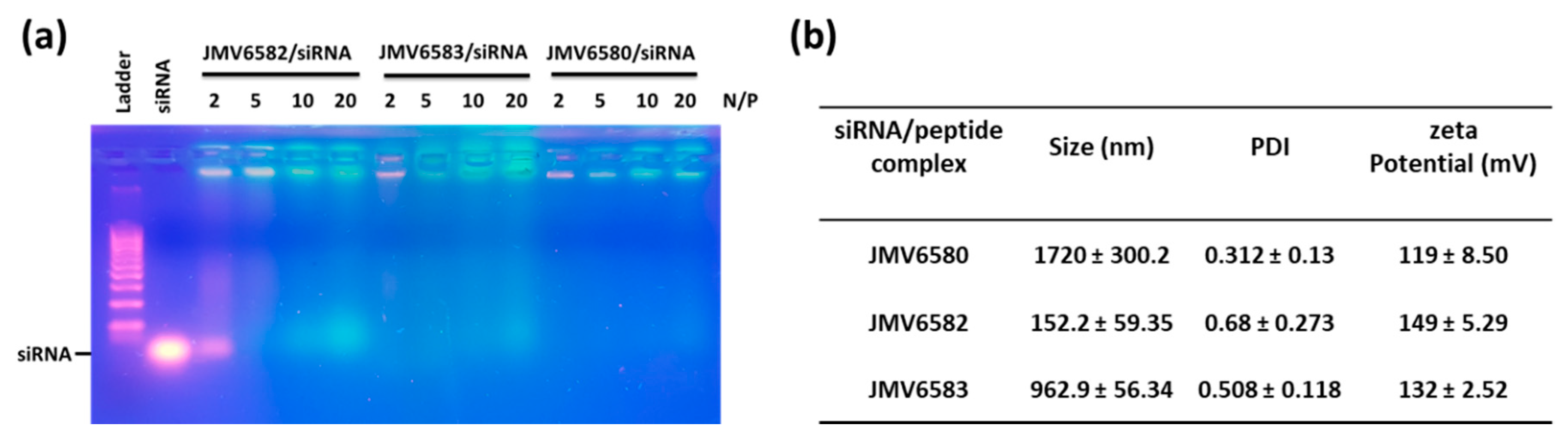
2.4. Gel Retardation Assay
2.5. Dynamic Light Scattering (DLS) and Zeta Potential Measurements
2.6. Cell Culture
2.7. Cell Viability (MTT) Assay
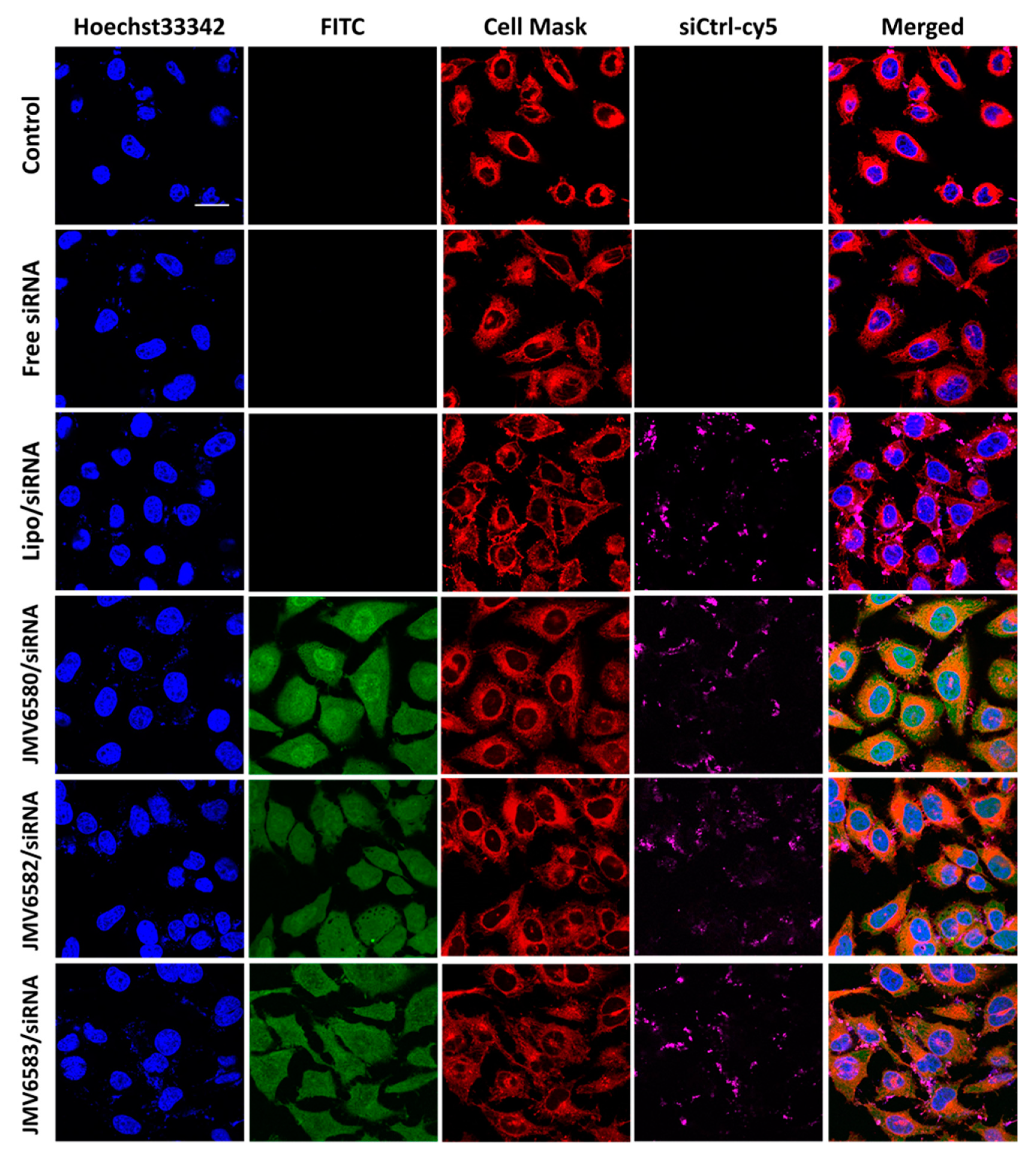
2.8. Cellular Uptake of FITC-Stapled Peptides
2.9. Imaging
2.10. Cell Luciferase Assay
2.11. Statistical Analysis
3. Results and Discussion
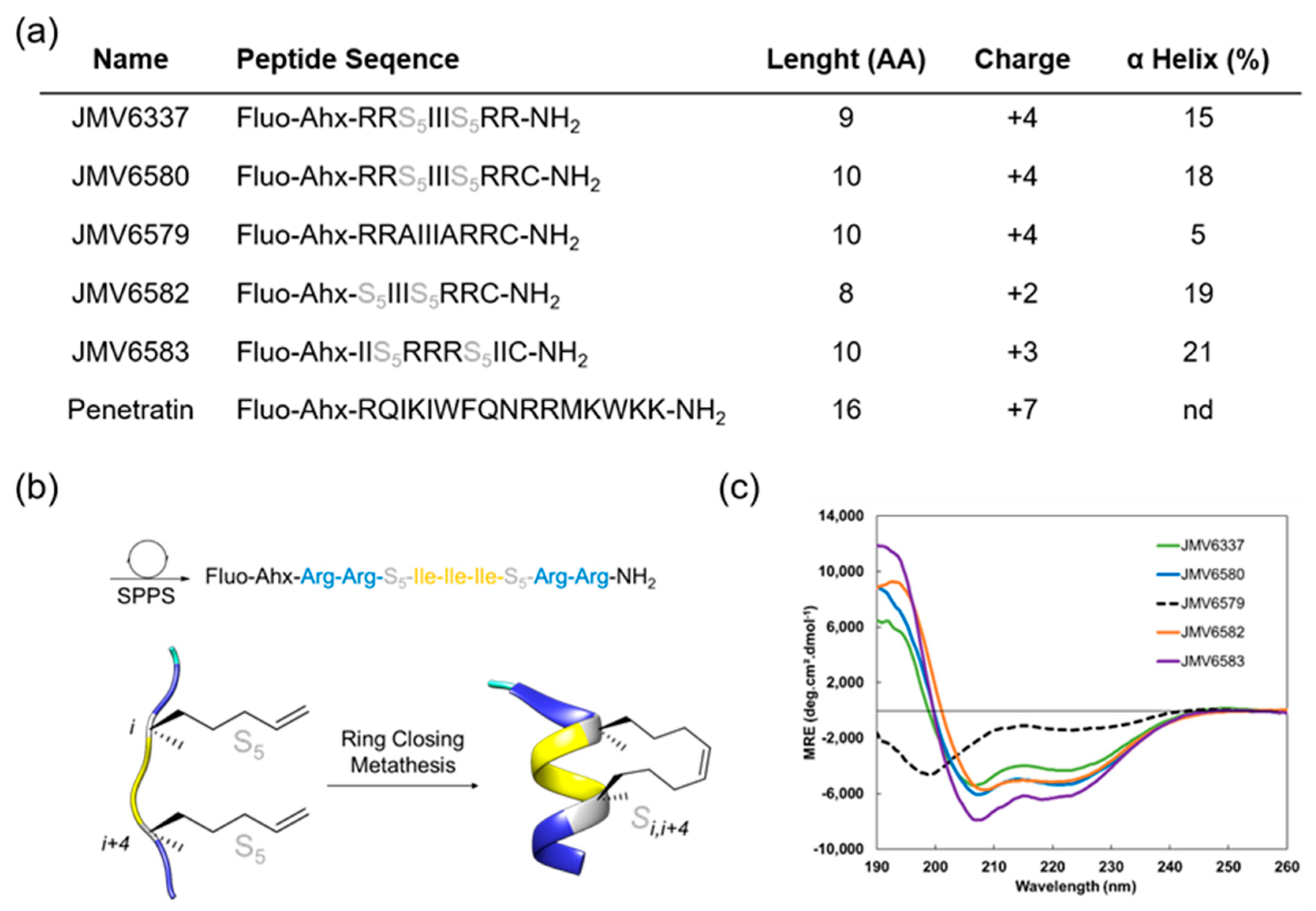
3.1. Design, Synthesis, and Characterization of the CPPs
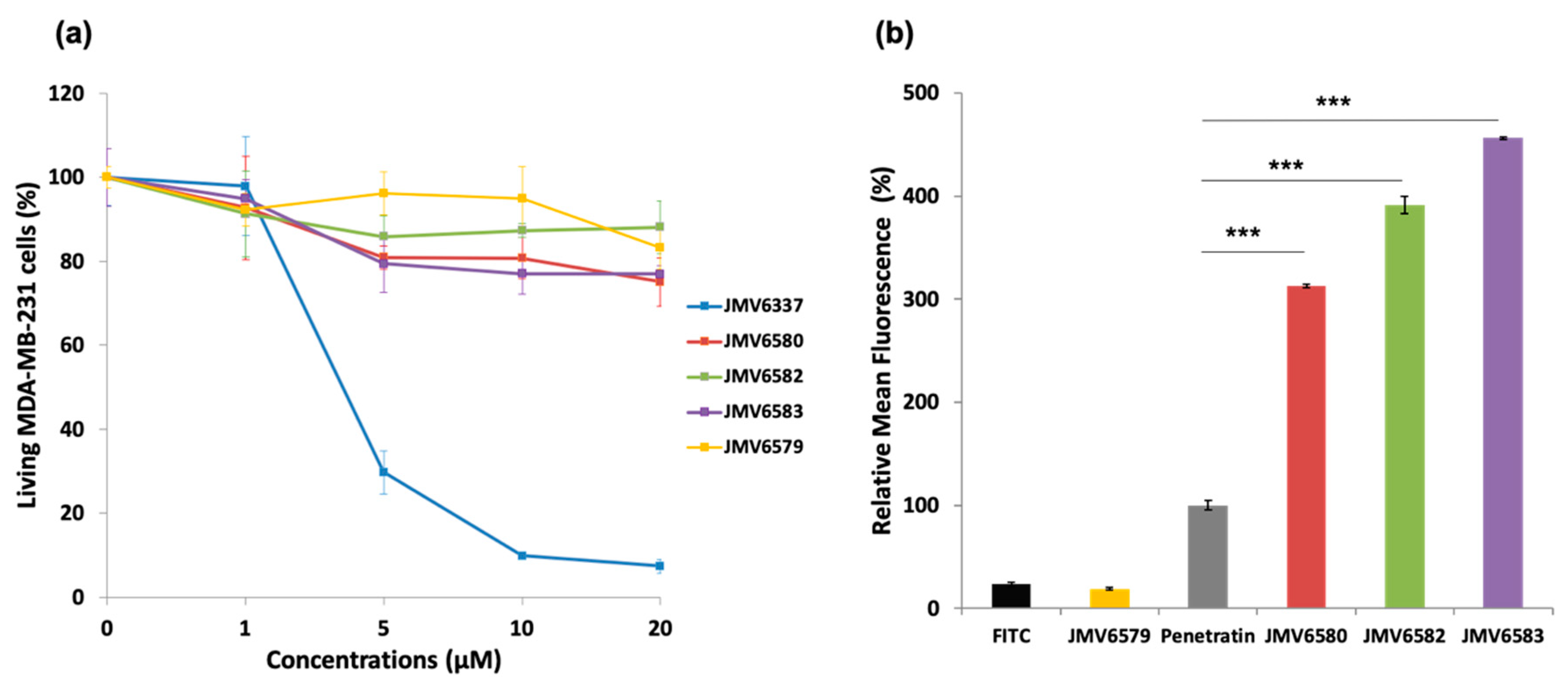
3.2. Cytotoxicity and Cellular Uptake Studies
3.3. siRNA Delivery Studies
3.4. Cellular Uptake
3.5. Inhibition of Luciferase Activity in MDA-MB-231-RFP-Luc Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turner, J.J.; Jones, S.W.; Moschos, S.A.; Lindsay, M.A.; Gait, M.J. MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity. Mol. Biosyst. 2007, 3, 43–50. [Google Scholar] [CrossRef]
- Reischl, D.; Zimmer, A. Drug delivery of siRNA therapeutics: Potentials and limits of nanosystems. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 8–20. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef]
- Laroui, N.; Cubedo, N.; Rossel, M.; Bettache, N. Improvement of Cell Penetrating Peptide for Efficient siRNA Targeting of Tumor Xenografts in Zbrafish Embryos. Adv. Ther. 2020, 3, 3. [Google Scholar] [CrossRef]
- Singh, T.; Murthy, A.S.N.; Yang, H.-J.; Im, J. Versatility of cell-penetrating peptides for intracellular delivery of siRNA. Drug Deliv. 2018, 25, 1996–2006. [Google Scholar] [CrossRef] [Green Version]
