
The Transcriptome of SH-SY5Y at Single-Cell Resolution: A CITE-Seq Data Analysis Workflow

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Hashing and Library Preparation for 10X Genomics Sequencing
2.3. Data Processing and Analysis
3. Results
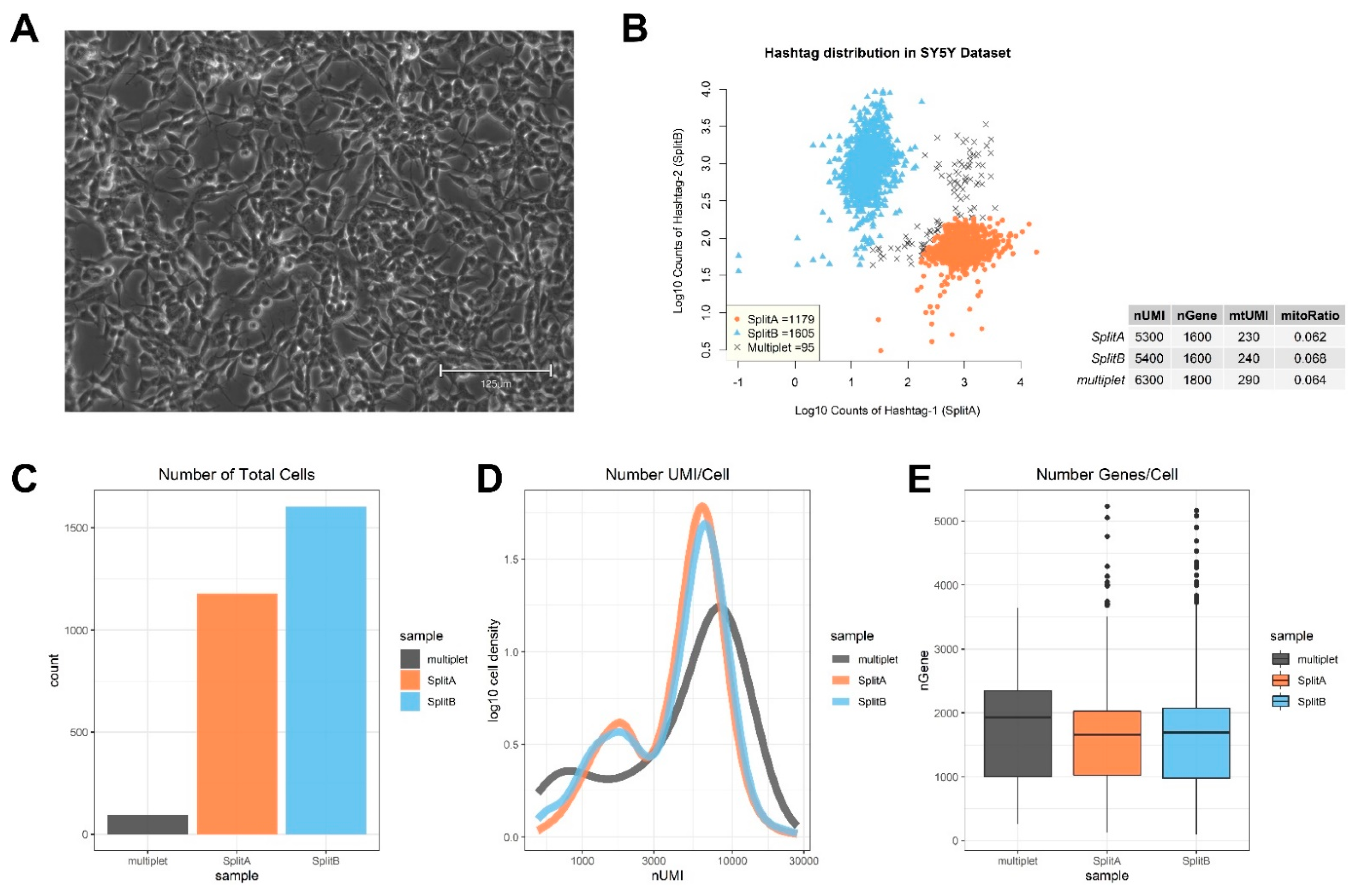
3.1. Quality Check and Cell Selection
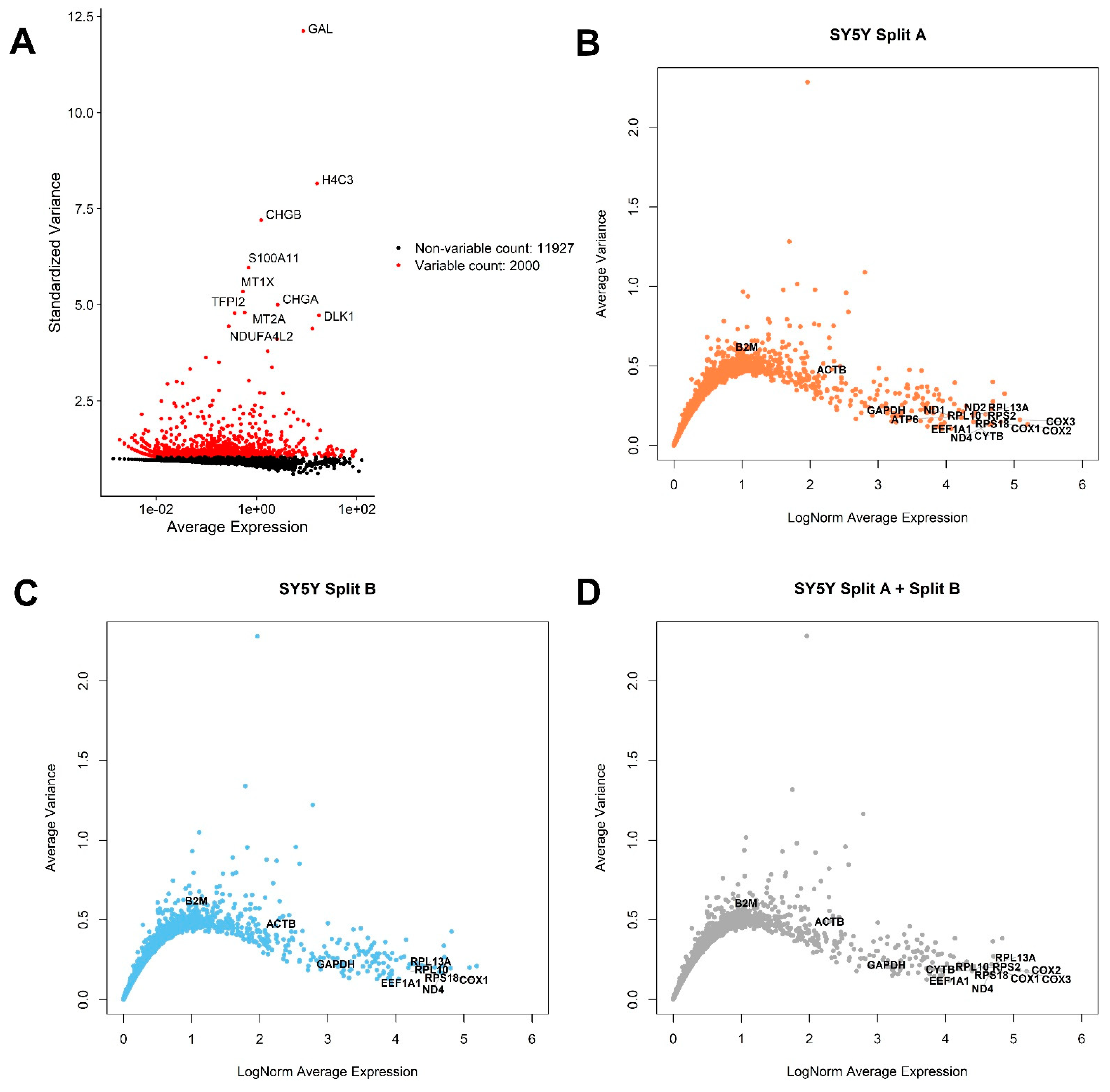
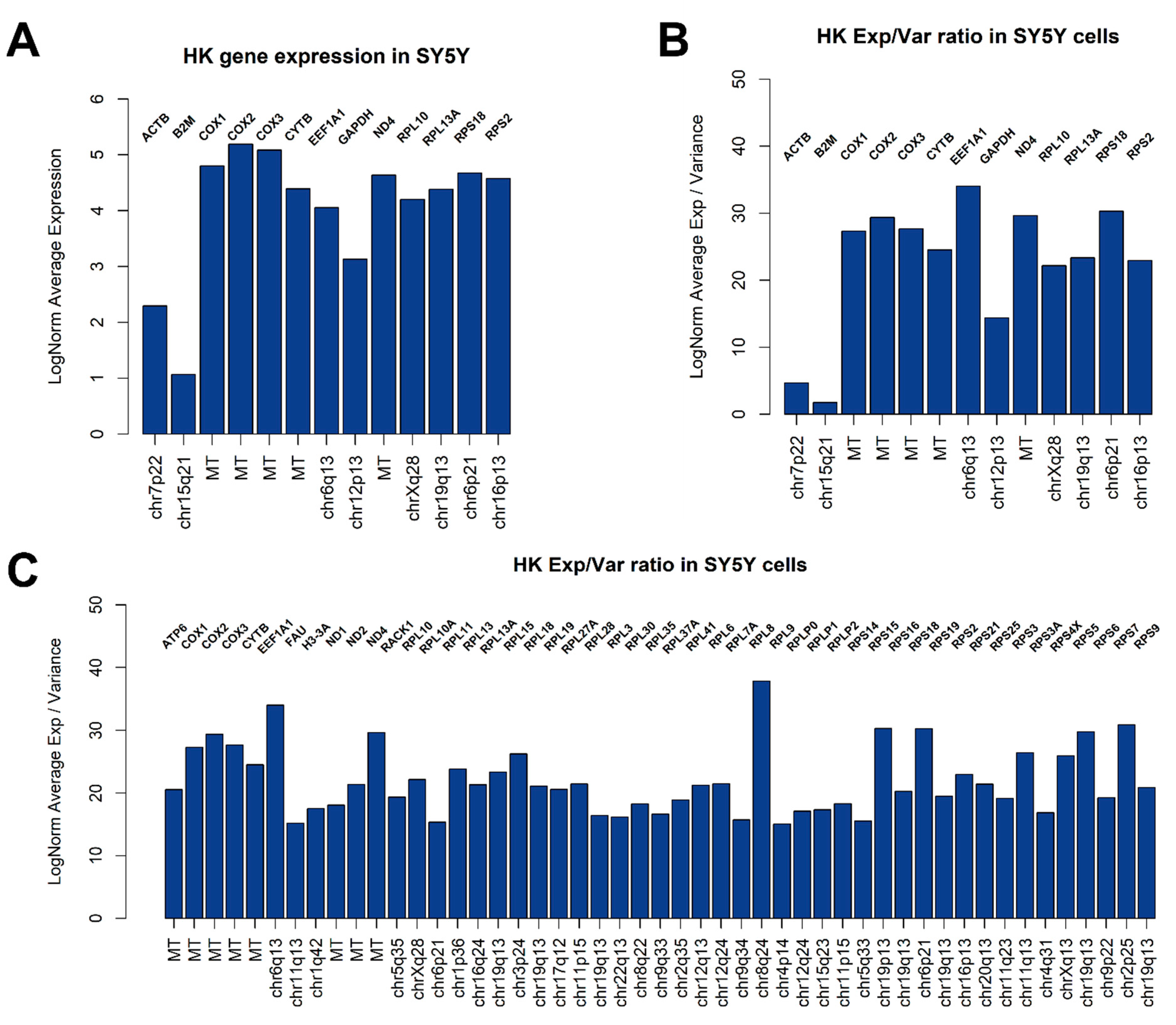
3.2. Global Gene Expression Analysis
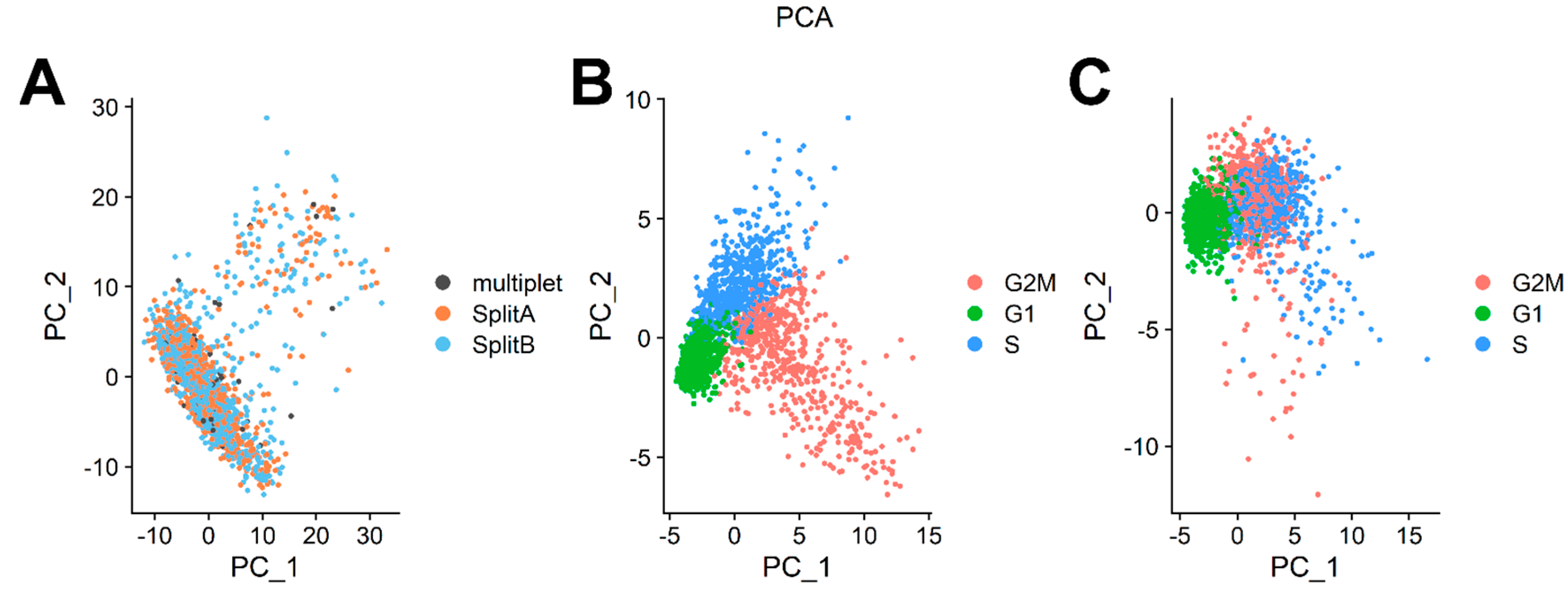
3.3. Dataset Dimensionality Reduction and Cell-Cycle Regression
3.4. Differential Expression and Cluster Analysis
3.5. Dissection of Cell Heterogeneity by Factorial Single-Cell Latent Variable Model Method
3.6. Use of Single-Cell Data for Co-Expression Network Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Antibody-Derived Tag |
| CITE-seq | Cellular Indexing of Transcriptomes and Epitopes by Sequencing |
| CSV | Comma-Separated Value |
| GRN | Gene Regulatory Network |
| HK | Housekeeping Gene |
| HTO | Hashtag Oligonucleotide |
| PCA | Principal Component Analysis |
| QC | Quality Check |
| scRNA-seq | Single-cell RNA-sequencing |
| TF | Transcription Factor |
| UMAP | Uniform Manifold Approximation and Projection |
| UMI | Unique Molecular Identifier |
References
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Angerer, P.; Simon, L.; Tritschler, S.; Wolf, F.A.; Fischer, D.; Theis, F.J. Single Cells Make Big Data: New Challenges and Opportunities in Transcriptomics. Curr. Opin. Syst. Biol. 2017, 4, 85–91. [Google Scholar] [CrossRef]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef] [PubMed]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e16. [Google Scholar] [CrossRef]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Häring, M.; Dyachuk, V.; et al. Spatiotemporal Structure of Cell Fate Decisions in Murine Neural Crest. Science 2019, 364. