
Bioactivity-Guided Synthesis: In Silico and In Vitro Studies of β-Glucosidase Inhibitors to Cope with Hepatic Cytotoxicity
 , , , , , ,
, , , , , ,
Abstract
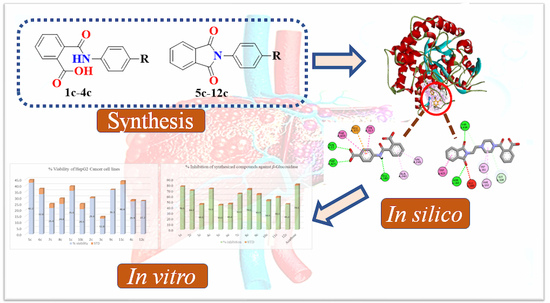
:
1. Introduction
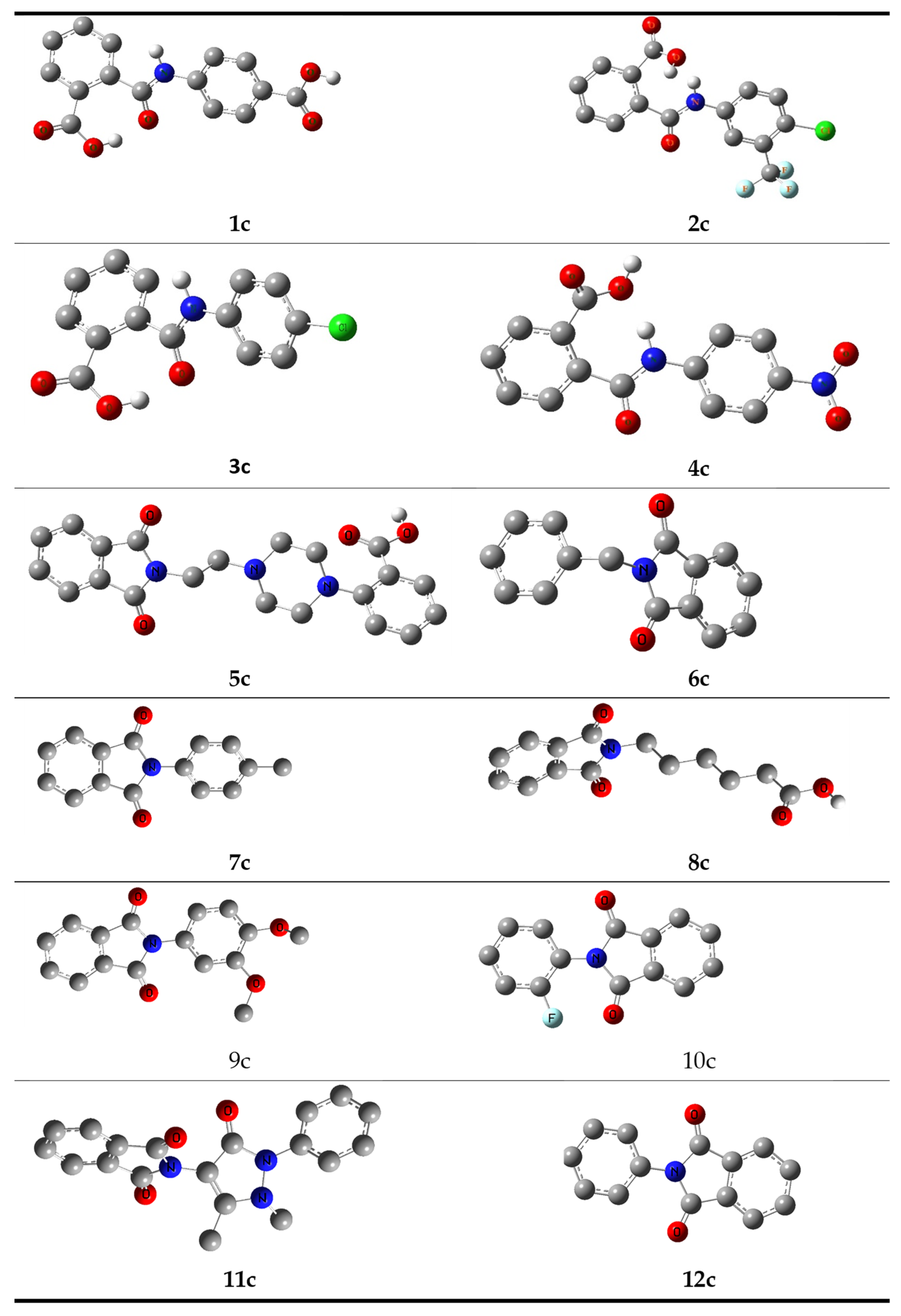
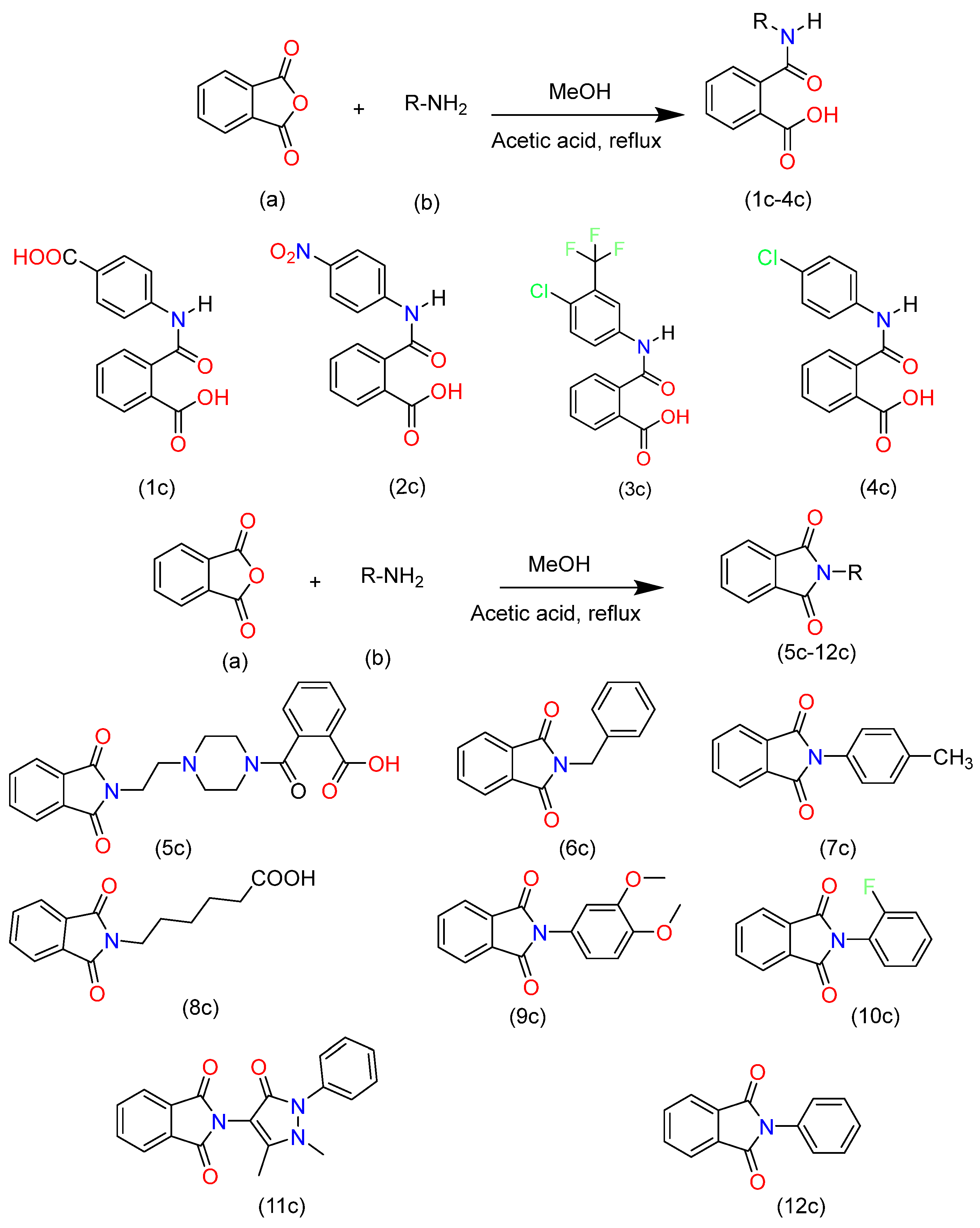
2. Synthesis and Characterization
Structure Optimization of Synthesized Compounds
3. Molecular Docking Study
3.1. ADME Analysis
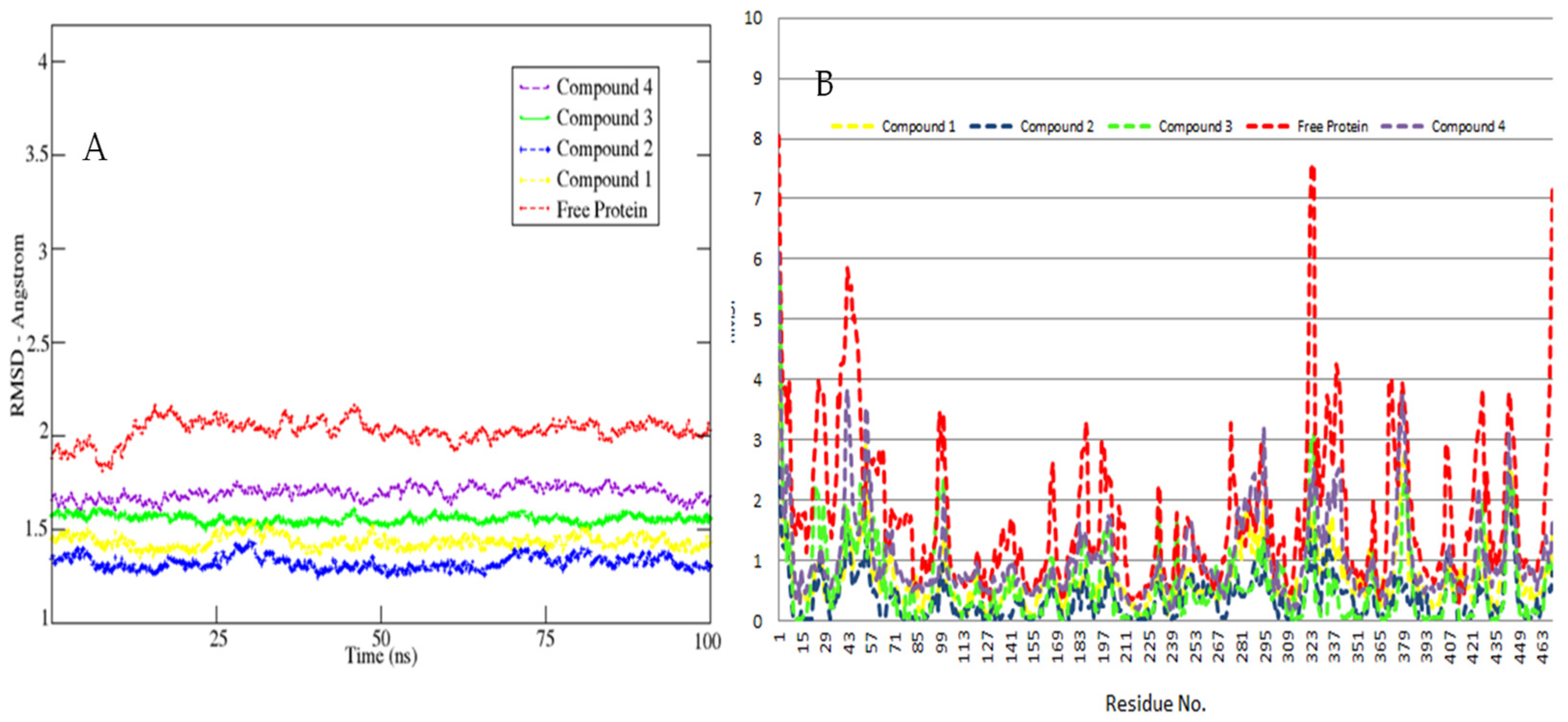
3.2. MD Simulations Study—Global Stability Indices
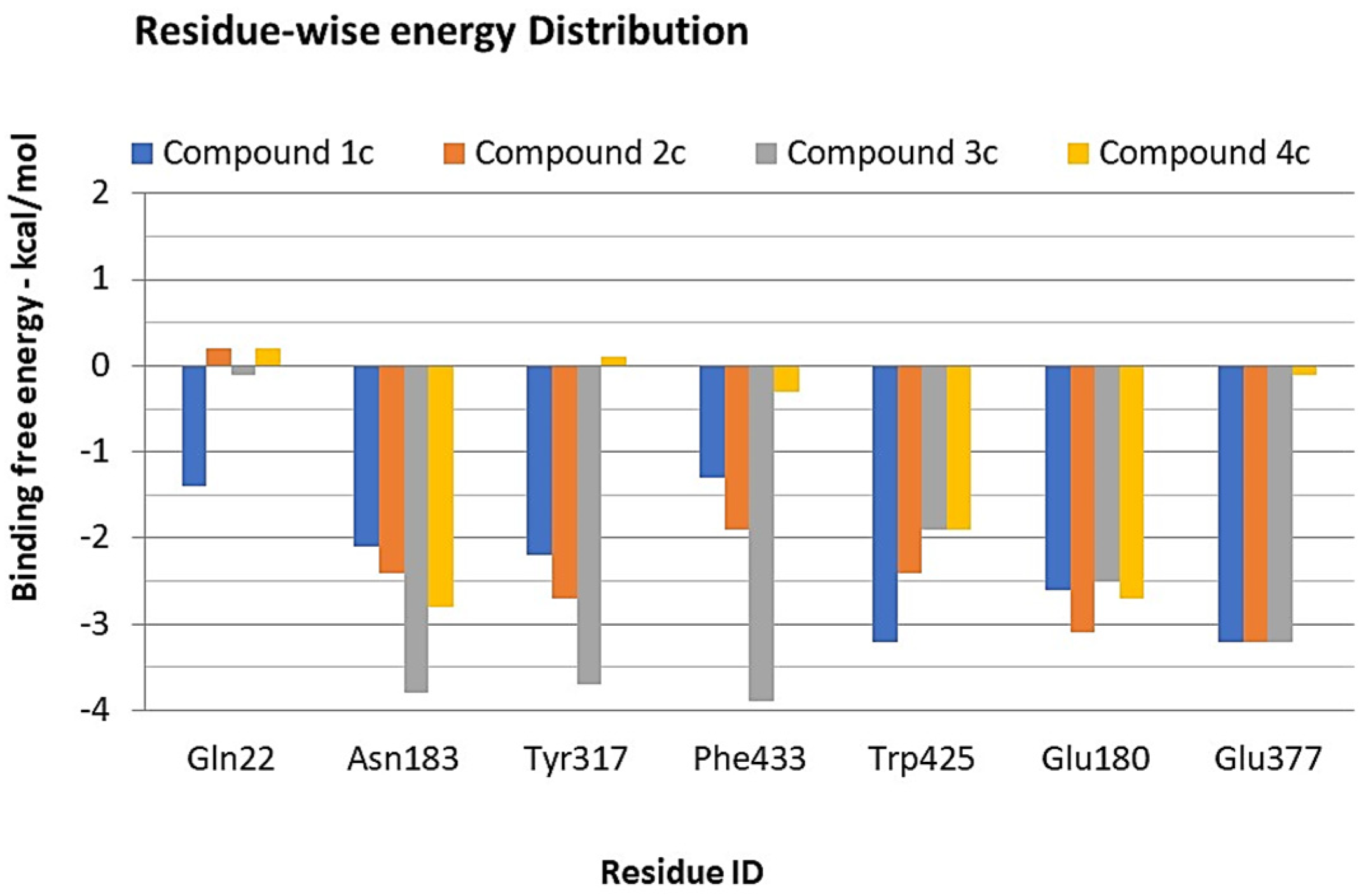
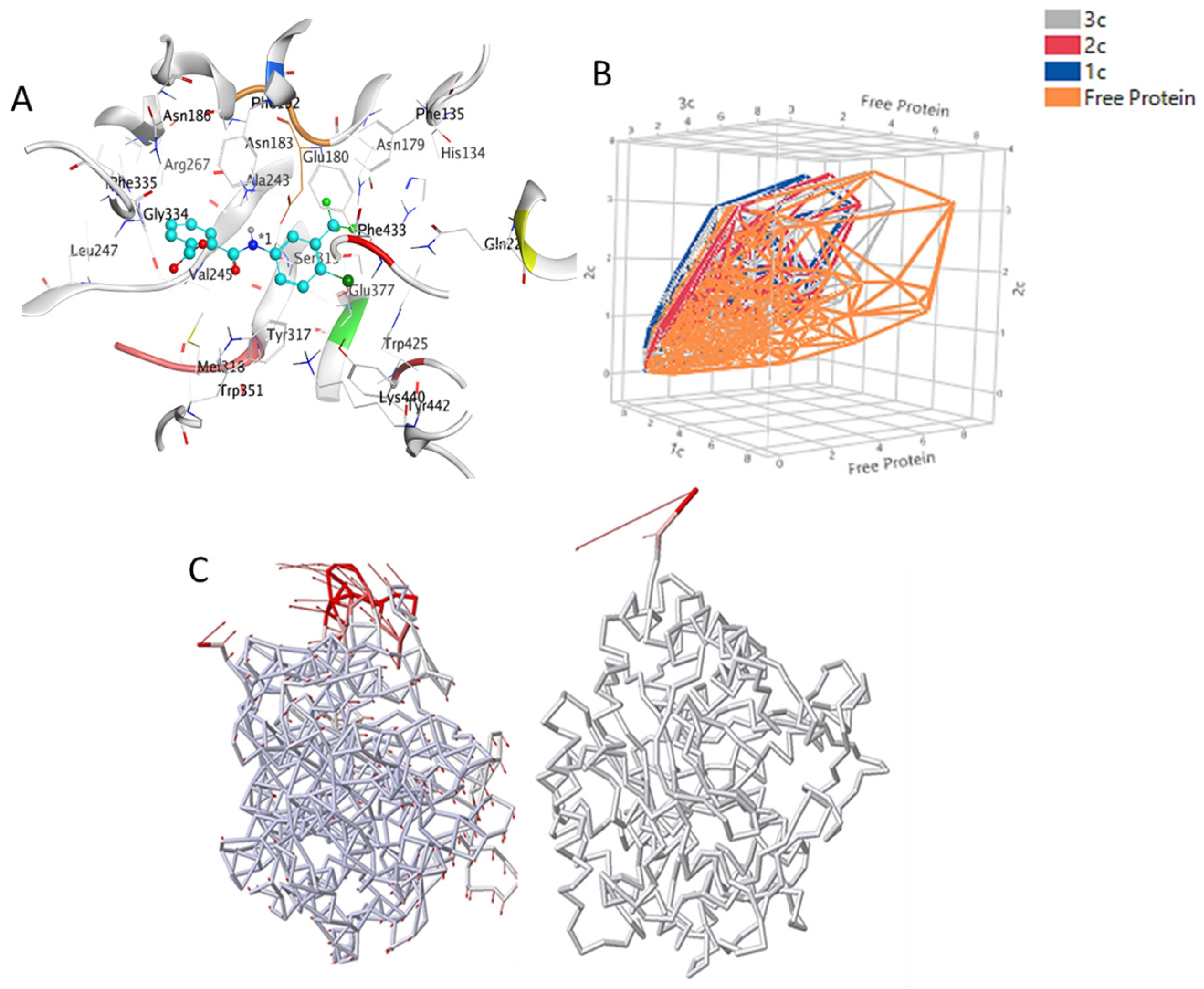
3.3. Hot Spot Regions in Binding
3.4. Binding Free Energy Calculations—MMPBSA Approach
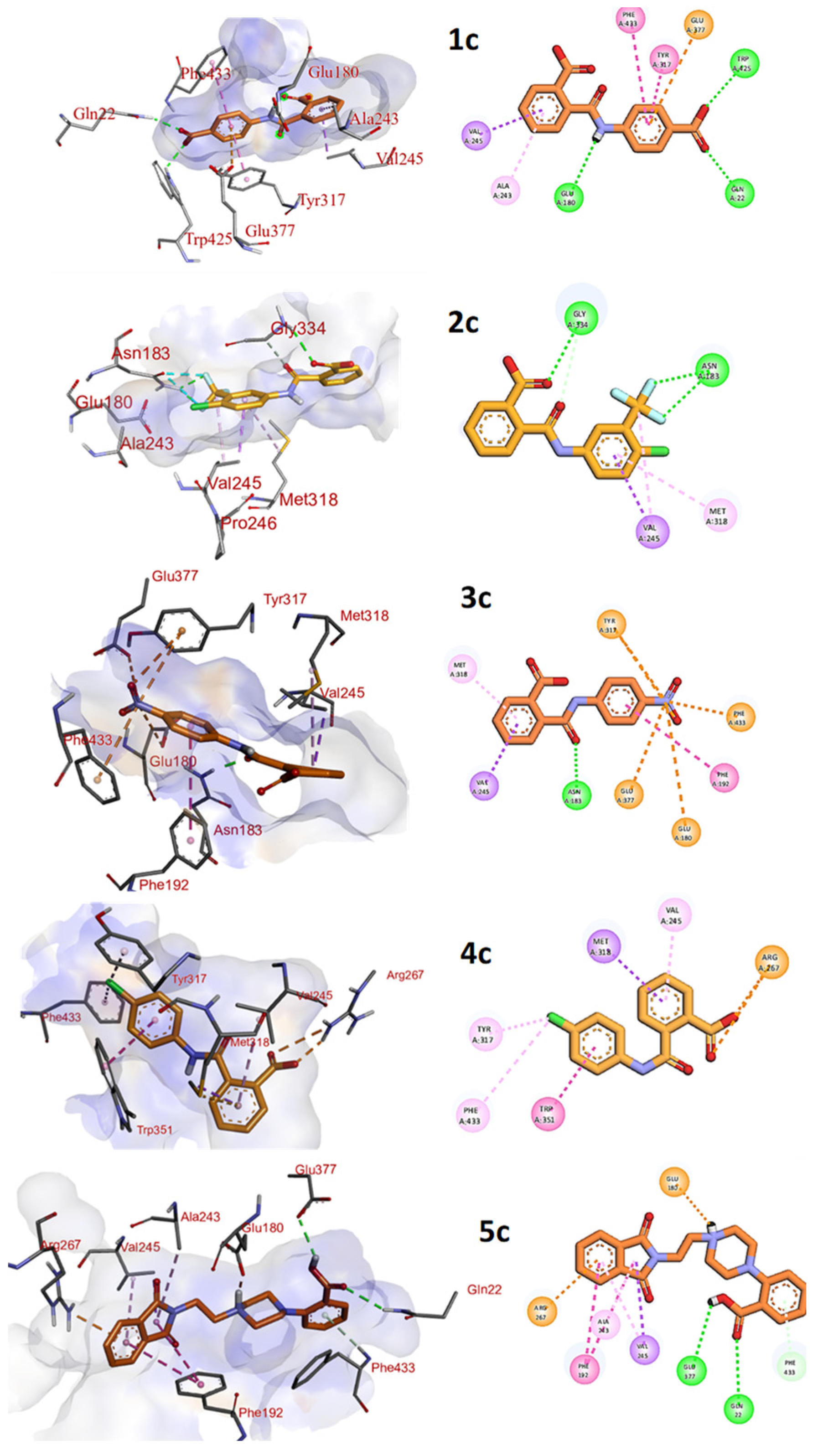
3.5. Interaction Pattern of Compounds 1c–3c and 5c within the Binding Pocket
3.6. In Vitro Studies
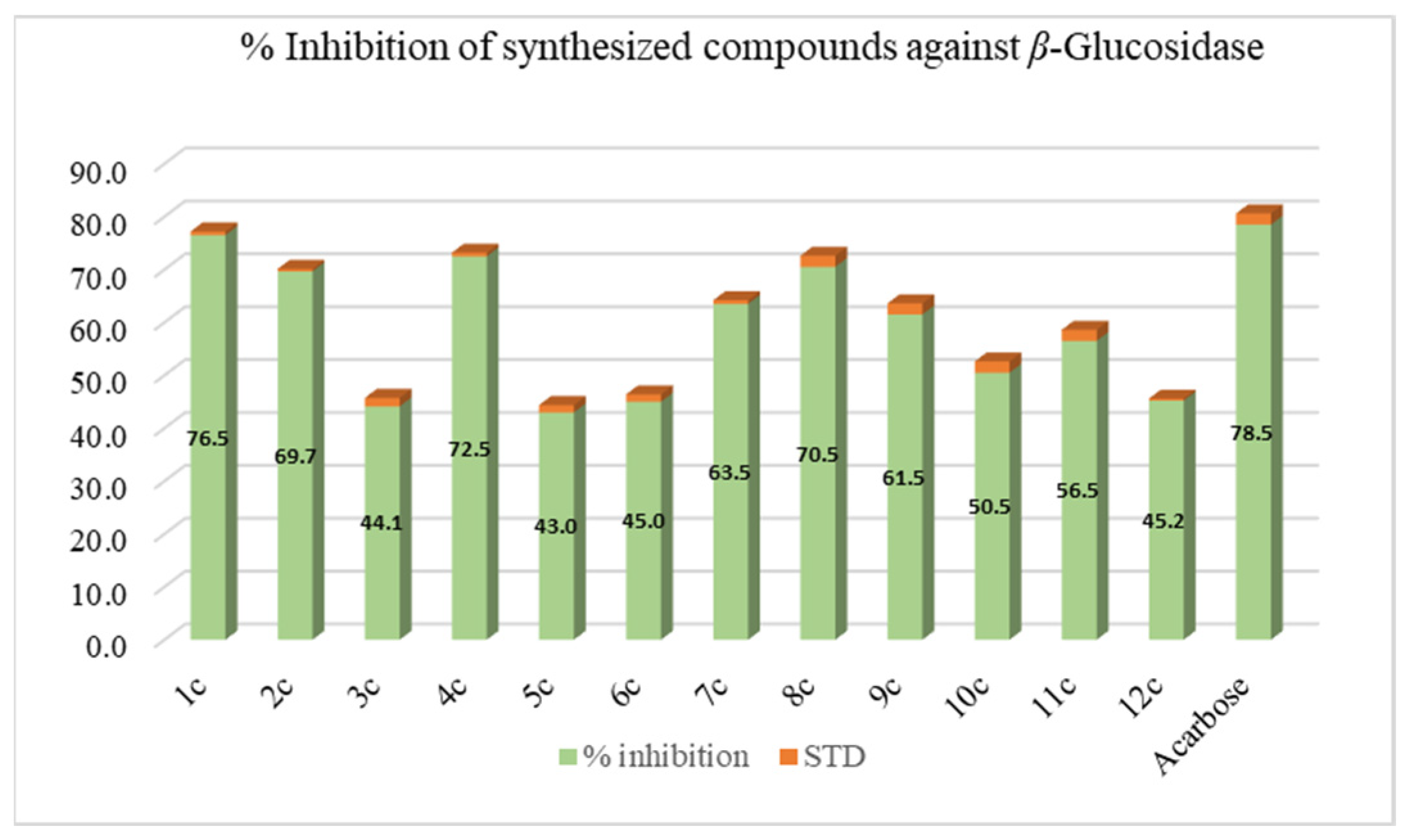
3.6.1. β-Glucosidase Inhibition
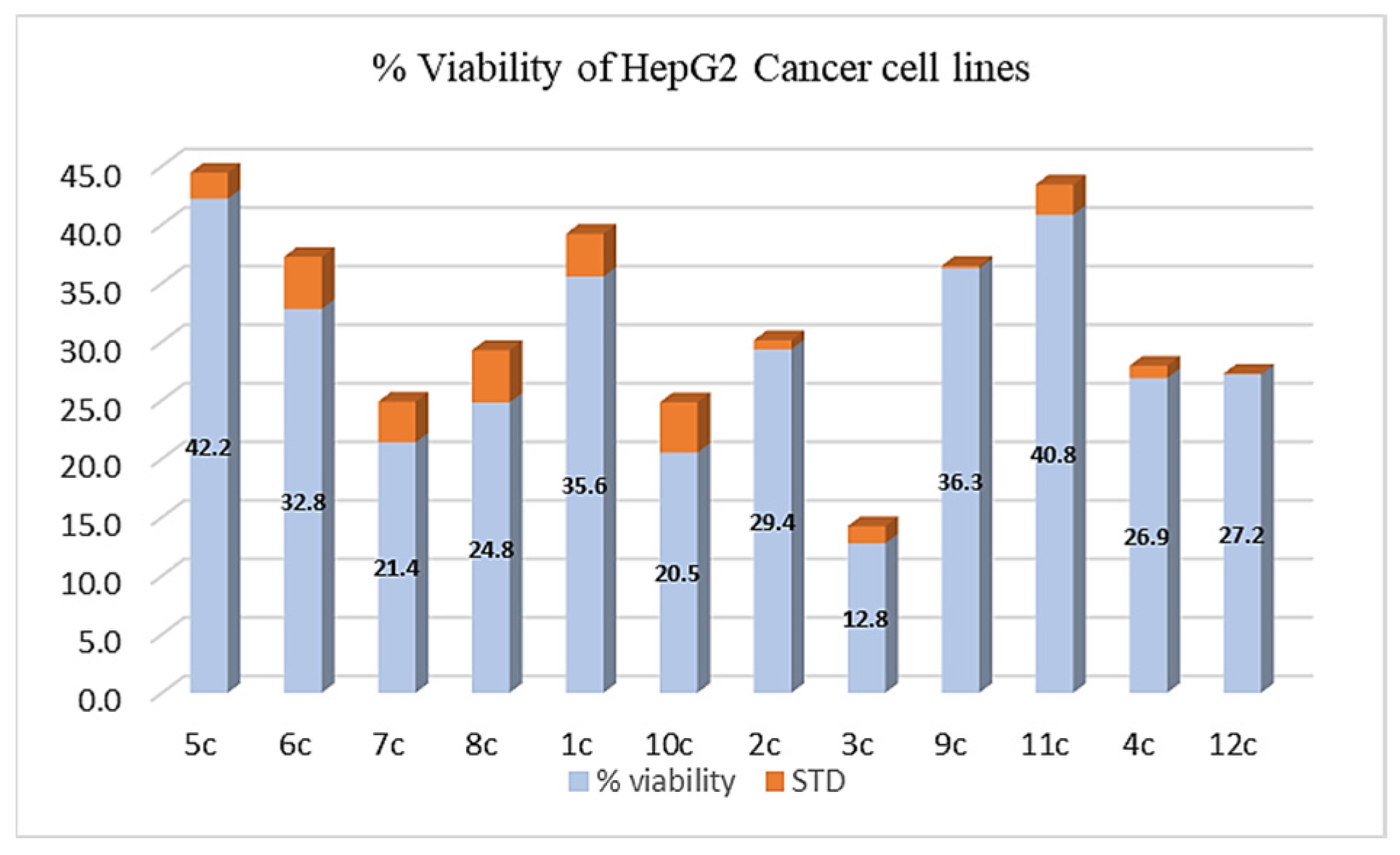
3.6.2. %Viability of the Synthesized Compounds against Human HepG2 Hepatic Cancer Cells
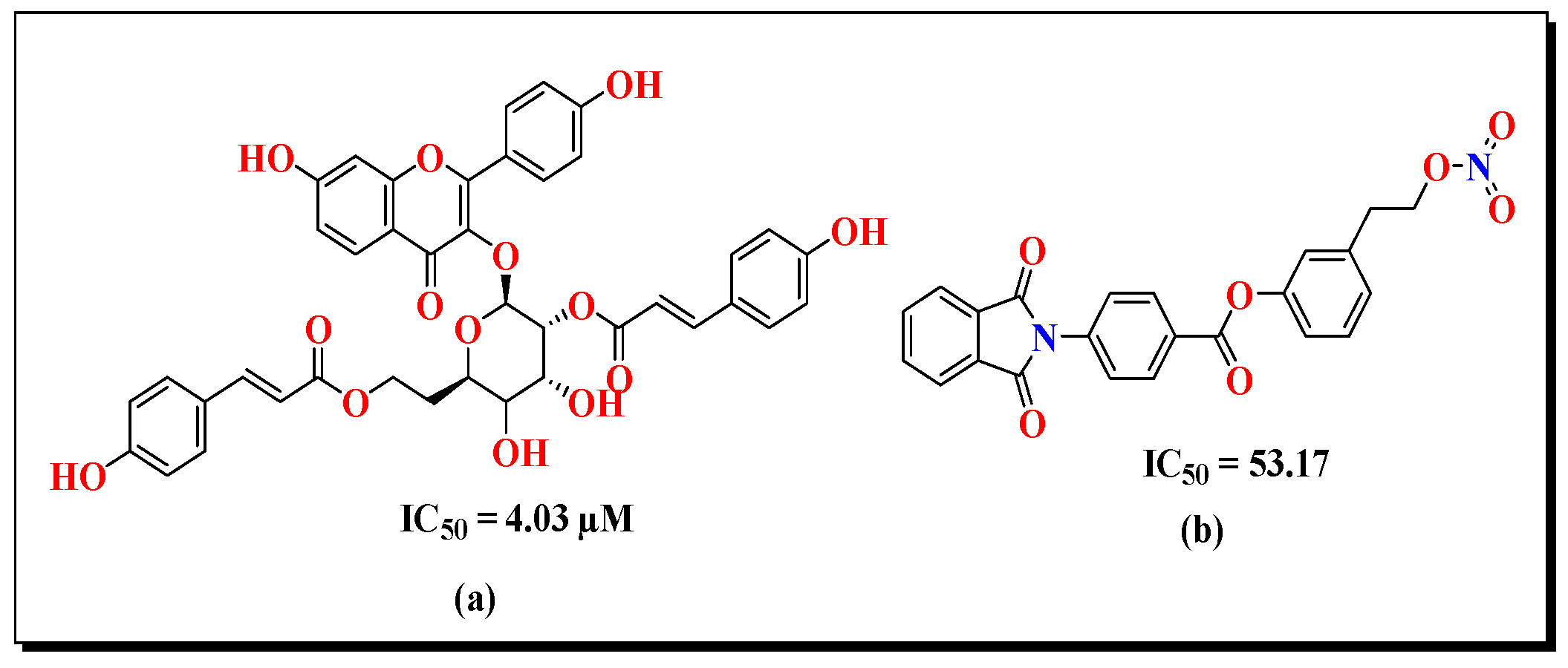
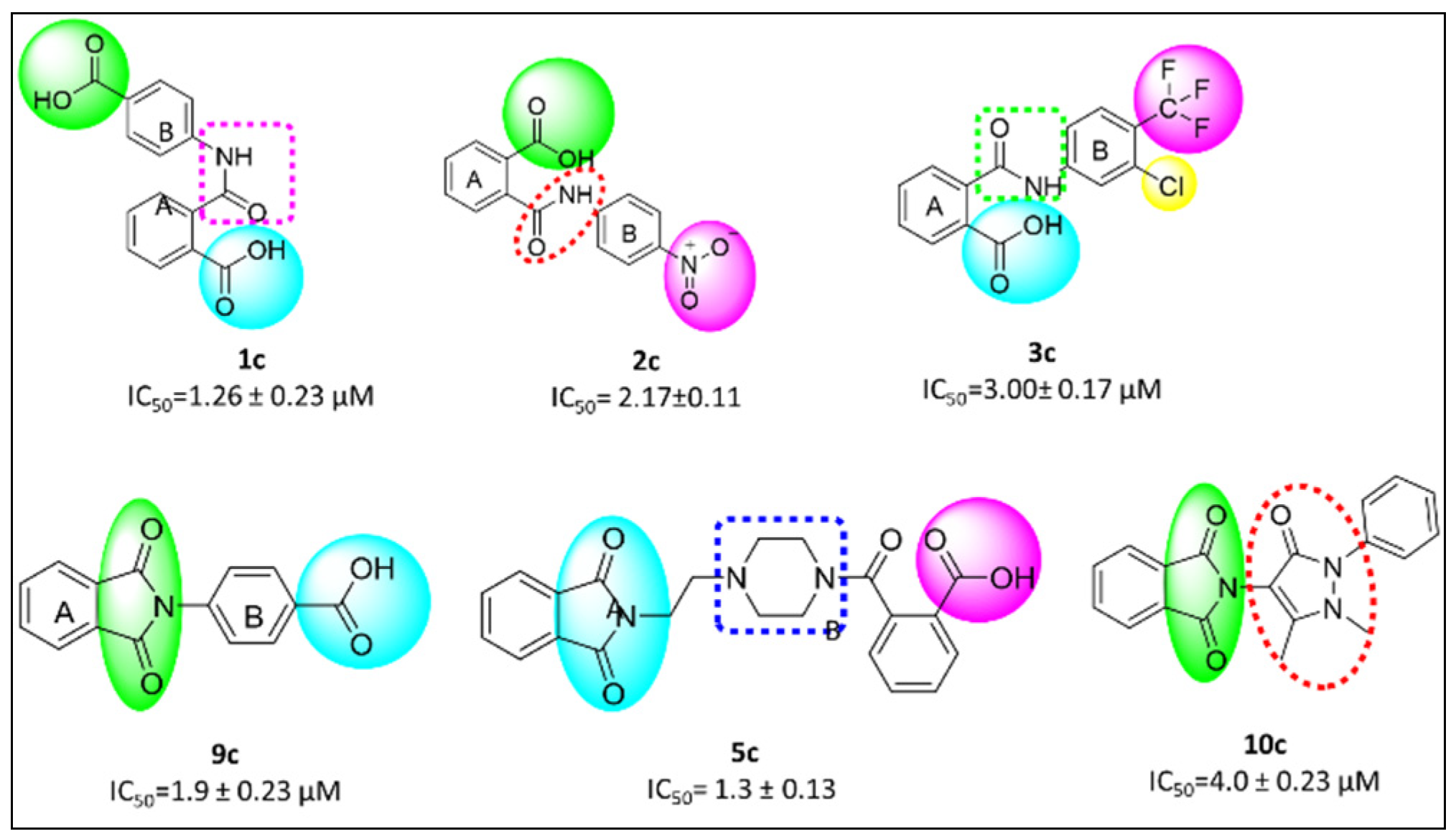
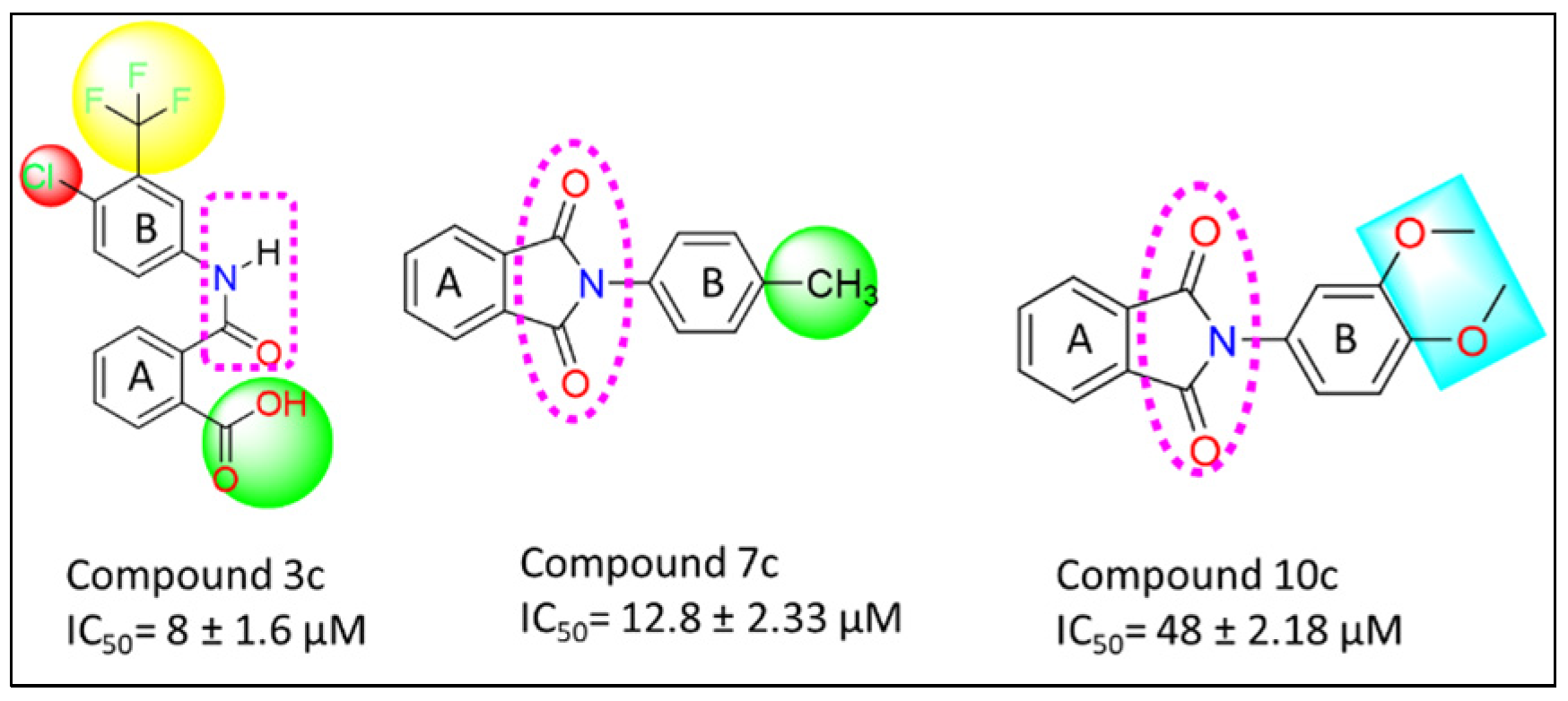
3.7. Structure Activity Relationship (SAR)
4. Materials and Methods
- Synthetic Scheme
- Molecular Docking studies
4.1. Molecular Dynamic Simulations
4.2. In-Vitro Studies
4.2.1. Enzyme Inhibition Assay
4.2.2. Cell Culture and Treatment
