

Nickel(II)-Catalyzed Formal [3+2] Cycloadditions between Indoles and Donor–Acceptor Cyclopropanes
 , , ,
, , ,  , , , , , and
, , , , , and
Abstract
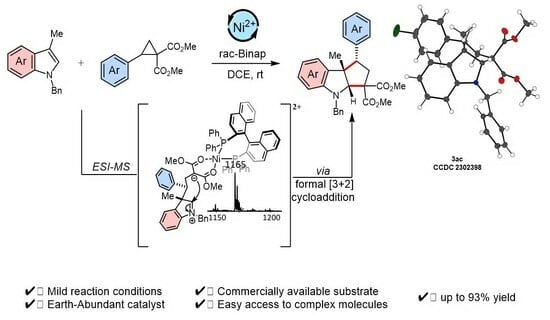
:
1. Introduction
2. Results and Discussion
 The results listed in Table 2 suggest that the rigidity and bite angle of bidentate phosphines might be crucial factors in the system’s reactivity. Ligands such as dppe (flexible ligand) and dppBz (rigid ligand) [69], with approximately 90° angles, exhibited a reduced yield and a significant increase in reaction time compared to the BINAP version, which features a greater bite angle (approximately 93°) than the previously mentioned ligands [70]. Ligands with larger angles, such as dppf (99°) [71], also fail to provide ideal conditions for this [3+2] cycloaddition. We primarily evaluated a possible enantioselective version of the cycloaddition using chiral bisphosphines such as (R)-BINAP, (R)-DM-BINAP, and (S)-DTBM-DEGPHOS. Among these, only the BINAP derivatives effectively catalyzed the reaction, yielding up to 81%, but not exceeding 40% ee in any case (see details in Table S5 in the Supplementary Materials). Further studies to develop a highly enantioselective version are under development.
The results listed in Table 2 suggest that the rigidity and bite angle of bidentate phosphines might be crucial factors in the system’s reactivity. Ligands such as dppe (flexible ligand) and dppBz (rigid ligand) [69], with approximately 90° angles, exhibited a reduced yield and a significant increase in reaction time compared to the BINAP version, which features a greater bite angle (approximately 93°) than the previously mentioned ligands [70]. Ligands with larger angles, such as dppf (99°) [71], also fail to provide ideal conditions for this [3+2] cycloaddition. We primarily evaluated a possible enantioselective version of the cycloaddition using chiral bisphosphines such as (R)-BINAP, (R)-DM-BINAP, and (S)-DTBM-DEGPHOS. Among these, only the BINAP derivatives effectively catalyzed the reaction, yielding up to 81%, but not exceeding 40% ee in any case (see details in Table S5 in the Supplementary Materials). Further studies to develop a highly enantioselective version are under development.3. Experimental Section
3.1. General Section
3.2. General Procedure for [3+2] Cycloaddition (Exemplified for the Synthesis of Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-1-(4-methoxyphenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3aa))
3.2.1. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-8b-methyl-1-phenyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ab)
3.2.2. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-1-(4-chlorophenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ac)

3.2.3. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-1-(4-bromophenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ad)
3.2.4. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-1-(4-fluorophenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ae)
3.2.5. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-1-(4-nitrophenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ag)
3.2.6. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-8b-methyl-1-((E)-styryl)-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ai)
3.2.7. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-7-methoxy-1-(4-methoxyphenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ca)
3.2.8. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-7-bromo-1-(4-methoxyphenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3da)
3.2.9. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-7-chloro-1-(4-methoxyphenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3ea)
