
Structural Analysis and Activity Correlation of Amphiphilic Cyclic Antimicrobial Peptides Derived from the [W4R4] Scaffold
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
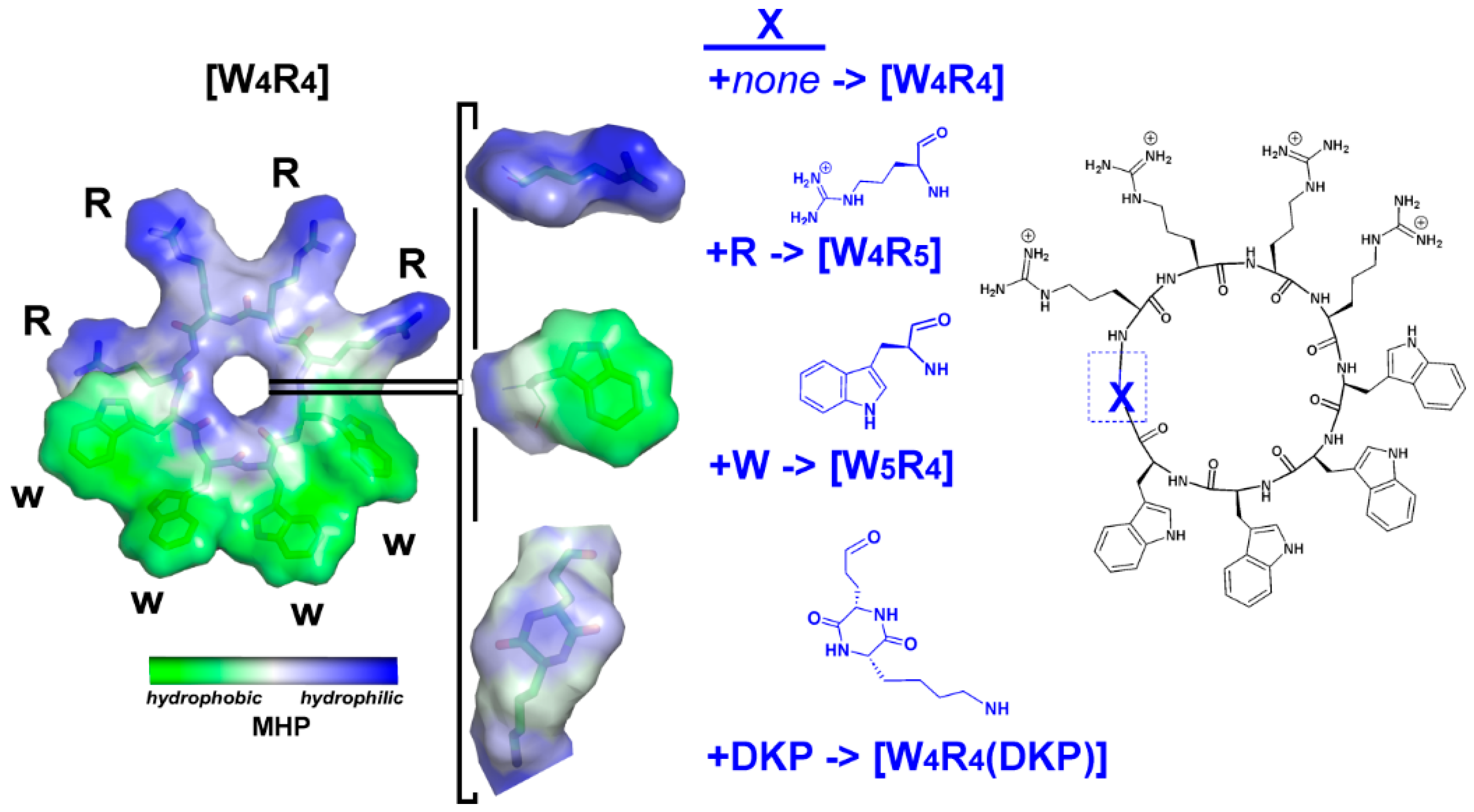
2.1. Design of This Study
2.2. Chemistry
2.3. Antibacterial Activity
2.4. Molecular Dynamics Simulations
2.4.1. Conformational Behavior of Cyclic Peptides in Water
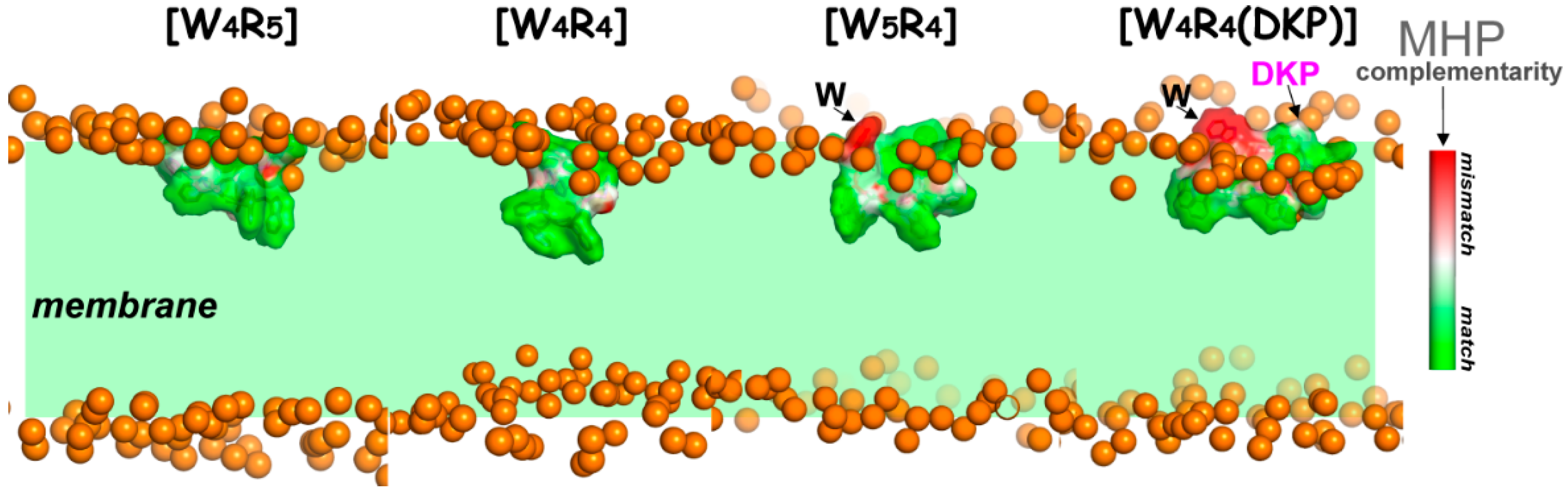
2.4.2. Peptide–Membrane Interactions in the Water–Lipid Environment
Conformational Behavior of Membrane-Embedded Peptides
Membrane-Binding Modes
Apolar Mode and Locked State of the DKP-Containing Peptide
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Chemical Synthesis of [W4R4(DKP)]
4.3. Antibacterial Assay
4.4. Molecular Dynamics Simulations
4.4.1. Preparation of the Starting Configurations
4.4.2. MD Protocols
4.4.3. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Khairkhah, N.; Namvar, A.; Bolhassani, A. Application of cell penetrating peptides as a Promising drug carrier to combat viral infections. Mol. Biotechnol. 2023, 65, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Vasconcelos, L.D.F.; Veiman, K.L.; Uusna, J.; Margus, H.; Garcia-Sosa, A.T.; Pooga, M.; Langel, Ü. Optimization of in vivo DNA delivery with NickFect peptide vectors. J. Control. Release 2016, 241, 135–143. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Tulp, I.; Langel, K.; Langel, Ü. Peptide-ligand binding modeling of siRNA with cell-penetrating peptides. Biomed. Res. Int. 2014, 2014, 257040. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-J.; Kim, D.-H.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial peptides: Their physicochemical properties and therapeutic application. Arch. Pharmacal. Res. 2012, 35, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane active peptides and their biophysical characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef]