- Determination of Cell Viability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Maiti, B.; Chanda, K. Diversity oriented synthesis of benzimidazole-based biheterocyclic molecules by combinatorial approach: A critical review. RSC Adv. 2016, 6, 50384–50413. [Google Scholar] [CrossRef]
- Refaat, H.M. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Han, C.; Zuo, D.; Li, Z.; Zhang, Q.; Zhai, Y.; Jiang, X.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and evaluation of benzimidazole carbamates bearing indole moieties for antiproliferative and antitubulin activities. Eur. J. Med. Chem. 2014, 87, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Daw, P.; Ben-David, Y.; Milstein, D. Direct synthesis of benzimidazoles by dehydrogenative coupling of aromatic diamines and alcohols catalyzed by cobalt. ACS Catal. 2017, 7, 7456–7460. [Google Scholar] [CrossRef]
- Sethi, P.; Bansal, Y.; Bansal, G. Synthesis and PASS-assisted evaluation of coumarin–benzimidazole derivatives as potential anti-inflammatory and anthelmintic agents. Med. Chem. Res. 2018, 27, 61–71. [Google Scholar] [CrossRef]
- Gremse, D.A. Lansoprazole: Pharmacokinetics, pharmacodynamics and clinical uses. Expert Opin. Pharmacother. 2001, 2, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Lavrador-Erb, K.; Ravula, S.B.; Yu, J.; Zamani-Kord, S.; Moree, W.J.; Petroski, R.E.; Wen, J.; Malany, S.; Hoare, S.R.; Madan, A. The discovery and structure–activity relationships of 2-(piperidin-3-yl)-1H-benzimidazoles as selective, CNS penetrating H1-antihistamines for insomnia. Bioorg. Med. Chem. Lett. 2010, 20, 2916–2919. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.K.; Ali, M.A.; Wei, A.C.; Choon, T.S.; Khaw, K.-Y.; Murugaiyah, V.; Osman, H.; Masand, V.H. Synthesis, characterization, and molecular docking analysis of novel benzimidazole derivatives as cholinesterase inhibitors. Bioorg. Chem. 2013, 49, 33–39. [Google Scholar] [CrossRef]
- Verma, S.; Nagarathnam, D.; Shao, J.; Zhang, L.; Zhao, J.; Wang, Y.; Li, T.; Mull, E.; Enyedy, I.; Wang, C. Substituted aminobenzimidazole pyrimidines as cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1973–1977. [Google Scholar] [CrossRef]
- Tantray, M.A.; Khan, I.; Hamid, H.; Alam, M.S.; Dhulap, A.; Kalam, A. Synthesis of benzimidazole-based 1, 3, 4-oxadiazole-1, 2, 3-triazole conjugates as glycogen synthase kinase-3β inhibitors with antidepressant activity in in vivo models. RSC Adv. 2016, 6, 43345–43355. [Google Scholar] [CrossRef]
- Bharadwaj, S.S.; Poojary, B.; Nandish, S.K.M.; Kengaiah, J.; Kirana, M.P.; Shankar, M.K.; Das, A.J.; Kulal, A.; Sannaningaiah, D. Efficient synthesis and in silico studies of the benzimidazole hybrid scaffold with the quinolinyloxadiazole skeleton with potential α-glucosidase inhibitory, anticoagulant, and antiplatelet activities for type-II diabetes mellitus management and treating thrombotic disorders. ACS Omega 2018, 3, 12562–12574. [Google Scholar] [PubMed]
