Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations

 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. HPMC MBGs Formulation
2.2. Scanning Electron Microscopy (SEM) Measurements
2.3. Electron Paramagnetic Resonance (EPR) Measurements
2.3.1. Spin-Probing
2.3.2. Spin-Labeling Lipase
2.3.3. Interpretation of the EPR Data
2.4. Small-Angle X-ray Scattering (SAXS) Measurements
3. Results
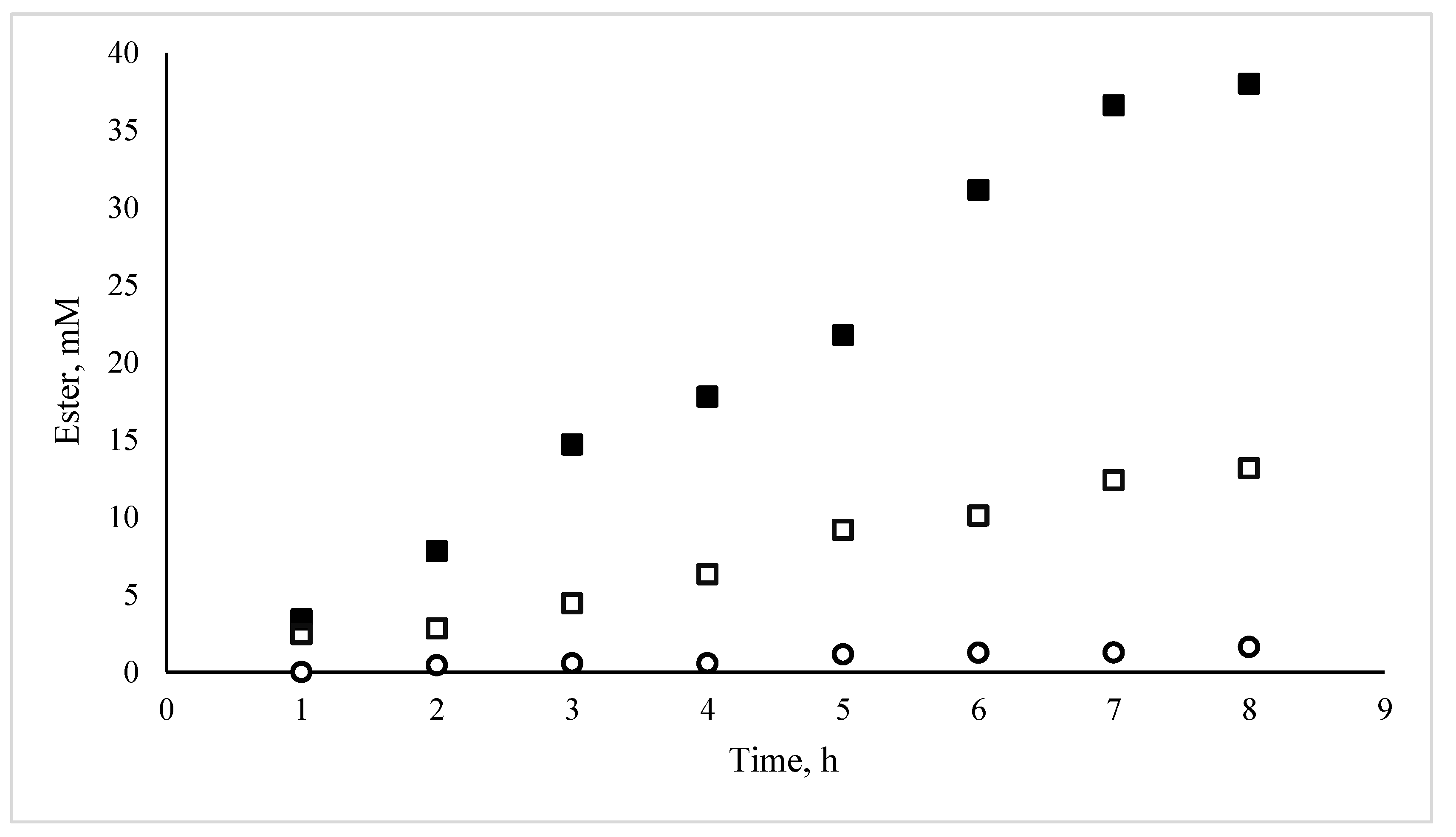
3.1. Catalytic Activity
3.2. Morphological Analysis
3.3. Interfacial Properties
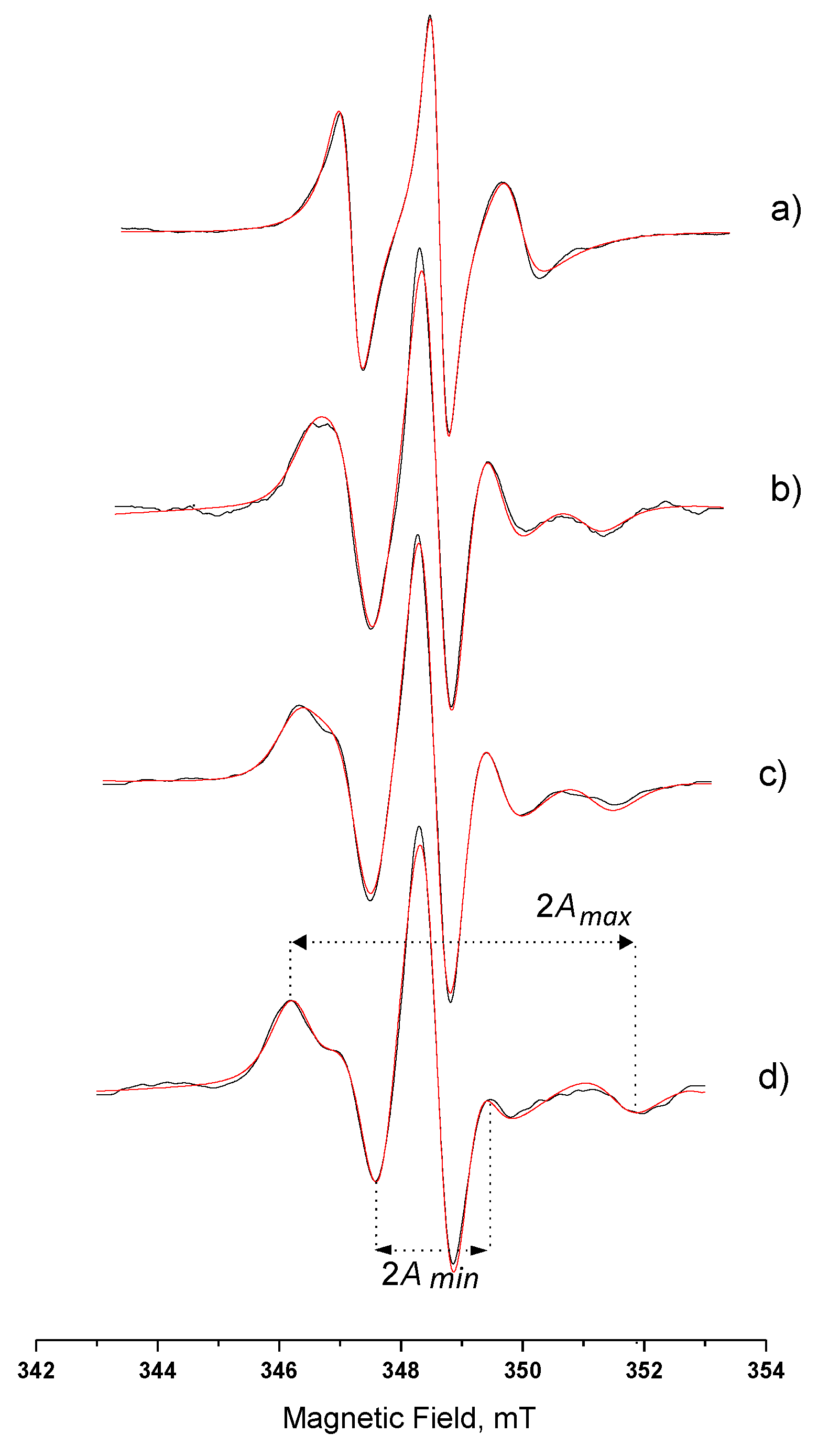
3.3.1. Hydrophilic Spin-Probe
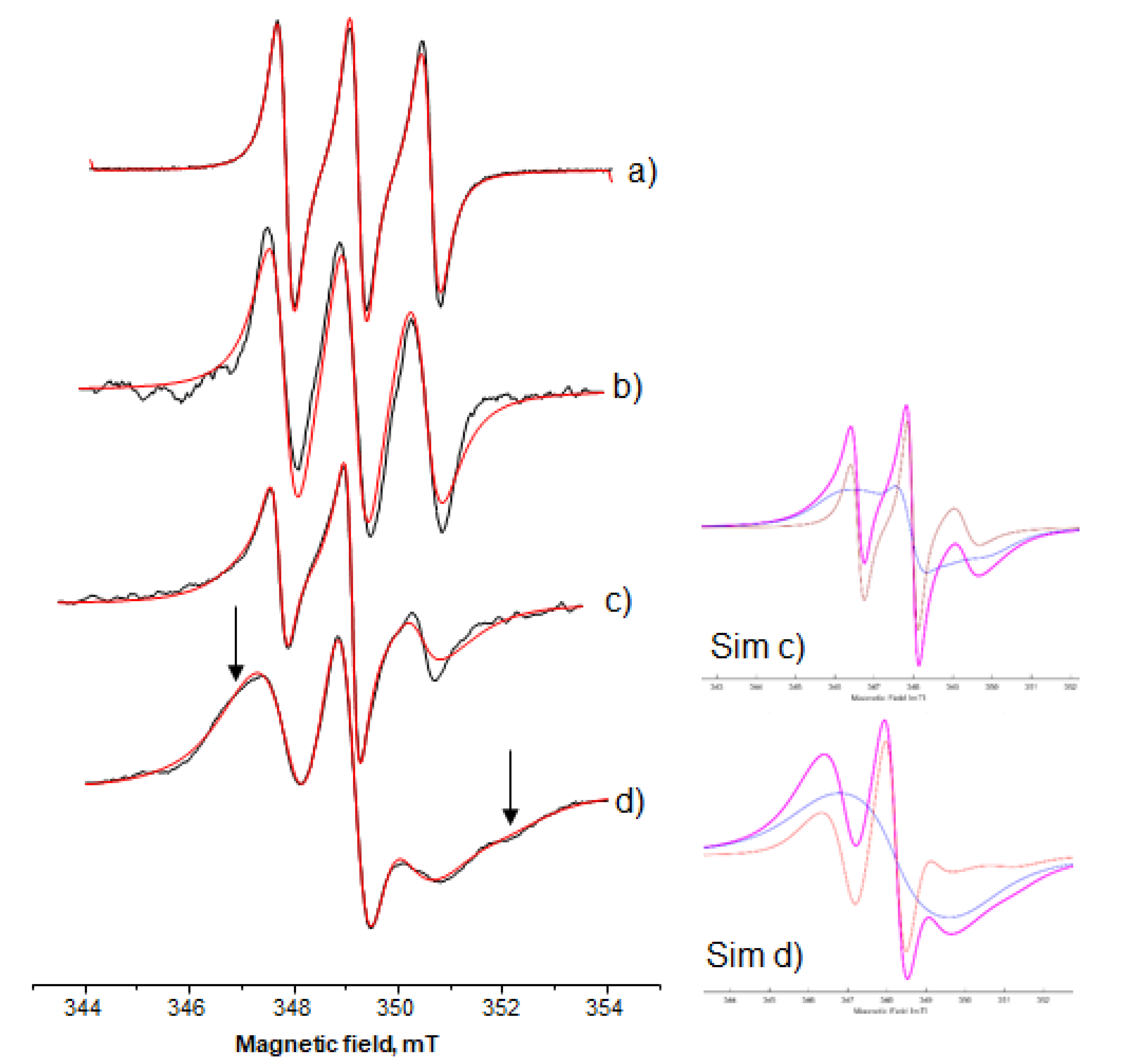
3.3.2. Amphiphilic Spin-Probes
3.3.3. Lipophilic Spin-Probes
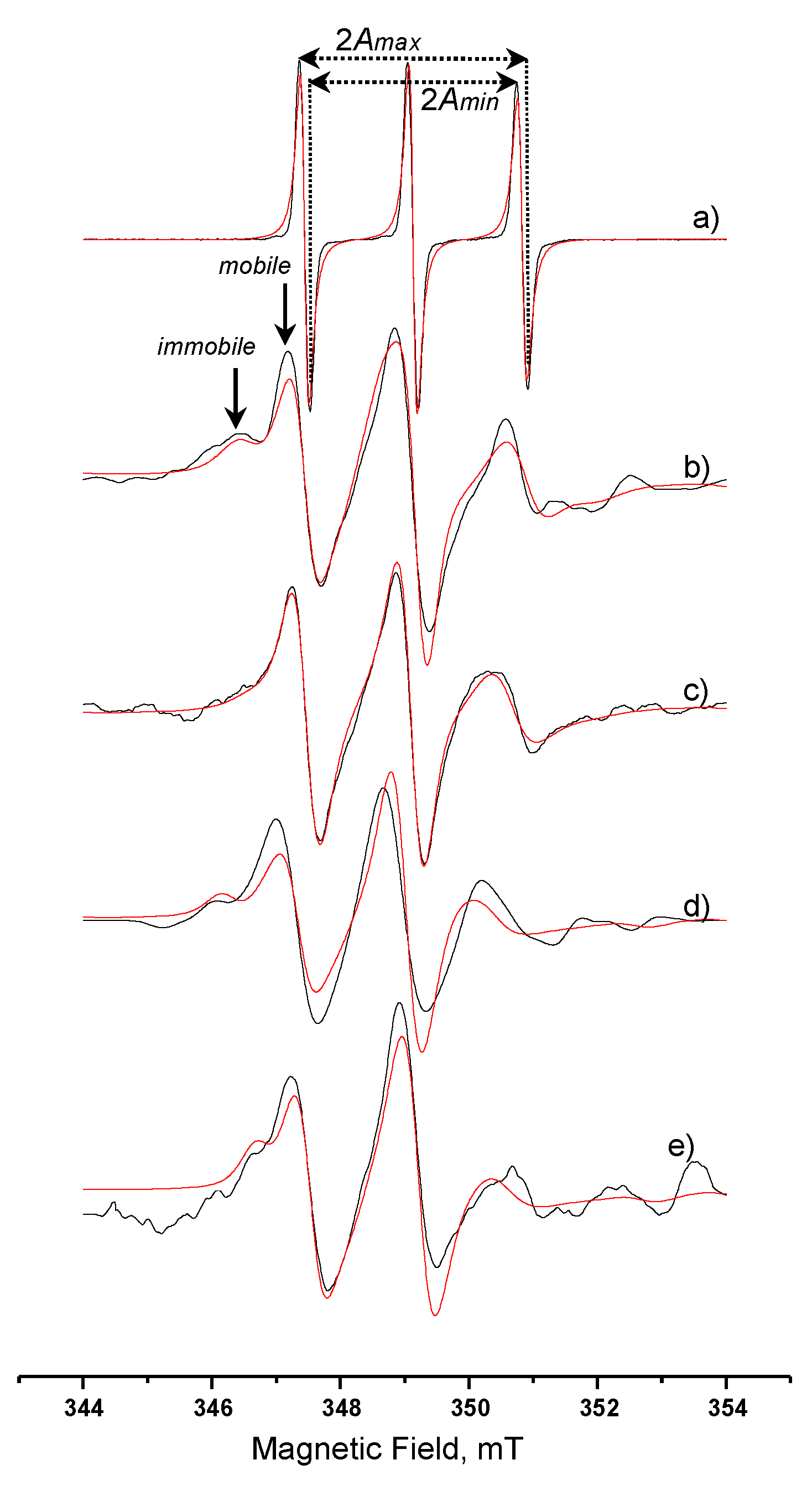
3.3.4. Spin-Labelled Lipase
3.4. Small-Angle X-ray Scattering (SAXS) Measurements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Zoumpanioti, M.; Karavas, E.; Skopelitis, C.; Stamatis, H.; Xenakis, A. Lecithin organogels as model carriers of pharmaceuticals. Progr. Colloid Polym. Sci. 2004, 123, 199–202. [Google Scholar]
- Avramiotis, S.; Papadimitriou, V.; Hatzara, E.; Bekiari, V.; Lianos, P.; Xenakis, A. Lecithin organogels used as bioactive compounds carriers. A microdomain properties investigation. Langmuir 2007, 23, 4438–4447. [Google Scholar] [CrossRef]
- De Souza Paglarini, C.; de Figueiredo Furtado, G.; Biachi, J.P.; Vidal, V.A.S.; Martini, S.; Forte, M.B.S.; Cunha, R.L.; Pollonio, M.A.R. Functional emulsion gels with potential application in meat products. J. Food Eng. 2018, 222, 29–37. [Google Scholar] [CrossRef]
- Huang, Y.; Mei, L.; Chen, X.; Wang, Q. Recent developments in food packaging based on nanomaterials. Nanomaterials 2018, 8, 830. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; An, S.; Won, K.; Kim, H.J.; Lee, S.H. Entrapment of enzymes into cellulose-biopolymer composite hydrogel beads using biocompatible ionic liquid. J. Mol. Catal. B Enzym. 2012, 75, 68–72. [Google Scholar] [CrossRef]