- Azmi, F.; Skwarczynski, M.; Toth, I. Towards the development of synthetic antibiotics: Designs inspired by natural antimicrobial peptides. Curr. Med. Chem. 2016, 23, 4610–4624. [Google Scholar] [CrossRef]
- Falanga, A.; Nigro, E.; De Biasi, M.G.; Daniele, A.; Morelli, G.; Galdiero, S.; Scudiero, O. Cyclic peptides as novel therapeutic microbicides: Engineering of human defensin mimetics. Molecules 2017, 22, 1217. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Erdem Büyükkiraz, M.; Kesmen, Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. Appl. Microbiol. 2022, 132, 1573–1596. [Google Scholar] [CrossRef]
- Jindal, M.; Le, C.; Mohd Yusof, M.; Sekaran, S. Net charge, hydrophobicity and specific amino acids contribute to the activity of antimicrobial peptides. J. Health Transl. Med. 2014, 17, 1–7. [Google Scholar]
- Mohammed, E.H.M.; Lohan, S.; Ghaffari, T.; Gupta, S.; Tiwari, R.K.; Parang, K. Membrane-active cyclic amphiphilic peptides: Broad-spectrum antibacterial activity alone and in combination with antibiotics. J. Med. Chem. 2022, 65, 15819–15839. [Google Scholar] [CrossRef] [PubMed]
- Sammes, P.G. Naturally occurring 2,5-dioxopiperazines and related compounds. Fortschr. Chem. Org. Naturst. 1975, 32, 51–118. [Google Scholar] [PubMed]
- De Rosa, S.; Mitova, M.; Tommonaro, G. Marine bacteria associated with sponge as source of cyclic peptides. Biomol. Eng. 2003, 20, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, X.; Xu, T.; Yang, X.; Liu, Y. Diketopiperazines from marine organisms. Chem. Biodivers. 2010, 7, 2809–2829. [Google Scholar] [CrossRef] [PubMed]
- Abderhalden, E.; Komm, E. The formation of diketopiperazines from polypeptides under various conditions. Z. Physiol. Chem. 1924, 139, 147–152. [Google Scholar] [CrossRef]