3.2.10. Dimethyl (1S*,3aR*,8bS*)-4-Benzyl-6-chloro-1-(4-methoxyphenyl)-8b-methyl-1,3a,4,8b-tetrahydrocyclopenta[b]indole-3,3(2H)-dicarboxylate (3fa)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaushik, N.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.; Verma, A.; Choi, E. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Gavia, D.J.; Tang, Y. Biosynthesis of Fungal Indole Alkaloids. Nat. Prod. Rep. 2014, 31, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed]
- Ishikura, M.; Abe, T.; Choshi, T.; Hibino, S. Simple Indole Alkaloids and Those with a Nonrearranged Monoterpenoid Unit. Nat. Prod. Rep. 2015, 32, 1389–1471. [Google Scholar] [CrossRef] [PubMed]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 2nd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2002. [Google Scholar] [CrossRef]
- Fattorusso, E.; Taglialetela-Scafati, O. Modern Alkaloids: Structure, Isolation, Synthesis and Biology; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Nakazawa, J.; Yajima, J.; Usui, T.; Ueki, M.; Takatsuki, A.; Imoto, M.; Toyoshima, Y.Y.; Osada, H. A Novel Action of Terpendole E on the Motor Activity of Mitotic Kinesin Eg5. Chem. Biol. 2003, 10, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Nagumo, Y.; Motoyama, T.; Hayashi, T.; Hirota, H.; Aono, H.; Kawatani, M.; Osada, H.; Usui, T. Structure-Activity Relationships of Terpendole E and Its Natural Derivatives. ChemistrySelect 2017, 2, 1533–1536. [Google Scholar] [CrossRef]
- Tiong, S.H.; Looi, C.Y.; Hazni, H.; Arya, A.; Paydar, M.; Wong, W.F.; Cheah, S.C.; Mustafa, M.R.; Awang, K. Antidiabetic and Antioxidant Properties of Alkaloids from Catharanthus roseus (L.) G. Don. Molecules 2013, 18, 9770–9784. [Google Scholar] [CrossRef] [PubMed]
- Omar, F.; Tareq, A.M.; Alqahtani, A.M.; Dhama, K.; Sayeed, M.A.; Emran, T.B.; Simal-Gandara, J. Plant-Based Indole Alkaloids: A Comprehensive Overview from a Pharmacological Perspective. Molecules 2021, 26, 2297. [Google Scholar] [CrossRef] [PubMed]
- Tillequin, F.; Koch, M.; Bert, M.; Sevenet, T. Plantes de Nouvelle Calédonie. LV. Isoborrévérine et Borrévérine, Alcaloïdes Bis-Indoliques de Flindersia Fournieri. J. Nat. Prod. 1979, 42, 92–95. [Google Scholar] [CrossRef]
- Maynart, G.; Pousset, J.L.; Mboup, S.; Denis, F. Antibacterial Effect of Borreverine, an Alkaloid Isolated from Borreria Verticillata (Rubiaceae). Comptes Rendus Seances Soc. Biol. Fil. 1980, 174, 925–928. [Google Scholar]
- Malona, J.A.; Colbourne, J.M.; Frontier, A.J. A General Method for the Catalytic Nazarov Cyclization of Heteroaromatic Compounds. Org. Lett. 2006, 8, 5661–5664. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Leonori, D. The First Calcium-Catalysed Nazarov Cyclisation. Chem. Commun. 2014, 50, 15171–15174. [Google Scholar] [CrossRef] [PubMed]
- Genesan, A.; Hoothcock, C.H. A Stereochemical Test of the Mechanism of Electrophilic Substitution in 3-Substituted Indoles. Tetrahedron Lett. 1993, 34, 439–440. [Google Scholar] [CrossRef]
- Kawahara, M.; Nishida, A.; Nakagawa, M. An Efficient Synthesis of Optically Active Physostigmine from Tryptophan via Alkylative Cyclization. Org. Lett. 2000, 2, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Petrini, M. Simplified Synthesis of 3-(1-Arylsulfonylalkyl) Indoles and Their Reaction with Reformatsky Reagents. J. Org. Chem. 2007, 72, 1863–1866. [Google Scholar] [CrossRef] [PubMed]
- Boal, B.W.; Schammel, A.W.; Garg, N.K. An Interrupted Fischer Indolization Approach toward Fused Indoline-Containing Natural Products. Org. Lett. 2009, 11, 3458–3461. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Webber, M.J.; List, B. The Catalytic Asymmetric Fischer Indolization. J. Am. Chem. Soc. 2011, 133, 18534–18537. [Google Scholar] [CrossRef]