- Vassiliadi, E.; Xenakis, A.; Zoumpanioti, M. Chitosan hydrogels: A new and simple matrix for lipase catalysed biosyntheses. Mol. Catal. 2018, 445, 206–212. [Google Scholar] [CrossRef]
- Laurienzo, P.; Malinconico, M.; Pizzano, R.; Manzo, C.; Piciocchi, N.; Sorrentino, A.; Volpe, M.G. Natural polysaccharide-based gels for dairy food preservation. J. Dairy Sci. 2006, 89, 2856–2864. [Google Scholar] [CrossRef] [Green Version]
- Bilal, M.; Iqbal, H.M.N. Naturally-derived biopolymers: Potential platforms for enzyme immobilization. Int. J. Biol. Macromol. 2019, 130, 462–482. [Google Scholar] [CrossRef]
- Turner, M.B.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Production of bioactive cellulose films reconstituted from ionic liquids. Biomacromolecules 2004, 5, 1379–1384. [Google Scholar] [CrossRef]
- Dalla-Vecchia, R.; Sebrão, D.; Nascimento, M.D.G.; Soldi, V. Carboxymethylcellulose and poly(vinyl alcohol) used as a film support for lipases immobilization. Process Biochem. 2005, 40, 2677–2682. [Google Scholar] [CrossRef]
- Arboleya, J.C.; Wilde, P.J. Competitive adsorption of proteins with methylcellulose and hydroxypropyl methylcellulose. Food Hydrocoll. 2005, 19, 485–491. [Google Scholar] [CrossRef]
- Pérez, O.E.; Sánchez, C.C.; Pilosof, A.M.R.; Rodríguez Patino, J.M. Kinetics of adsorption of whey proteins and hydroxypropyl-methyl-cellulose mixtures at the air-water interface. J. Colloid Interface Sci. 2009, 336, 485–496. [Google Scholar] [CrossRef]
- Rodriguez Patino, J.M.; Pilosof, A.M.R. Protein-polysaccharide interactions at fluid interfaces. Food Hydrocoll. 2011, 25, 1925–1937. [Google Scholar] [CrossRef]
- Pérez, O.E.; Carrera Sánchez, C.; Pilosof, A.M.R.; Rodríguez Patino, J.M. Impact of hydroxypropylmethylcellulose on whey protein concentrate spread film at the air-water interface: Structural and surface dilatational characteristics. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 465, 1–10. [Google Scholar] [CrossRef]