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Chung, W.; Lee, H.-O.; Jeong, D.E.; Jo, A.; Lim, J.E.; Hong, J.H.; Nam, D.-H.; Jeong, B.C.; Park, S.H.; et al. Single-Cell RNA Sequencing Reveals the Tumor Microenvironment and Facilitates Strategic Choices to Circumvent Treatment Failure in a Chemorefractory Bladder Cancer Patient. Genome Med. 2020, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Curr. Protoc. Mol. Biol. 2013, 103, 4.19.1–4.19.8. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Pervolarakis, N.; Nee, K.; Kessenbrock, K. Experimental Considerations for Single-Cell RNA Sequencing Approaches. Front. Cell Dev. Biol. 2018, 6, 108. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M.; Smibert, P.; Satija, R. Cell Hashing with Barcoded Antibodies Enables Multiplexing and Doublet Detection for Single Cell Genomics. Genome Biol. 2018, 19, 224. [Google Scholar] [CrossRef] [Green Version]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and Growth, Tumorigenicity, and Cytogenetics of Human Neuroblastoma Cells in Continuous Culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar] [PubMed]
- Mercatelli, D.; Balboni, N.; Palma, A.; Aleo, E.; Sanna, P.P.; Perini, G.; Giorgi, F.M. Single-Cell Gene Network Analysis and Transcriptional Landscape of MYCN-Amplified Neuroblastoma Cell Lines. Biomolecules 2021, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Harenza, J.L.; Diamond, M.A.; Adams, R.N.; Song, M.M.; Davidson, H.L.; Hart, L.S.; Dent, M.H.; Fortina, P.; Reynolds, C.P.; Maris, J.M. Transcriptomic Profiling of 39 Commonly-Used Neuroblastoma Cell Lines. Sci. Data 2017, 4, 170033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-Cell RNA-Seq Reveals Changes in Cell Cycle and Differentiation