- Ghosh, B.; Balhara, R.; Jindal, G.; Mukherjee, S. Catalytic Enantioselective Desymmetrizing Fischer Indolization through Dynamic Kinetic Resolution. Angew. Chem. Int. Ed. 2021, 60, 9086–9092. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.F.; Kim, S.G.; Sinz, C.J.; Xiao, W.J.; MacMillan, D.W.C. Enantioselective Organocatalytic Construction of Pyrroloindolines by a Cascade Addition-Cyclization Strategy: Synthesis of (-)-Flustramine B. Proc. Natl. Acad. Sci. USA 2004, 101, 5482–5487. [Google Scholar] [CrossRef]
- Xu, B.; Guo, Z.L.; Jin, W.Y.; Wang, Z.P.; Peng, Y.G.; Guo, Q.X. Multistep One-Pot Synthesis of Enantioenriched Polysubstituted Cyclopenta[b] Indoles. Angew. Chem. Int. Ed. 2012, 51, 1059–1062. [Google Scholar] [CrossRef]
- Repka, L.M.; Ni, J.; Reisman, S.E. Enantioselective Synthesis of Pyrroloindolines by a Formal [3 + 2] Cycloaddition Reaction. J. Am. Chem. Soc. 2010, 132, 14418–14420. [Google Scholar] [CrossRef]
- Han, B.; Xiao, Y.; Yao, Y.; Chen, Y. Lewis Acid Catalyzed Intramolecular Direct Ene Reaction of Indoles. Angew. Chem. 2010, 122, 10387–10389. [Google Scholar] [CrossRef]
- Baran, P.S.; Guerrero, C.A.; Ambhaikar, N.B.; Hafensteiner, B.D. Short, Enantioselective Total Synthesis of Stephacidin A. Angew. Chem. Int. Ed. 2005, 44, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Nazaré, M.; Schneider, C.; Lindenschmidt, A.; Will, D.W. A Flexible, Palladium-Catalyzed Indole and Azaindole Synthesis by Direct Annulation of Chloroanilines and Chloroaminopyridines with Ketones. Angew. Chem. Int. Ed. 2004, 43, 4526–4528. [Google Scholar] [CrossRef]
- Barluenga, J.; Jiménez-Aquino, A.; Aznar, F.; Valdés, C. Modular Synthesis of Indoles from Imines and O-Dihaloarenes or o-Chlorosulfonates by a Pd-Catalyzed Cascade Process. J. Am. Chem. Soc. 2009, 131, 4031–4041. [Google Scholar] [CrossRef]
- Vickerman, K.L.; Stanley, L.M. Catalytic, Enantioselective Synthesis of Polycyclic Nitrogen, Oxygen, and Sulfur Heterocycles via Rh-Catalyzed Alkene Hydroacylation. Org. Lett. 2017, 19, 5054–5057. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Cheng, Q.Q.; Deng, Y.; Arman, H.; Doyle, M.P. Highly Regio- and Enantioselective Formal [3 + 2]-Annulation of Indoles with Electrophilic Enol Carbene Intermediates. Org. Lett. 2016, 18, 4550–4553. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, W.B.; Tu, H.F.; You, S.L. Ligand-Enabled Ir-Catalyzed Intermolecular Diastereoselective and Enantioselective Allylic Alkylation of 3-Substituted Indoles. Chem. Sci. 2015, 6, 4525–4529. [Google Scholar] [CrossRef] [PubMed]
- Mietke, T.; Cruchter, T.; Larionov, V.A.; Faber, T.; Harms, K.; Meggers, E. Asymmetric Nazarov Cyclizations Catalyzed by Chiral-at-Metal Complexes. Adv. Synth. Catal. 2018, 360, 2093–2100. [Google Scholar] [CrossRef]
- Zi, W.; Wu, H.; Toste, F.D. Gold(I)-Catalyzed Dearomative Rautenstrauch Rearrangement: Enantioselective Access to Cyclopenta[ b ]Indoles. J. Am. Chem. Soc. 2015, 137, 3225–3228. [Google Scholar] [CrossRef]
- Scarpi, D.; Petrović, M.; Fiser, B.; Gómez-Bengoa, E.; Occhiato, E.G. Construction of Cyclopenta[b]Indol-1-Ones by a Tandem Gold(I)-Catalyzed Rearrangement/Nazarov Reaction and Application to the Synthesis of Bruceolline H. Org. Lett. 2016, 18, 3922–3925. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.Y.; Wei, Y.; Tang, X.Y.; Shi, M. Catalyst-Dependent Stereodivergent and Regioselective Synthesis of Indole-Fused Heterocycles through Formal Cycloadditions of Indolyl-Allenes. J. Am. Chem. Soc. 2015, 137, 8131–8137. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.Q.; Deng, Y.; Lankelma, M.; Doyle, M.P. Cycloaddition Reactions of Enoldiazo Compounds. Chem. Soc. Rev. 2017, 46, 5425–5443. [Google Scholar] [CrossRef] [PubMed]