- La Motta, C.; Sartini, S.; Salerno, S.; Simorini, F.; Taliani, S.; Marini, A.M.; Da Settimo, F.; Marinelli, L.; Limongelli, V.; Novellino, E. Acetic acid aldose reductase inhibitors bearing a five-membered heterocyclic core with potent topical activity in a visual impairment rat model. J. Med. Chem. 2008, 51, 3182–3193. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Rahim, F.; Taha, M.; Ullah, H.; Wadood, A.; Nawaz, M.; Khan, F.; Wahab, Z.; Shah, S.A.A.; Rehman, A.U. Synthesis, in vitro urease inhibitory potential and molecular docking study of Benzimidazole analogues. Bioorg. Chem. 2019, 89, 103024. [Google Scholar] [CrossRef]
- Uslu, A.G.; Maz, T.G.; Nocentini, A.; Banoglu, E.; Supuran, C.T.; Çalışkan, B. Benzimidazole derivatives as potent and isoform selective tumor-associated carbonic anhydrase IX/XII inhibitors. Bioorg. Chem. 2020, 95, 103544. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kong, D.; Cheng, H.; Tan, L.; Zhang, Z.; Zhuang, X.; Long, H.; Zhou, Y.; Xu, Y.; Yang, X. New benzimidazole-2-urea derivates as tubulin inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4250–4253. [Google Scholar] [CrossRef] [PubMed]
- Menteşe, E.; Emirik, M.; Sökmen, B.B. Design, molecular docking and synthesis of novel 5, 6-dichloro-2-methyl-1H-benzimidazole derivatives as potential urease enzyme inhibitors. Bioorg. Chem. 2019, 86, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Wang, G.