Programs upon Aging of Hematopoietic Stem Cells. Genome Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The Dynamics and Regulators of Cell Fate Decisions Are Revealed by Pseudotemporal Ordering of Single Cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Buettner, F.; Pratanwanich, N.; McCarthy, D.J.; Marioni, J.C.; Stegle, O. F-ScLVM: Scalable and Versatile Factor Analysis for Single-Cell RNA-Seq. Genome Biol. 2017, 18, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. WikiPathways: Connecting Communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. Corto: A Lightweight R Package for Gene Network Inference and Master Regulator Analysis. Bioinformatics 2020, 36, 3916–3917. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- DePasquale, E.A.K.; Schnell, D.J.; Van Camp, P.-J.; Valiente-Alandí, Í.; Blaxall, B.C.; Grimes, H.L.; Singh, H.; Salomonis, N. DoubletDecon: Deconvoluting Doublets from Single-Cell RNA-Sequencing Data. Cell Rep. 2019, 29, 1718–1727.e8. [Google Scholar] [CrossRef] [PubMed]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, K.M.; Gomez, L.A.; Rios, C.; Garbayo, E.; Raval, A.P.; Perez-Pinzon, M.A.; Schiller, P.C. EF1alpha and RPL13a Represent Normalization Genes Suitable for RT-QPCR Analysis of Bone Marrow Derived Mesenchymal Stem Cells. BMC Mol. Biol. 2010, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Romani, C.; Calza, S.; Todeschini, P.; Tassi, R.A.; Zanotti, L.; Bandiera, E.; Sartori, E.; Pecorelli, S.; Ravaggi, A.; Santin, A.D.; et al. Identification of Optimal Reference Genes for Gene Expression Normalization in a Wide Cohort of Endometrioid Endometrial Carcinoma Tissues. PLoS ONE 2014, 9, e113781. [Google Scholar] [CrossRef] [Green Version]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef]