- Awata, A.; Arai, T. PyBidine/Copper Catalyst: Asymmetric Exo’-Selective [3+2] Cycloaddition Using Imino Ester and Electrophilic Indole. Angew. Chem. Int. Ed. Engl. 2014, 53, 10462–10465. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Ehmke, V.; Michael O’Keefe, B.; Bringley, D.A. Palladium-Catalyzed Dearomative Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2014, 136, 8213–8216. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Doyle, M.P. Asymmetric [3 + n]-Cycloaddition Reactions of Donor-Acceptor Cyclopropanes. ChemCatChem 2023, 15, e202301090. [Google Scholar] [CrossRef]
- Schneider, T.F.; Kaschel, J.; Werz, D.B. A New Golden Age for Donor-Acceptor Cyclopropanes. Angew. Chem.-Int. Ed. 2014, 53, 5504–5523. [Google Scholar] [CrossRef]
- Cavitt, M.A.; Phun, L.H.; France, S. Intramolecular Donor-Acceptor Cyclopropane Ring-Opening Cyclizations. Chem. Soc. Rev. 2014, 43, 804–818. [Google Scholar] [CrossRef]
- Meazza, M.; Guo, H.; Rios, R. Synthetic Applications of Vinyl Cyclopropane Opening. Org. Biomol. Chem. 2017, 15, 2479–2490. [Google Scholar] [CrossRef]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2021, 121, 227–263. [Google Scholar] [CrossRef]
- Liu, J.; Liu, R.; Wei, Y.; Shi, M. Recent Developments in Cyclopropane Cycloaddition Reactions. Trends Chem. 2019, 1, 779–793. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, X.; Feng, X. Asymmetric Catalytic Reactions of Donor–Acceptor Cyclopropanes. Angew. Chem. Int. Ed. 2021, 60, 9192–9204. [Google Scholar] [CrossRef] [PubMed]
- Harrington, P.; Kerr, M.A. The High Pressure Reaction of Cyclopropanes with Indoles Catalyzed by Ytterbium Triflate. Tetrahedron Lett. 1997, 38, 5949–5952. [Google Scholar] [CrossRef]
- Kerr, M.A.; Keddy, R.G. The Annulation of 3-Alkylindoles with 1,1-Cyclopropanediesters. Tetrahedron Lett. 1999, 40, 5671–5675. [Google Scholar] [CrossRef]
- England, D.B.; Kuss, T.D.O.; Keddy, R.G.; Kerr, M.A. Cyclopentannulation of 3-Alkylindoles: A Synthesis of a Tetracyclic Subunit of the Kopsane Alkaloids. J. Org. Chem. 2001, 66, 4704–4709. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Xu, H.; Liao, S.; Xie, Z.; Tang, Y. Copper-Catalyzed Highly Enantioselective Cyclopentannulation of Indoles with Donor-Acceptor Cyclopropanes. J. Am. Chem. Soc. 2013, 135, 7851–7854. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Lopez, K.; Gurung, R.; Arman, H.; Doyle, M.P. Stereoretentive Catalytic [3 + 2]-Cycloaddition/Rearrangement/Decarboxylation Reactions of Indoles with Non-Racemic Donor-Acceptor Cyclopropanes. ACS Catal. 2023, 13, 1621–1629. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Tong, F.; Sun, B.B.; Fan, W.T.; Chen, J.B.; Hu, D.; Wang, X.W. Pd-Catalyzed Asymmetric Dearomative Cycloaddition for Construction of Optically Active Pyrroloindoline and Cyclopentaindoline Derivatives: Access to 3a-Aminopyrroloindolines. J. Org. Chem. 2018, 83, 2882–2891. [Google Scholar] [CrossRef] [PubMed]
- Laugeois, M.; Ling, J.; Férard, C.; Michelet, V.; Ratovelomanana-Vidal, V.; Vitale, M.R. Palladium(0)-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles with Vinylcyclopropanes: An Entry to Stereodefined 2,3-Fused Cyclopentannulated Indoline Derivatives. Org. Lett. 2017, 19, 2266–2269. [Google Scholar] [CrossRef]
- Sun, M.; Zhu, Z.Q.; Gu, L.; Wan, X.; Mei, G.J.; Shi, F. Catalytic Asymmetric Dearomative [3 + 2] Cycloaddition of Electron-Deficient Indoles with All-Carbon 1,3-Dipoles. J. Org. Chem. 2018, 83, 2341–2348. [Google Scholar] [CrossRef]
- Trost, B.M.; Bai, W.J.; Hohn, C.; Bai, Y.; Cregg, J.J. Palladium-Catalyzed Asymmetric Allylic Alkylation of 3-Substituted 1 H-Indoles and Tryptophan Derivatives with Vinylcyclopropanes. J. Am. Chem. Soc. 2018, 140, 6710–6717. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Zhu, Y.M.; Yang, P.J.; Wang, S.; Wang, S.; Liu, Z.; Yang, G. Copper(I)-Catalyzed Kinetic Resolution of N-Sulfonylaziridines with Indoles: Efficient Construction of Pyrroloindolines. J. Am. Chem. Soc. 2015, 137, 10088–10091. [Google Scholar] [CrossRef]