- Fathi, M.; Martín, Á.; McClements, D.J. Nanoencapsulation of food ingredients using carbohydrate based delivery systems. Trends Food Sci. Technol. 2014, 39, 18–39. [Google Scholar] [CrossRef]
- Raghavendra, T.; Sayania, D.; Madamwar, D. Synthesis of the “green apple ester” ethyl valerate in organic solvents by Candida rugosa lipase immobilized in MBGs in organic solvents: Effects of immobilization and reaction parameters. J. Mol. Catal. B Enzym. 2010, 63, 31–38. [Google Scholar] [CrossRef]
- Nagayama, K.; Karaiwa, K.; Doi, T.; Imai, M. Esterification activity and stability of Candida rugosa lipase in AOT microemulsion-based organogels. Biochem. Eng. J. 1998, 2, 121–126. [Google Scholar] [CrossRef]
- Quellet, C.; Eicke, H.-F. Mutual gelation of gelatin and water-in-oil microemulsions. Chimia 1986, 40, 233–238. [Google Scholar]
- Haering, G.; Luisi, P.L. Hydrocarbon Gels from Water-in-Oil Microemulsions. J. Phys. Chem. 1986, 16, 5892–5895. [Google Scholar] [CrossRef]
- Pastou, A.; Stamatis, H.; Xenakis, A. Microemulsion-based organogels containing lipase: Application in the synthesis of esters. Progr. Colloid Polym. Sci. 2000, 115, 192–195. [Google Scholar]
- Delimitsou, C.; Zoumpanioti, M.; Xenakis, A.; Stamatis, H. Activity and stability studies of Mucor miehei lipase immobilized in novel microemulsion-based organogels. Biocatal. Biotransform. 2002, 20, 319–327. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Merianou, E.; Karandreas, T.; Stamatis, H.; Xenakis, A. Esterification of phenolic acids catalyzed by lipases immobilized in organogels. Biotechnol. Lett. 2010, 32, 1457–1462. [Google Scholar] [CrossRef] [Green Version]
- Zoumpanioti, M.; Stamatis, H.; Xenakis, A. Microemulsion-based organogels as matrices for lipase immobilization. Biotechnol. Adv. 2010, 28, 395–406. [Google Scholar] [CrossRef]