- Konate, K.; Crombez, L.; Deshayes, S.; Decaffmeyer, M.; Thomas, A.; Brasseur, R.; Aldrian, G.; Heitz, F.; Divita, G. Insight into the cellular uptake mechanism of a secondary amphipathic cell-penetrating peptide for siRNA delivery. Biochemistry 2010, 49, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.; Lundqvist, M.; Liedberg, B.; Jonsson, B.-H.; Ederth, T. Secondary structure in de novo designed peptides induced by electrostatic interaction with a lipid bilayer membrane. Langmuir 2010, 26, 6437–6448. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Bauzá, A.; Girardet, C.; Alves, I.D.; LeComte, S.; Illien, F.; Cardon, S.; Chaianantakul, N.; Pallerla, M.; Burlina, F.; et al. Ionpair-pi interactions favor cell penetration of arginine/tryptophan-rich cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183098. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Y.A.; Raschle, P.S.; Wennemers, H. Effect of Preorganized Charge-Display on the Cell-Penetrating Properties of Cationic Peptides. Angew. Chem. Int. Ed. Engl. 2017, 56, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Vezenkov, L.L.; Martin, V.; Bettache, N.; Simon, M.; Messerschmitt, A.; Legrand, B.; Bantignies, J.-L.; Subra, G.; Maynadier, M.; Bellet, V.; et al. Ribbon-like Foldamers for Cellular Uptake and Drug Delivery. Chembiochem 2017, 18, 2110–2114. [Google Scholar] [CrossRef]
- Yamashita, H.; Misawa, T.; Oba, M.; Tanaka, M.; Naito, M.; Kurihara, M.; Demizu, Y. Development of helix-stabilized cell-penetrating peptides containing cationic alpha,alpha-disubstituted amino acids as helical promoters. Bioorg. Med. Chem. 2017, 25, 1846–1851. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Oba, M.; Misawa, T.; Tanaka, M.; Hattori, T.; Naito, M.; Kurihara, M.; Demizu, Y. A Helix-Stabilized Cell-Penetrating Peptide as an Intracellular Delivery Tool. Chembiochem 2016, 17, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, E.H.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Miller, J.S.; Blackwell, H.E.; Grubbs, R.H. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. J. Am. Chem. Soc. 1996, 118, 9606–9614. [Google Scholar] [CrossRef]
- Miller, J.S.; Grubbs, R.H. Synthesis of Conformationally Restricted Amino-Acids and Peptides Employing Olefin Metathesis. J. Am. Chem. Soc. 1995, 117, 5855–5856. [Google Scholar] [CrossRef]
- Bird, G.H.; Bernal, F.; Pitter, K.; Walensky, L.D. Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzym. 2008, 446, 369–386. [Google Scholar]
- Chu, Q.; Moellering, R.E.; Hilinski, G.J.; Kim, H.K.; Grossmann, T.N.; Yeh, J.T.-H.; Verdine, G.L. Towards understanding cell penetration by stapled peptides. Medchemcomm 2015, 6, 111–119. [Google Scholar] [CrossRef]
- Schafmeister, E.C.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Verdine, L.G.; Hilinski, G.J. Stapled peptides for intracellular drug targets. Methods Enzym. 2012, 503, 3–33. [Google Scholar]
- Walensky, D.L.; Bird, G.H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [Green Version]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walensky, L.D.; Pitter, K.; Morash, J.; Oh, K.J.; Barbuto, S.; Fisher, J.; Smith, E.; Verdine, G.L.; Korsmeyer, S.J. A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell 2006, 24, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.S.; Spring, D.R. Using Peptidomimetics and Constrained Peptides as Valuable Tools for Inhibiting Protein(-)Protein Interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef]