- Huang, Y.M.; Zheng, C.W.; Pan, L.; Jin, Q.W.; Zhao, G. Palladium(II)-Catalyzed Formal [3 + 2] Cycloaddition of Aziridines with 3-Substituted Indoles: Synthesis of Enantioenriched Pyrroloindolines. J. Org. Chem. 2015, 80, 10710–10718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Z.; Wu, H.H.; Zhang, J. Ni(ClO4)2-Catalysed Regio- and Diastereoselective [3 + 2] Cycloaddition of Indoles and Aryl Oxiranyl-Dicarboxylates/Diketones: A Facile Access to Furo[3,4-b]Indoles. Chem. Commun. 2012, 48, 1817–1819. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.M. Catalysis without Precious Metals; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2010. [Google Scholar]
- Wender, P.A.; Miller, B.L. Synthesis at the Molecular Frontier. Nature 2009, 460, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A. Toward the Ideal Synthesis and Molecular Function through Synthesis-Informed Design. Nat. Prod. Rep. 2014, 31, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Gaich, T.; Baran, P.S. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef] [PubMed]
- Sibi, M.P.; Ma, Z.; Jasperse, C.P. Enantioselective addition of nitrones to activated cyclopropanes. J. Am. Chem. Soc. 2005, 127, 5764–5765. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.; Sun, X.L.; Tang, Y. Highly enantioselective and diastereoselective cycloaddition of cyclopropanes with nitrones and its application in the kinetic resolution of 2-substituted cyclopropane-1,1-dicarboxylates. Angew. Chem. Int. Ed. 2007, 46, 3918–3921. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Li, J.; Ling, L.; Liao, S.H.; Sun, X.L.; Li, Y.X.; Wang, L.J.; Tang, Y. Highly enantioselective [3 + 3] cycloaddition of aromatic azomethine imines with cyclopropanes directed by π-π Stacking interactions. Angew. Chem. Int. Ed. 2013, 52, 1452–1456. [Google Scholar] [CrossRef]
- Suo, J.J.; Liu, W.; Du, J.; Ding, C.H.; Hou, X.L. Diastereo- and Enantioselective Palladium-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles. Chem. Asian J. 2018, 13, 959–963. [Google Scholar] [CrossRef]
- Chen, W.; Xia, Y.; Lin, L.; Yuan, X.; Guo, S.; Liu, X.; Feng, X. Asymmetric Synthesis of Furo[3,4-b]Indoles by Catalytic [3+2] Cycloaddition of Indoles with Epoxides. Chem. Eur. J. 2015, 21, 15104–15107. [Google Scholar] [CrossRef]
- Dixon, N.E.; Jackson, W.G.; Lawrance, G.A.; Sargeson, A.M. Labile (Trifluoromethanesulfonato) Cobalt (III) Amine Complexes. Inorg. Chem. 1981, 20, 470–476. [Google Scholar] [CrossRef]
- Moran, J.; Smith, A.G.; Carris, R.M.; Johnson, S.; Krische, M.J. Polarity Inversion of Donor-Acceptor Cyclopropanes: Disubstituted. J. Am. Chem. Soc. 2011, 133, 18618–18621. [Google Scholar] [CrossRef] [PubMed]
- Nájera, C.; de Gracia Retamosa, M.; Sansano, J.M.; de Cozar, A.; Cossío, F.P. Enantioselective Synthesis of Polysubstituted Prolines by Binap-Silver-Catalyzed 1,3-Dipolar Cycloadditions. Tetrahedron Asymmetry 2008, 19, 2913–2923. [Google Scholar] [CrossRef]
- Clevenger, A.L.; Stolley, R.M.; Staudaher, N.D.; Al, N.; Rheingold, A.L.; Vanderlinden, R.T.; Louie, J. Comprehensive Study of the Reactions between Chelating Phosphines and Ni(Cod)2. Organometallics 2018, 37, 3259–3268. [Google Scholar] [CrossRef]
- Spielvogel, D.J.; Davis, W.M.; Buchwald, S.L. Preparation, Crystal Structure Analysis, and Catalytic Application of [(S)-BINAP]Ni(COD) and [(S)-BINAP]NiBr2. Organometallics 2002, 21, 3833–3836. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Reek, J.N.H. The Bite Angle Makes the Catalyst. Pure Appl. Chem. 1999, 71, 1443–1452. [Google Scholar] [CrossRef]
- CCDC 2302398 Contains the Supplementary Crystallographic Data for Compound 3ac. These Data Can Be Obtained Free of Charge from the Cambridge Crystallographic Data Centre via. Available online: www.ccdc.cam.ac.uk/data_request/cif (accessed on 15 March 2024).