- Griffith, O.H.; Jost, P.C. Lipid Spin Labels in Biological Membranes. In Spin Labeling; Academic Press: Cambridge, MA, USA, 1976; Volume 1, pp. 453–523. [Google Scholar]
- Papadimitriou, V.; Sotiroudis, T.G.; Xenakis, A. Olive Oil Microemulsions: Enzymatic Activities and Structural Characteristics. Langmuir 2007, 23, 2071–2077. [Google Scholar] [CrossRef]
- Fanun, M.; Papadimitriou, V.; Xenakis, A. Characterization of Cephalexin Loaded Nonionic Microemulsions. J. Colloid Interface Sci. 2011, 361, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Knauer, B.R.; Napier, J.J. The Nitrogen Hyperfine Splitting Constant of the Nitroxide Functional Group as a Solvent Polarity Parameter. The Relative Importance for a Solvent Polarity Parameter of Its Being a Cybotactic Probe vs. Its Being a Model Process. J. Am. Chem. Soc. 1976, 98, 4395–4400. [Google Scholar] [CrossRef]
- Marsh, D. Spin-Label EPR for Determining Polarity and Proticity in Biomolecular Assemblies: Transmembrane Profiles. Appl. Magn. Reson. 2010, 37, 435–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Hemminga, M.A.; Berliner, L. ESR Spectroscopy in Membrane Biophysics; Biological Magnetic Resonance; Springer US: Boston, MA, USA, 2007; Volume 27. [Google Scholar]
- Etienne, E.; Le Breton, N.; Martinho, M.; Mileo, E.; Belle, V. SimLabel: A graphical user interface to simulate continuous wave EPR spectra from site-directed spin labeling experiments. Magn. Reson. Chem. 2017, 55, 714–719. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Karali, M.; Xenakis, A.; Stamatis, H. Lipase biocatalytic processes in surfactant free microemulsion-like ternary systems and related organogels. Enzym. Microb. Technol. 2006, 39, 531–539. [Google Scholar] [CrossRef]
- Stamatis, H.; Xenakis, A. Biocatalysis using microemulsion-based polymer gels containing lipase. J. Mol. Catal. B Enzym. 1999, 6, 399–406. [Google Scholar] [CrossRef]