-F.; He, P.-L.; Huang, W.-G.; Zhu, F.-H.; Gao, H.-Y.; Tang, W.; Luo, Y.; Feng, C.-L.; Shi, L.-P. Synthesis and anti-hepatitis B virus activity of novel benzimidazole derivatives. J. Med. Chem. 2006, 49, 4790–4794. [Google Scholar] [CrossRef] [PubMed]
- Welsh, A.; Rylands, L.-i.; Arion, V.B.; Prince, S.; Smith, G.S. Synthesis and antiproliferative activity of benzimidazole-based, trinuclear neutral cyclometallated and cationic, N^ N-chelated ruthenium (ii) complexes. Dalton Trans. 2020, 49, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-H.; Hsu, S.-M.; Kuo, Y.-C.; Liu, C.-Y.; Hsieh, C.-Y.; Twu, Y.-C.; Wang, C.-K.; Wang, Y.-H.; Liao, Y.-J. Treatment with low-dose sorafenib in combination with a novel benzimidazole derivative bearing a pyrolidine side chain provides synergistic anti-proliferative effects against human liver cancer. Rsc Adv. 2017, 7, 16253–16263. [Google Scholar] [CrossRef]
- Dadwal, S.; Kumar, M.; Bhalla, V. “Metal-Free” Nanoassemblies of AIEE-ICT-Active Pyrazine Derivative: Efficient Photoredox System for the Synthesis of Benzimidazoles. J. Org. Chem. 2020, 85, 13906–13919. [Google Scholar] [CrossRef]
- Zuo, M.; Guo, W.; Pang, Y.; Guo, R.; Hou, C.; Sun, S.; Wu, H.; Sun, Z.; Chu, W. Direct synthesis of 2-substituted benzimidazoles via dehydrogenative coupling of aromatic-diamine and primary alcohol catalyzed by a Co complex. New J. Chem. 2020, 44, 14490–14495. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Nguyen, X.-T.T.; Nguyen, T.-L.H.; Tran, P.H. Synthesis of benzoxazoles, benzimidazoles, and benzothiazoles using a Brønsted acidic ionic liquid gel as an efficient heterogeneous catalyst under a solvent-free condition. ACS Omega 2019, 4, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, R.; Liu, Q.; Wang, Z.; Liu, Y.; Wang, Q. Formation of Amidinyl Radicals via Visible-Light-Promoted Reduction of N-Phenyl Amidoxime Esters and Application to the Synthesis of 2-Substituted Benzimidazoles. J. Org. Chem. 2019, 84, 8646–8660. [Google Scholar] [CrossRef] [PubMed]
- Kovvuri, J.; Nagaraju, B.; Kamal, A.; Srivastava, A.K. An efficient synthesis of 2-substituted benzimidazoles via photocatalytic condensation of o-phenylenediamines and aldehydes. ACS Comb. Sci. 2016, 18, 644–650. [Google Scholar] [CrossRef]
- Totir, M.; Echols, N.; Nanao, M.; Gee, C.L.; Moskaleva, A.; Gradia, S.; Iavarone, A.T.; Berger, J.M.; May, A.P.; Zubieta, C. Macro-to-micro structural proteomics: Native source proteins for high-throughput crystallization. PLoS ONE 2012, 7, e32498. [Google Scholar] [CrossRef]
- Badieyan, S.; Bevan, D.R.; Zhang, C. Probing the active site chemistry of β-glucosidases along the hydrolysis reaction pathway. Biochemistry 2012, 51, 8907–8918. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen bond: Its role beyond drug–target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Hashmi, M.A.; Tehseen, Y.; Khan, A.; Khan, S.S.; Iqbal, J.; Perveen, S.; Khan, S.; Farooq, U.; Ahmad, V.U. Antidiabetic flavonol glycosides from Eryngium caeruleum. Rec. Nat. Prod. 2017, 11, 229–234. [Google Scholar]
- Wang, T.; Zhang, Y.H.; Ji, H.; Chen, Y.P.; Peng, S.X. Synthesis and bioactivity of novel phthalimide derivatives. Chin. Chem. Lett. 2008, 19, 26–28. [Google Scholar] [CrossRef]
- Prasad, H.N.; Ananda, A.; Najundaswamy, S.; Nagashree, S.; Mallesha, L.; Dayananda, B.; Jayanth, H.; Mallu, P. Design, synthesis and molecular docking studies of novel piperazine metal complexes as potential antibacterial candidate against MRSA. J. Mol. Struct. 2021, 1232, 130047. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Frisch, A. Gaussian 09W Reference; Gaussian, Inc.: Wallingford, CT, USA, 2009; Volume 470, 25p. [Google Scholar]
- Zaman, Z.; Khan, S.; Nouroz, F.; Farooq, U.; Urooj, A. Targeting protein tyrosine phosphatase to unravel possible inhibitors for Streptococcus pneumoniae using molecular docking, molecular dynamics simulations coupled with free energy calculations. Life Sci. 2021, 264, 118621. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Li, T.; Ahmad Khan, M.K.; Rasul, A.; Nawaz, F.; Sun, M.; Zheng, Y.; Ma, T. Alantolactone induces apoptosis in HepG2 cells through GSH depletion, inhibition of STAT3 activation, and mitochondrial dysfunction. BioMed Res. Int. 2013, 2013, 719858. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆G Kcal/mol | ∆Eelec | ∆Evdw | ∆EGAS (elec + vdw) | PB-SOL (Polar + np) | ∆G(Total) Kcal/mol | |