- Dawson, D.D.; Oswald, V.F.; Borovik, A.S.; Jarvo, E.R. Identification of the Active Catalyst for Nickel-Catalyzed Stereospecific Kumada Coupling Reactions of Ethers. Chem. Eur. J. 2020, 26, 3044–3048. [Google Scholar] [CrossRef]
- Evans, D.A.; Thomson, R.J.; Franco, F. Ni(II) Tol-BINAP-Catalyzed Enantioselective Michael Reactions of β-Ketoesters and Unsaturated N-Acylthiazolidinethiones. J. Am. Chem. Soc. 2005, 127, 10816–10817. [Google Scholar] [CrossRef]
- Pohlhaus, P.D.; Sanders, S.D.; Parsons, A.T.; Li, W.; Johnson, J.S. Scope and Mechanism for Lewis Acid-Catalyzed Cycloadditions of Aldehydes and Donor-Acceptor Cyclopropanes: Evidence for a Stereospecific Intimate Ion Pair Pathway. J. Am. Chem. Soc. 2008, 130, 8642–8650. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Johnson, J.S.; Parsons, A.T.; Pohlhaus, P.D.; Sanders, S.D. Complexity-Building Annulations of Strained Cycloalkanes and C=O π Bonds. J. Org. Chem. 2010, 75, 6317–6325. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S. Reactive Intermediates: MS Investigations in Solution; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2009. [Google Scholar] [CrossRef]
- Santos, L.S.; Metzger, J.O. Study of Homogeneously Catalyzed Ziegler-Natta Polymerization of Ethene by ESI-MS. Angew. Chem.-Int. Ed. 2006, 45, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Sathish, M.; Nachtigall, F.M.; Santos, L.S. Enantioselective Imine Reduction of Dihydro-β-Carbolines by Fe-Thiosquaramide Catalyst. Org. Lett. 2022, 24, 7627–7631. [Google Scholar] [CrossRef] [PubMed]
- Gozzo, F.C.; Santos, L.S.; Augusti, R.; Consorti, C.S.; Dupont, J.; Eberlin, M.N. Gaseous Supramolecules of Imidazolium Ionic Liquids: “Magic” Numbers and Intrinsic Strengths of Hydrogen Bonds. Chem. Eur. J. 2004, 10, 6187–6193. [Google Scholar] [CrossRef] [PubMed]
- Ortega, D.E.; Cortés-Arriagada, D.; Trofymchuk, O.S.; Nachtigall, F.M.; Santos, L.S.; Rojas, R.S.; Toro-Labbé, A. Mechanistic Study of the Competitiveness between Branched and Linear Polyethylene Production on: N-Arylcyano-β-Diketiminate Nickel Hydride. Polym. Chem. 2020, 11, 6640–6649. [Google Scholar] [CrossRef]
- Trofymchuk, O.S.; Ortega, D.E.; Cortés-Arriagada, D.; Pereira, A.; Daniliuc, C.G.; Klitzke, C.F.; Santos, L.S.; Rojas, R.S. Neutral and Cationic Methallyl Nickel Complexes in Alkene Activation: A Combined DFT, ESI-MS and Chemometric Approach. Catal. Sci. Technol. 2021, 11, 7475–7485. [Google Scholar] [CrossRef]
- Escobar, M.A.; Trofymchuk, O.S.; Rodriguez, B.E.; Lopez-Lira, C.; Tapia, R.; Daniliuc, C.; Berke, H.; Nachtigall, F.M.; Santos, L.S.; Rojas, R.S. Lewis Acid Enhanced Ethene Dimerization and Alkene Isomerization-ESI-MS Identification of the Catalytically Active Pyridyldimethoxybenzimidazole Nickel(II) Hydride Species. ACS Catal. 2015, 5, 7338–7342. [Google Scholar] [CrossRef]
- Bao, M.; Doyle, M.P. Stereoretentive Catalytic [3 + 2]/[3 + 3]-Cycloaddition of Nonracemic Donor–Acceptor Cyclopropanes: Synthesis of Substituted Pyrrolidines and 1,2-Oxazinanes. Org. Lett. 2023, 25, 3029–3033. [Google Scholar] [CrossRef]
- Augustin, A.U.; Merz, J.L.; Jones, P.G.; Mlostoń, G.; Werz, D.B. (4 + 3)-Cycloaddition of Donor-Acceptor Cyclopropanes with Thiochalcones: A Diastereoselective Access to Tetrahydrothiepines. Org. Lett. 2019, 21, 9405–9409. [Google Scholar] [CrossRef]