- Gardini, E.; Giorgi, F.M.; Decherchi, S.; Cavalli, A. Spathial: An R Package for the Evolutionary Analysis of Biological Data. Bioinformatics 2020, 36, 4664–4667. [Google Scholar] [CrossRef]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene Regulatory Network Inference Resources: A Practical Overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef]
- Mercatelli, D.; Bortolotti, M.; Giorgi, F.M. Transcriptional Network Inference and Master Regulator Analysis of the Response to Ribosome-Inactivating Proteins in Leukemia Cells. Toxicology 2020, 441, 152531. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Alvarez, M.J.; Talos, F.; Dhruv, H.; Rieckhof, G.E.; Iyer, A.; Diefes, K.L.; Aldape, K.; Berens, M.; Shen, M.M.; et al. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell 2014, 159, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Saito, M.; Bisikirska, B.C.; Alvarez, M.J.; Lim, W.K.; Rajbhandari, P.; Shen, Q.; Nemenman, I.; Basso, K.; Margolin, A.A.; et al. Genome-Wide Identification of Post-Translational Modulators of Transcription Factor Activity in Human B Cells. Nat. Biotechnol. 2009, 27, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable Transcript Quantification by Real-Time Reverse Transcriptase-Polymerase Chain Reaction in Primary Neuroblastoma Using Normalization to Averaged Expression Levels of the Control Genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Hoerndli, F.J.; Toigo, M.; Schild, A.; Götz, J.; Day, P.J. Reference Genes Identified in SH-SY5Y Cells Using Custom-Made Gene Arrays with Validation by Quantitative Polymerase Chain Reaction. Anal. Biochem. 2004, 335, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Jögi, A.; Vallon-Christersson, J.; Holmquist, L.; Axelson, H.; Borg, A.; Påhlman, S. Human Neuroblastoma Cells Exposed to Hypoxia: Induction of Genes Associated with Growth, Survival, and Aggressive Behavior. Exp. Cell Res. 2004, 295, 469–487. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Aiken, J.; Buscaglia, G.; Bates, E.A.; Moore, J.K. The α-Tubulin Gene TUBA1A in Brain Development: A Key Ingredient in the Neuronal Isotype Blend. J. Dev. Biol. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Limpt, V.A.E.; Chan, A.J.; Van Sluis, P.G.; Caron, H.N.; Van Noesel, C.J.M.; Versteeg, R. High Delta-like 1 Expression in a Subset of Neuroblastoma Cell Lines Corresponds to a Differentiated Chromaffin Cell Type. Int. J. Cancer 2003, 105, 61–69. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 Differentially Modulate Cell Survival by Interacting with Distinct RAGE (Receptor for Advanced Glycation End Products) Immunoglobulin Domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Kamal, A.; Burrows, F.J.; Evers, B.M.; Chung, D.H. Inhibition of Neuroblastoma Xenograft Growth by Hsp90 Inhibitors. Anticancer Res. 2006, 26, 1903–1908. [Google Scholar]
- Sasaki, H.; Sato, Y.; Kondo, S.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y. Expression of the Prothymosin Alpha MRNA Correlated with That of N-Myc in Neuroblastoma. Cancer Lett. 2001, 168, 191–195. [Google Scholar] [CrossRef]
- Michniewicz, F.; Saletta, F.; Rouaen, J.; Hewavisenti, R.; Mercatelli, D.; Cirillo, G.; Giorgi, F.M.; Trahair, T.; Ziegler, D.; Vittorio, O. Copper: An Intracellular Achilles Heel Allowing the Targeting of Epigenetics, Kinase Pathways and Cell Metabolism in Cancer Therapeutics. ChemMedChem 2021. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Shin, J.W.; Rood, J.E.; Hupalowska, A.; Human Cell Atlas Standards and Technology Working Group; Regev, A.; Heyn, H. Building a High-Quality Human Cell Atlas. Nat. Biotechnol. 2021, 39, 149–153. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, J.; Jia, P.; Zhao, Z. ScRNASeqDB: A Database for RNA-Seq Based Gene Expression Profiles in Human Single Cells. Genes 2017, 8, 368. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a Human Cell Landscape at Single-Cell Level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.A.; Papas, B.N.; Trotter, K.W.; Archer, T.K. Single-Cell RNA Sequencing Reveals a Heterogeneous Response to Glucocorticoids in Breast Cancer Cells. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Osorio, D.; Yu, X.; Yu, P.; Serpedin, E.; Cai, J.J. Single-Cell RNA Sequencing of a European and an African Lymphoblastoid Cell Line. Sci. Data 2019, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Fuhr, V.; Vafadarnejad, E.; Dietrich, O.; Arampatzi, P.; Riedel, A.; Saliba, A.-E.; Rosenwald, A.; Rauert-Wunderlich, H. Time-Resolved ScRNA-Seq Tracks the Adaptation of a Sensitive MCL Cell Line to Ibrutinib Treatment. Int. J. Mol. Sci. 2021, 22, 2276. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercatelli, D.; Balboni, N.; Giorgio, F.D.; Aleo, E.; Garone, C.; Giorgi, F.M. The Transcriptome of SH-SY5Y at Single-Cell Resolution: A CITE-Seq Data Analysis Workflow. Methods Protoc. 2021, 4, 28. https://doi.org/10.3390/mps4020028
Mercatelli D, Balboni N, Giorgio FD, Aleo E, Garone C, Giorgi FM. The Transcriptome of SH-SY5Y at Single-Cell Resolution: A CITE-Seq Data Analysis Workflow. Methods and Protocols. 2021; 4(2):28. https://doi.org/10.3390/mps4020028
Chicago/Turabian StyleMercatelli, Daniele, Nicola Balboni, Francesca De Giorgio, Emanuela Aleo, Caterina Garone, and Federico Manuel Giorgi. 2021. "The Transcriptome of SH-SY5Y at Single-Cell Resolution: A CITE-Seq Data Analysis Workflow" Methods and Protocols 4, no. 2: 28. https://doi.org/10.3390/mps4020028