- Itabaiana, I.; Gonçalves, K.M.; Zoumpanioti, M.; Leal, I.C.R.; Miranda, L.S.M.E.; Xenakis, A.; De Souza, R.O.M.A. Microemulsion-based organogels as an efficient support for lipase-catalyzed reactions under continuous-flow conditions. Org. Process Res. Dev. 2014, 18, 1372–1376. [Google Scholar] [CrossRef]
- Blattner, C.; Zoumpanioti, M.; Kröner, J.; Schmeer, G.; Xenakis, A.; Kunz, W. Biocatalysis using lipase encapsulated in microemulsion-based organogels in supercritical carbon dioxide. J. Supercrit. Fluids 2006, 36, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Dandavate, V.; Madamwar, D. Reusability of surfactant-coated Candida rugosa lipase immobilized in gelatin microemulsion-based organogels for ethyl isovalerate synthesis. J. Microbiol. Biotechnol. 2008, 18, 735–741. [Google Scholar]
- Zhang, W.W.; Wang, N.; Zhang, L.; Wu, W.X.; Hu, C.L.; Yu, X.Q. Effects of additives on lipase immobilization in microemulsion-based organogels. Appl. Biochem. Biotechnol. 2014, 172, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Moulik, S.P.; Paul, B.K. Structure, dynamics and transport properties of micro emulsions. Adv. Colloid Interface Sci. 1998, 78, 99–195. [Google Scholar] [CrossRef]
- González-Blanco, C.; Rodríguez, L.J.; Velázquez, M.M. Effect of the solvent on the water properties of water/oil microemulsions. J. Colloid Interface Sci. 1999, 211, 380–866. [Google Scholar] [CrossRef]
- Haering, G.; Luisi, P.L.; Hauser, H. Characterization by electron spin resonance of reversed micelles consisting of the ternary system AOT-isooctane-water. J. Phys. Chem. 1988, 92, 3574–3581. [Google Scholar] [CrossRef]
- Schreier, S.; Polnaszek, C.F.; Smith, I.C.P. Spin labels in membranes problems in practice. BBA Rev. Biomembr. 1978, 515, 395–436. [Google Scholar] [CrossRef]