- Chagarovskiy, A.O.; Ivanova, O.A.; Rakhmankulov, E.R.; Budynina, E.M.; Trushkov, I.V.; Melnikov, M.Y. Lewis Acid-Catalyzed Isomerization of 2-Arylcyclopropane-1,1- Dicarboxylates: A New Efficient Route to 2-Styrylmalonates. Adv. Synth. Catal. 2010, 352, 3179–3184. [Google Scholar] [CrossRef]
- Xie, M.S.; Zhao, G.F.; Qin, T.; Suo, Y.B.; Qu, G.R.; Guo, H.M. Thiourea Participation in [3 + 2] Cycloaddition with Donor-Acceptor Cyclopropanes: A Domino Process to 2-Amino-Dihydrothiophenes. Chem. Commun. 2019, 55, 1580–1583. [Google Scholar] [CrossRef]
- Pradhan, T.R.; Kim, H.W.; Park, J.K. Harnessing the Polarizability of Conjugated Alkynes toward [2 + 2] Cycloaddition, Alkenylation, and Ring Expansion of Indoles. Org. Lett. 2018, 20, 5286–5290. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Kanno, A.; Hanzawa, Y. Synthesis of 2,3-Disubstituted Indoles by a Rhodium-Catalyzed Aromatic Amino-Claisen Rearrangement of N-Propargyl Anilines. Angew. Chem.—Int. Ed. 2007, 46, 3931–3933. [Google Scholar] [CrossRef]
- Bergès, J.; García, B.; Muñiz, K. An Electrophilic Bromine Redox Catalysis for the Synthesis of Indole Alkaloid Building Blocks by Selective Aliphatic C−H Amination. Angew. Chem.—Int. Ed. 2018, 57, 15891–15895. [Google Scholar] [CrossRef]
- Varlet, T.; Bouchet, D.; Van Elslande, E.; Masson, G. Decatungstate-Photocatalyzed Dearomative Hydroacylation of Indoles: Direct Synthesis of 2-Acylindolines. Chem.—A Eur. J. 2022, 28, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.; Chakraborti, G.; Dash, J. Reductive Aromatization of Oxindoles to 3-Substituted Indoles. Tetrahedron Lett. 2020, 61, 152109. [Google Scholar] [CrossRef]
- Yu, L.; Zhong, Y.; Yu, J.; Gan, L.; Cai, Z.; Wang, R.; Jiang, X. Highly Diastereoselective Oxa-[3+3] Cyclization with C,N-Cyclic Azomethine Imines: Via the Copper-Catalyzed Aerobic Oxygenated CC Bond of Indoles. Chem. Commun. 2018, 54, 2353–2356. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.W.; Ko, T.Y.; Jang, M.J.; Jang, S.S. Silver(I)-Mediated C-H Amination of 2-Alkenylanilines: Unique Solvent-Dependent Migratory Aptitude. Adv. Synth. Catal. 2015, 357, 227–234. [Google Scholar] [CrossRef]
- Pan, S.; Ryu, N.; Shibata, T. Ir(I)-Catalyzed C-H Bond Alkylation of C2-Position of Indole with Alkenes: Selective Synthesis of Linear or Branched 2-Alkylindoles. J. Am. Chem. Soc. 2012, 134, 17474–17477. [Google Scholar] [CrossRef]
- Goudreau, R.; Marcoux, D.; Charette, B. General Method for the Synthesis of Phenyliodonium Ylides from Malonate Esters: Easy Access to 1, 1-Cyclopropane Diesters. J. Org. Chem. 2009, 74, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Novikov, R.A.; Tarasova, A.V.; Korolev, V.A.; Timofeev, V.P.; Tomilov, Y.V. A New Type of Donor-Acceptor Cyclopropane Reactivity: The Generation of Formal 1,2- and 1,4-Dipoles. Angew. Chem.—Int. Ed. 2014, 53, 3187–3191. [Google Scholar] [CrossRef] [PubMed]