- Bouclier, C.; Simon, M.; Laconde, G.; Pellerano, M.; Diot, S.; Lantuejoul, S.; Busser, B.; Vanwonterghem, L.; Vollaire, J.; Josserand, V.; et al. Stapled peptide targeting the CDK4/Cyclin D interface combined with Abemaciclib inhibits KRAS mutant lung cancer growth. Theranostics 2020, 10, 2008–2028. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.; Yu, J.; Lee, H.N.; Lee, C.; Oh, D.; Lee, D.-K.; Lee, C.; Lee, Y.; Yu, J. Construction of histidine-containing hydrocarbon stapled cell penetrating peptides for in vitro and in vivo delivery of siRNAs. Chem. Sci. 2018, 9, 3820–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, Q.; Bhattacharya, S.; Waheed, A.A.; Tong, X.; Hong, A.; Heck, S.; Curreli, F.; Goger, M.; Cowburn, D.; et al. A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J. Mol. Biol. 2008, 378, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Park, J.; Ryu, J.; Kim, K.-A.; Lee, H.J.; Bahn, J.H.; Han, K.; Choi, E.Y.; Lee, K.S.; Kwon, H.Y.; Choi, S.Y. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol. 2002, 83 Pt 5, 1173–1181. [Google Scholar] [CrossRef]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin Story: An Overview. Methods Mol. Biol. 2015, 1324, 29–37. [Google Scholar] [PubMed]
- Matsumoto, R.; Okochi, M.; Shimizu, K.; Kanie, K.; Kato, R.; Honda, H. Effects of the properties of short peptides conjugated with cell-penetrating peptides on their internalization into cells. Sci. Rep. 2015, 5, 12884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åmand, H.L.; Nordén, B.; Fant, K. Functionalization with C-terminal cysteine enhances transfection efficiency of cell-penetrating peptides through dimer formation. Biochem. Biophys. Res. Commun. 