- Caldararu, H.; Timmins, G.S.; Gilbert, B.C. The Structure of Gelatin-Water/Oil Microemulsion Sols and Gels. An EPR Spin-Probe and Spin-Labelling Study. Phys. Chem. Chem. Phys. 1999, 1, 5689–5695. [Google Scholar] [CrossRef]
- Alberghina, L.; Lotti, M. [14] Cloning, sequencing, and expression of Candida rugosa lipases. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 284, pp. 246–260. [Google Scholar]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Bouthillier, F.; Smith, P.; Harrison, D.; Rubin, B.; Cygler, M. Insights into interfacial activation from an open structure of Candida rugosa lipase. J. Biol. Chem. 1993, 268, 12843–12847. [Google Scholar]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Yang, L.; Vincent, C.; Lei, X.; Ottaviani, M.F.; Ananthapadmanabhan, K.P. A comparison between interactions of triglyceride oil and mineral oil with proteins and their ability to reduce cleanser surfactant-induced irritation. Int. J. Cosmet. Sci. 2015, 37, 371–378. [Google Scholar] [CrossRef]
- Bösecke, P.; Diat, O. Small-angle X-ray scattering at the ESRF high-brilliance beamline. J. Appl. Crystallogr. 1997, 30, 867–871. [Google Scholar] [CrossRef] [Green Version]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Crystallogr. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Hammouda, B.; Ho, D.L.; Kline, S. Insight into clustering in poly(ethylene oxide) solutions. Macromolecules 2004, 37, 6932–6937. [Google Scholar] [CrossRef]
- Bhattacharjee, S.M.; Giacometti, A.; Maritan, A. Flory theory for polymers. J. Phys. Condens. Matter 2013, 25, 503101. [Google Scholar] [CrossRef]
- Atkinson, P.J.; Heenan, R.K.; Grimson, M.J.; Howe, A.M.; Robinson, B.H. Structure of microemulsion-based organo-gels. Progr. Colloid Polym. Sci. 1990, 23, 1807–1809. [Google Scholar]
- Petit, C.; Zemb, T.; Pileni, M.P. Structural Study of Microemulsion-Based Gels at the Saturation Point. Langmuir 1991, 7, 223–231. [Google Scholar] [CrossRef]