- De Carvalho, M.P.; Abraham, W.-R. Antimicrobial and biofilm inhibiting diketopiperazines. Curr. Med. Chem. 2012, 19, 3564–3577. [Google Scholar] [CrossRef]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Bojarska, J.; Mieczkowski, A.; Ziora, Z.M.; Skwarczynski, M.; Toth, I.; Shalash, A.O.; Parang, K.; El-Mowafi, S.A.; Mohammed EH, M.; Elnagdy, S.; et al. Cyclic Dipeptides: The biological and structural landscape with special focus on the anti-cancer proline-based scaffold. Biomolecules 2021, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Lohan, S.; Mandal, D.; Choi, W.; Konshina, A.G.; Tiwari, R.K.; Efremov, R.G.; Maslennikov, I.; Parang, K. Small amphiphilic peptides: Activity against a broad range of drug-resistant bacteria and structural insight into membranolytic properties. J. Med. Chem. 2022, 65, 665–687. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell Jr, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. Naccess; Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993; Volume 2. [Google Scholar]
- Koromyslova, A.D.; Chugunov, A.O.; Efremov, R.G. Deciphering fine molecular details of proteins’ Structure and function with a protein surface topography (PST) Method. J. Chem. Inf. Model. 2014, 54, 1189–1199. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (µg/mL) a | ||||||
|---|---|---|---|---|---|---|
| [W4R4] | [W4R5] | [W5R4] | [W4R4(DKP)] | Meropenem | Daptomycin | |
| MRSA (ATCC BAA-1556) (LAC clone) | 4 | 4 | 8 | 32 | 2 | ND |
| Klebsiella pneumoniae (ATCC BAA 1705) | 32 | 32 | 128 | 64 | 16 | ND |
| Pseudomonas aeruginosa (ATCC 27883) | 64 | 32 | 128 | 64 | 1 | ND |
| E. coli (ATCC 25922) | 16 | 16 | 64 | 32 | 1 | ND |
| S. aureus (ATCC 29213) | 8 | 4 | ND b | 32 | ND | 1 |
| E. faecium (ATCC 700221) | 4 | 4 | ND | 16 | ND | 2 |
| E. faecalis (ATCC 29212) | 16 | 8 | ND | 64 | ND | 16 |
| S. pneumoniae (ATCC 51938) | 4 | 2 | ND | 4 | ND | 8 |
| Bacillus subitilis (ATCC 6633) | 1 | 4 | ND | 64 | ND | 0.5 |
| A-Mode a: | [W4R5] | [W4R4] | [W5R4] | [W4R4(DKP)] |
|---|---|---|---|---|
| Number of 200 ns-MD runs | 6 from 16 | 3 from 12 | 2 from 12 | 2 from 12 |
| %MD states in 200 ns/400 ns-MD runs | 20.4/30.0 | 6.7/11.0 | 6.3/10.8 | 0.8/2.8 |