- Perreault, C.; Goudreau, S.R.; Zimmer, L.E.; Charette, A.B. Cycloadditions of Aromatic Azomethine Imines with 1,1-Cyclopropane Diesters. Org. Lett. 2008, 10, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, R.; Tiwari, D.P.; Saha, A.; Ghorai, M.K. Diastereoselective Synthesis of Functionalized Tetrahydrocarbazoles via a Domino-Ring Opening-Cyclization of Donor-Acceptor Cyclopropanes with Substituted 2-Vinylindoles. Org. Lett. 2014, 16, 3954–3957. [Google Scholar] [CrossRef]
- Parsons, A.T.; Campbell, M.J.; Johnson, J.S. Diastereoselective Synthesis of Tetrahydrofurans via Palladium(0)-Catalyzed [3 + 2] Cycloaddition of Vinylcyclopropanes and Aldehydes. Org. Lett. 2008, 10, 2541–2544. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | [M] (X mol%) | Solvent | t (h) | Conv. (%) 2 | Yield (%) 3 | dr4 |
| 1 | Cu(OTf)2 (20) | DCE | 24 | >99 | 79 | 5:1 |
| 2 | Co(ClO4)2·6H2O (20) | DCE | 24 | - | 42 | 6:1 |
| 3 5 | Zn(ClO4)2·6H2O (20) | DCE | 48 | 63 | 28 | 6.4:1 |
| 4 | Ni(ClO4)2·6H2O (20) | DCE | 24 | >99 | 95 | 6:1 |
| 5 | Ni(ClO4)2·6H2O (10) | DCE | 48 | 50 | 48 | 6:1 |
| 6 | Ni(OAc)2·4H2O (10) | DCE | 24 | 15 | 9.3 | 5:1 |
| 7 | NiSO4·6H2O (10) | DCE | 24 | 86 | 56 | 4:1 |
| 8 | Ni(NO3)2·6H2O (10) | DCE | 24 | 91 | 48 | 9:1 |
| 9 | NiCl2 (10) | DCE | 24 | 57 | 55 | 3.6:1 |
| 10 | Ni(OTf)2 (10) | DCE | 24 | >99 | 78 | 5:1 |
| 11 | Ni(OTf)2 (10) | DCM | 24 | 73 | 47 | 7.7:1 |
| 12 | Ni(OTf)2 (10) | CHCl3 | 24 | 85 | 25 | 1:1 |
| 13 | Ni(OTf)2 (10) | Toluene | 24 | 76 | 29 | 1:1 |
| 14 | Ni(OTf)2 (10) | CH3CN | 48 | <5 | - | - |
| 15 | Ni(OTf)2 (10) | AcOEt | 48 | <5 | - | - |
 | |||||
|---|---|---|---|---|---|
| Entry | [Ni]/Ligand | t (h) | Conv. (%) 2 | Yield (%) 3 | dr4 |
| 1 5 | Ni(OTf)2/Bipy | 144 | <5 | - | - |
| 2 5 | Ni(OTf)2/Phen | 72 | 26 | 12 | 1:1 |
| 3 | Ni(OTf)2/dppe | 144 | 7.5 | - | - |
| 4 | Ni(OTf)2/dFppe | 144 | 24 | 13 | 4:1 |
| 5 5 | Ni(OTf)2/L2-PrPr2 | 72 | <5 | - | - |
| 6 5 | Ni(OTf)2/L3-PrPr2 | 72 | <5 | - | - |
| 7 | Ni(ClO4)2·6H2O/Phen | 96 | 63 | 16 | 4.4:1 |
| 8 | Ni(ClO4)2·6H2O/dppe | 96 | - | 19 | 5.7:1 |
| 9 | Ni(ClO4)2·6H2O/dFppe | 168 | - | 43 | 5.5:1 |
| 10 | Ni(ClO4)2·6H2O/dppBz | 120 | 70 | 53 | 5.5:1 |
| 11 5 | Ni(ClO4)2·6H2O/dppf | 144 | <5 | - | - |
| 12 | Ni(ClO4)2·6H2O/rac-BINAP | 5 | >99 | 73 | 6:1 |
| 13 6 | Ni(ClO4)2·6H2O/rac-BINAP | 5 | >99 | 52 | 3:1 |
| 14 5 | Ni(ClO4)2·6H2O/rac-BINAP | 5 | >99 | 28 | 4:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quezada, V.; Castroagudín, M.; Verdugo, F.; Ortiz, S.; Zaragoza, G.; Nachtigall, F.M.; Reis, F.A.A.; Castro-Alvarez, A.; Santos, L.S.; Nelson, R. Nickel(II)-Catalyzed Formal [3+2] Cycloadditions between Indoles and Donor–Acceptor Cyclopropanes. Molecules 2024, 29, 1604. https://doi.org/10.3390/molecules29071604
Quezada V, Castroagudín M, Verdugo F, Ortiz S, Zaragoza G, Nachtigall FM, Reis FAA, Castro-Alvarez A, Santos LS, Nelson R. Nickel(II)-Catalyzed Formal [3+2] Cycloadditions between Indoles and Donor–Acceptor Cyclopropanes. Molecules. 2024; 29(7):1604. https://doi.org/10.3390/molecules29071604
Chicago/Turabian StyleQuezada, Víctor, Mariña Castroagudín, Felipe Verdugo, Sergio Ortiz, Guillermo Zaragoza, Fabiane M. Nachtigall, Francisco A. A. Reis, Alejandro Castro-Alvarez, Leonardo S. Santos, and Ronald Nelson. 2024. "Nickel(II)-Catalyzed Formal [3+2] Cycloadditions between Indoles and Donor–Acceptor Cyclopropanes" Molecules 29, no. 7: 1604. https://doi.org/10.3390/molecules29071604