- Hauser, H.; Haering, G.; Pande, A.; Luisi, P.L. Interaction of water with sodium bis(2-ethyl-1-hexyl) sulfosuccinate in reversed micelles. J. Phys. Chem. 1989, 93, 7869–7876. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MBG | HPMC | H2O | µE |
|---|---|---|---|
| % w/w | |||
| System A | 18 | 71 | 11 |
| System B | 28 | 55 | 17 |
| System C | 44 | 43 | 13 |
| Hydrophilic Probe Hydroxy-TEMPO | ||
|---|---|---|
| System | τR, ns | AN, 10−4 T |
| Water | 0.03 ± 0.00 | 17.49 ± 0.04 |
| System A* | 0.07 ± 0.01 | 17.29 ± 0.11 |
| System B* | 0.15 ± 0.01 | 17.25 ± 0.03 |
| System C* | 0.40 ± 0.01 | 17.07 ± 0.04 |
| System C** | 0.40 ± 0.06 | 17.07 ± 0.03 |
| (AOT) = 0.1 Μ µE wo = 15 | 0.13 ± 0.01 | 16.25 ± 0.02 |
| (AOT) = 0.2 Μ, µE wo = 7.5 | 0.21 ± 0.01 | 15.84 ± 0.01 |
| Probe incorporated in the HPMC-based MBGs via the AOT microemulsion | ||
| System A | 0.06 ± 0.01 | 17.31 ± 0.01 |
| System B | 0.14 ± 0.01 | 17.21 ± 0.02 |
| System C | 0.40 ± 0.01 | 17.14 ± 0.01 |
| System C† | 0.40 ± 0.06 | 17.11 ± 0.05 |
| Probe incorporated in the HPMC-based MBG via the HPMC/water mixture | ||
| System A | 0.08 ± 0.01 | 17.35 ± 0.04 |
| System B | 0.15 ± 0.02 | 17.22 ± 0.03 |
| System C | 0.38 ± 0.03 | 17.11 ± 0.02 |
| Amphiphilic Probes | ||||||
|---|---|---|---|---|---|---|
| System | 5 DSA | 16 DSA | ||||
| τR, ns | S | AN, 10−4 T | τR, ns | S | AN, 10−4 T | |
| µE wo = 15 | 3.45 ± 0.10 | 0.29 ± 0.02 | 15.06 ± 0.03 | 0.07 ± 0.01 | 0.06 ± 0.01 | 14.39 ± 0.08 |
| μE wo = 7.5 | 2.30 ± 0.08 | 0.14 ± 0.01 | 14.01 ± 0.05 | 0.09 ± 0.02 | 0.09 ± 0.01 | 14.26 ± 0.06 |
| System A | 6.02 ± 0.47 | 0.49 ± 0.03 | 15.08 ± 0.12 | 0.95 ± 0.01 | 0.05 ± 0.01 | 14.97 ± 0.02 |
| System B | 6.57 ± 0.05 | 0.55 ± 0.01 | 15.37 ± 0.05 | 1.39 ± 0.05 | 0.07 ± 0.01 | 14.85 ± 0.01 |
| System C | 6.96 ± 0.14 | 0.61 ± 0.03 | 15.69 ± 0.17 | 3.31 ± 0.04 | 0.17 ± 0.01 | 14.21 ± 0.14 |