|---|---|---|---|---|---|---|
| 1c | −7.98 | −6.27 | −23.10 | −29.37 | 11.14 | −18.23 |
| 2c | −7.80 | −7.04 | −23.22 | −30.26 | 12.70 | −17.56 |
| 3c | −8.58 | −7.21 | −22.98 | −30.19 | 10.12 | −20.07 |
| 5c | −7.71 | −6.52 | −20.19 | −26.71 | 11.37 | −14.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khushal, A.; Farooq, U.; Khan, S.; Rasul, A.; Wani, T.A.; Zargar, S.; Shahzad, S.A.; Bukhari, S.M.; Khan, N.A. Bioactivity-Guided Synthesis: In Silico and In Vitro Studies of β-Glucosidase Inhibitors to Cope with Hepatic Cytotoxicity. Molecules 2023, 28, 6548. https://doi.org/10.3390/molecules28186548
Khushal A, Farooq U, Khan S, Rasul A, Wani TA, Zargar S, Shahzad SA, Bukhari SM, Khan NA. Bioactivity-Guided Synthesis: In Silico and In Vitro Studies of β-Glucosidase Inhibitors to Cope with Hepatic Cytotoxicity. Molecules. 2023; 28(18):6548. https://doi.org/10.3390/molecules28186548
Chicago/Turabian StyleKhushal, Aneela, Umar Farooq, Sara Khan, Azhar Rasul, Tanveer A. Wani, Seema Zargar, Sohail Anjum Shahzad, Syed Majid Bukhari, and Nazeer Ahmad Khan. 2023. "Bioactivity-Guided Synthesis: In Silico and In Vitro Studies of β-Glucosidase Inhibitors to Cope with Hepatic Cytotoxicity" Molecules 28, no. 18: 6548. https://doi.org/10.3390/molecules28186548