2012, 418, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Van Asbeck, A.H.; Beyerle, A.; McNeill, H.; Bovee-Geurts, P.H.; Lindberg, S.; Verdurmen, W.P.; Hällbrink, M.; Langel, Ü.; Heidenreich, O.; Brock, R. Molecular Parameters of siRNA-Cell Penetrating Peptide Nanocomplexes for Efficient Cellular Delivery. ACS Nano 2013, 7, 3797–3807. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L.; Hilinski, G.J. All-hydrocarbon stapled peptides as Synthetic Cell-Accessible Mini-Proteins. Drug Discov. Today Technol. 2012, 9, e1–e70. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, M.; Laroui, N.; Heyraud, M.; Laconde, G.; Ali, L.M.A.; Bourbiaux, K.; Subra, G.; Vezenkov, L.L.; Legrand, B.; Amblard, M.; et al. Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery. Nanomaterials 2020, 10, 2334. https://doi.org/10.3390/nano10122334
Simon M, Laroui N, Heyraud M, Laconde G, Ali LMA, Bourbiaux K, Subra G, Vezenkov LL, Legrand B, Amblard M, et al. Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery. Nanomaterials. 2020; 10(12):2334. https://doi.org/10.3390/nano10122334
Chicago/Turabian StyleSimon, Matthieu, Nabila Laroui, Marianne Heyraud, Guillaume Laconde, Lamiaa M. A. Ali, Kevin Bourbiaux, Gilles Subra, Lubomir L. Vezenkov, Baptiste Legrand, Muriel Amblard, and et al. 2020. "Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery" Nanomaterials 10, no. 12: 2334. https://doi.org/10.3390/nano10122334