| Binding depth in µs-MD (Å) b | 15.2 ± 1.8/ 12.9 ± 1.9 | 14.9 ± 2.4/ 11.9 ± 2.2 | 15.8 ± 2.0/ 14.2 ± 2.0 | 16.4 ± 1.8/ 15.2 ± 2.1 |
| Stability of deep membrane binding in µs-MD c | + | + | − | − |
| Conformational behavior in water: | ||||
| RMSD, Å d | 1.7 ± 0.5 | 0.9 ± 0.3 | 1.6 ± 0.7 | 2.1 ± 0.8 |
| Backbone plasticity: number of h-bonds e | 0.7 ± 0.8 | 0.7 ± 0.7 | 1.3 ± 1.0 | 1.3 ± 0.8 |
| Peptide | SAR-Profile | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Activity a | Modeling b | ||||||||
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | ||
| MRSA | K. pneumoniae | P. aeruginosa | E. coli | HC50 | Backbone plasticity | Binding Depth | Inserted W-motif | ||
| [W4R7] | ○ c | ○ | ○ | ○ | ○ | ||||
| +R | [W4R6] | ○ | ○ | ● | ○ | ○ | |||
| ↑ d | [W4R5] | ● | ● | ○ | ● | ● | ● | ● | ● |
| [W4R4] | ● | ● | ○ | ● | ○ | ● | ● | ○ | |
| ↓ | [W5R4] | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ○ |
| +W | [W6R4] | ○ | ○ | ○ | ○ | ○ | |||
| [W7R4] | ○ | ○ | ○ | ○ | ○ | ||||
| [W4R4(DKP)] | ○ | ○ | ○ | ○ | ○ | ○ | ○ | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Mowafi, S.A.; Konshina, A.G.; Mohammed, E.H.M.; Krylov, N.A.; Efremov, R.G.; Parang, K. Structural Analysis and Activity Correlation of Amphiphilic Cyclic Antimicrobial Peptides Derived from the [W4R4] Scaffold. Molecules 2023, 28, 8049. https://doi.org/10.3390/molecules28248049
El-Mowafi SA, Konshina AG, Mohammed EHM, Krylov NA, Efremov RG, Parang K. Structural Analysis and Activity Correlation of Amphiphilic Cyclic Antimicrobial Peptides Derived from the [W4R4] Scaffold. Molecules. 2023; 28(24):8049. https://doi.org/10.3390/molecules28248049
Chicago/Turabian StyleEl-Mowafi, Shaima A., Anastasia G. Konshina, Eman H. M. Mohammed, Nikolay A. Krylov, Roman G. Efremov, and Keykavous Parang. 2023. "Structural Analysis and Activity Correlation of Amphiphilic Cyclic Antimicrobial Peptides Derived from the [W4R4] Scaffold" Molecules 28, no. 24: 8049. https://doi.org/10.3390/molecules28248049