| Spin-Labelled Lipase from Candida rugosa—Iodoacetamide Tempo | |||||
|---|---|---|---|---|---|
| τR, ns | S | AN, 10−4T | |||
| Free spin label in water | 0.06 ± 0.01 | 0.01 ± 0.01 | 17.48 ± 0.09 | ||
| Spin-labelled Candida rugosa in aqueous solution | |||||
| Two components | Immobile | – | mobile | ||
| 10.83 ns (44%) | – | 1.66 ns (56%) | 0.41 ± 0.03 | 17.23 ± 0.14 | |
| Spin-labelled Candida rugosa in AOT microemulsion, wo = 15 | |||||
| Two components | Immobile | – | mobile | ||
| 8.92 ns (20%) | – | 1.78 ns (80%) | 0.14 ± 0.06 | 16.00 ± 0.03 | |
| Spin-labelled Candida rugosa in HPMC-based MBGs | |||||
| Two components | Immobile | – | mobile | ||
| System A | 19.85 ns (45%) | – | 3.16 ns (55%) | 0.35 ± 0.04 | 16.48 ± 0.13 |
| System B | 21.87 ns (43%) | – | 3.31 ns (57%) | 0.39 ± 0.05 | 16.73 ± 0.08 |
| Parameters | System C* | System C** | System C‡ | System C† |
|---|---|---|---|---|
| Correlation length (Å) | 44.6 | 44.1 | 66.9 | 71.6 |
| Porod exponent (n) | 4.5 | 4.5 | 4.8 | 5.1 |
| Lorentzian exponent (m) | 3.0 | 3.2 | 2.8 | 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassiliadi, E.; Mitsou, E.; Avramiotis, S.; Chochos, C.L.; Pirolt, F.; Medebach, M.; Glatter, O.; Xenakis, A.; Zoumpanioti, M. Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations. Nanomaterials 2020, 10, 2204. https://doi.org/10.3390/nano10112204
Vassiliadi E, Mitsou E, Avramiotis S, Chochos CL, Pirolt F, Medebach M, Glatter O, Xenakis A, Zoumpanioti M. Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations. Nanomaterials. 2020; 10(11):2204. https://doi.org/10.3390/nano10112204
Chicago/Turabian StyleVassiliadi, Evdokia, Evgenia Mitsou, Spyridon Avramiotis, Christos L. Chochos, Franz Pirolt, Martin Medebach, Otto Glatter, Aristotelis Xenakis, and Maria Zoumpanioti. 2020. "Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations" Nanomaterials 10, no. 11: 2204. https://doi.org/10.3390/nano10112204