
Efficiency of the Enzymatic Conversion of Flavone Glycosides Isolated from Carrot Leaves and Anti-Inflammatory Effects of Enzyme-Treated Carrot Leaves

Abstract
:1. Introduction
2. Results and Discussion
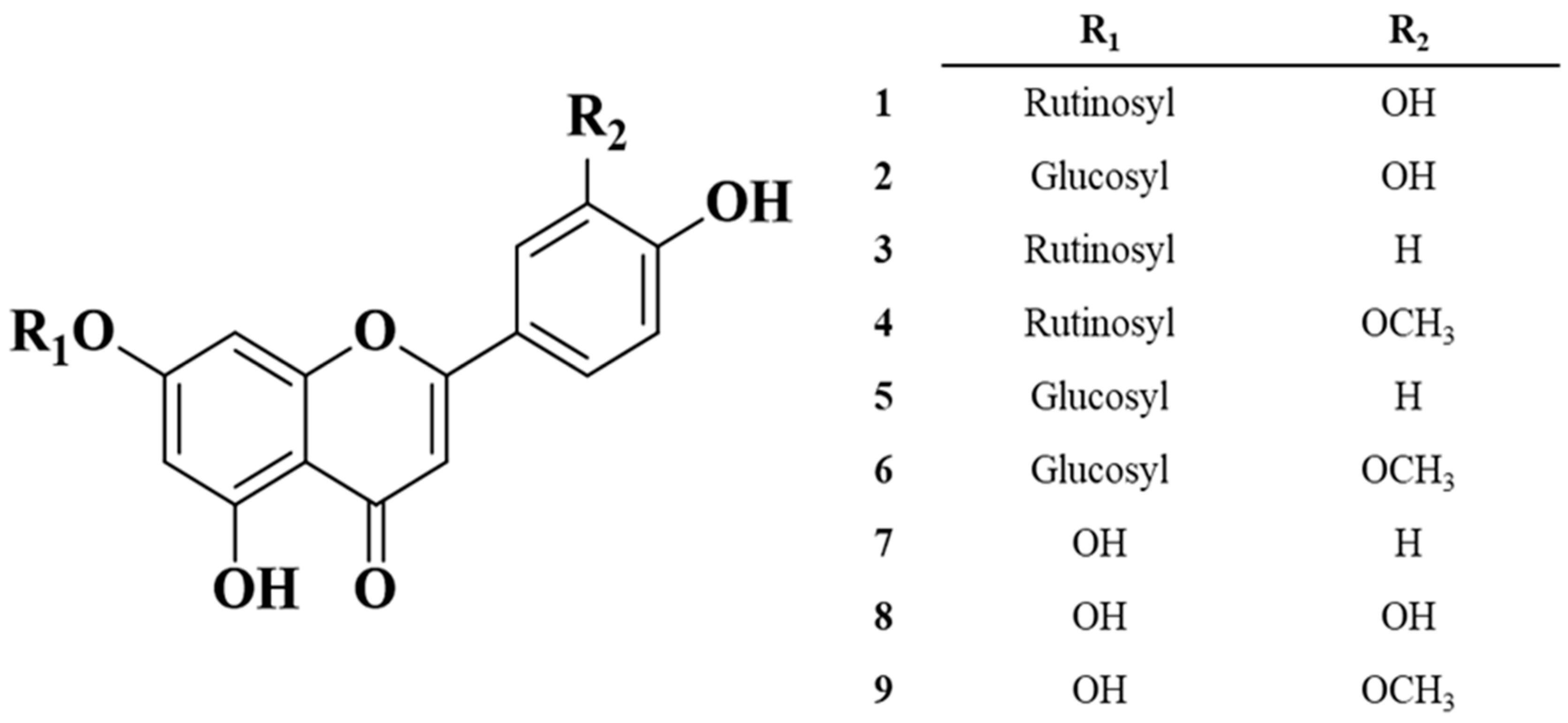
2.1. Structure Elucidation of the Isolated Compounds
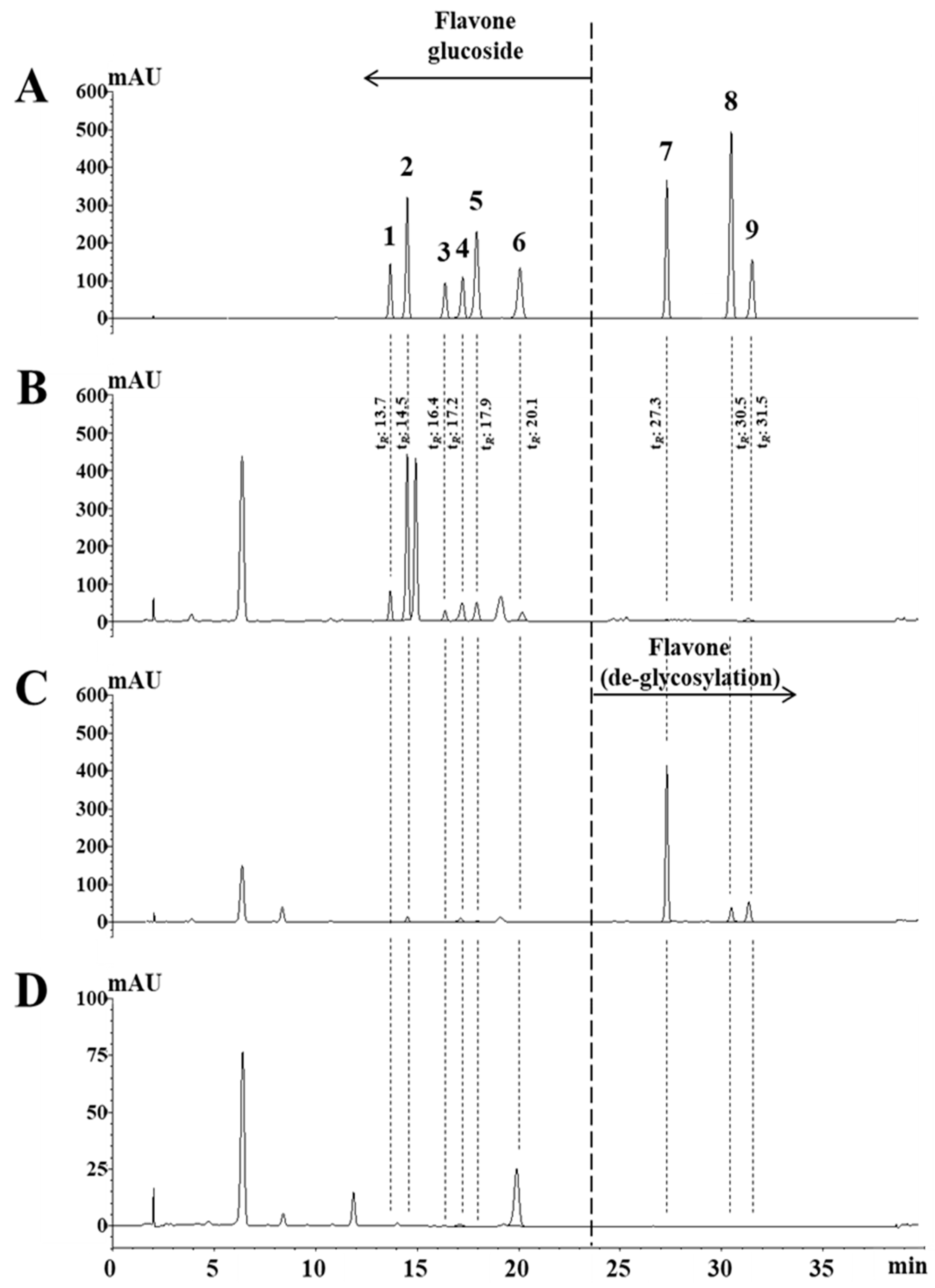
2.2. Optimization of Analytical Conditions and Quantitation
2.3. Validation of the Assay Method
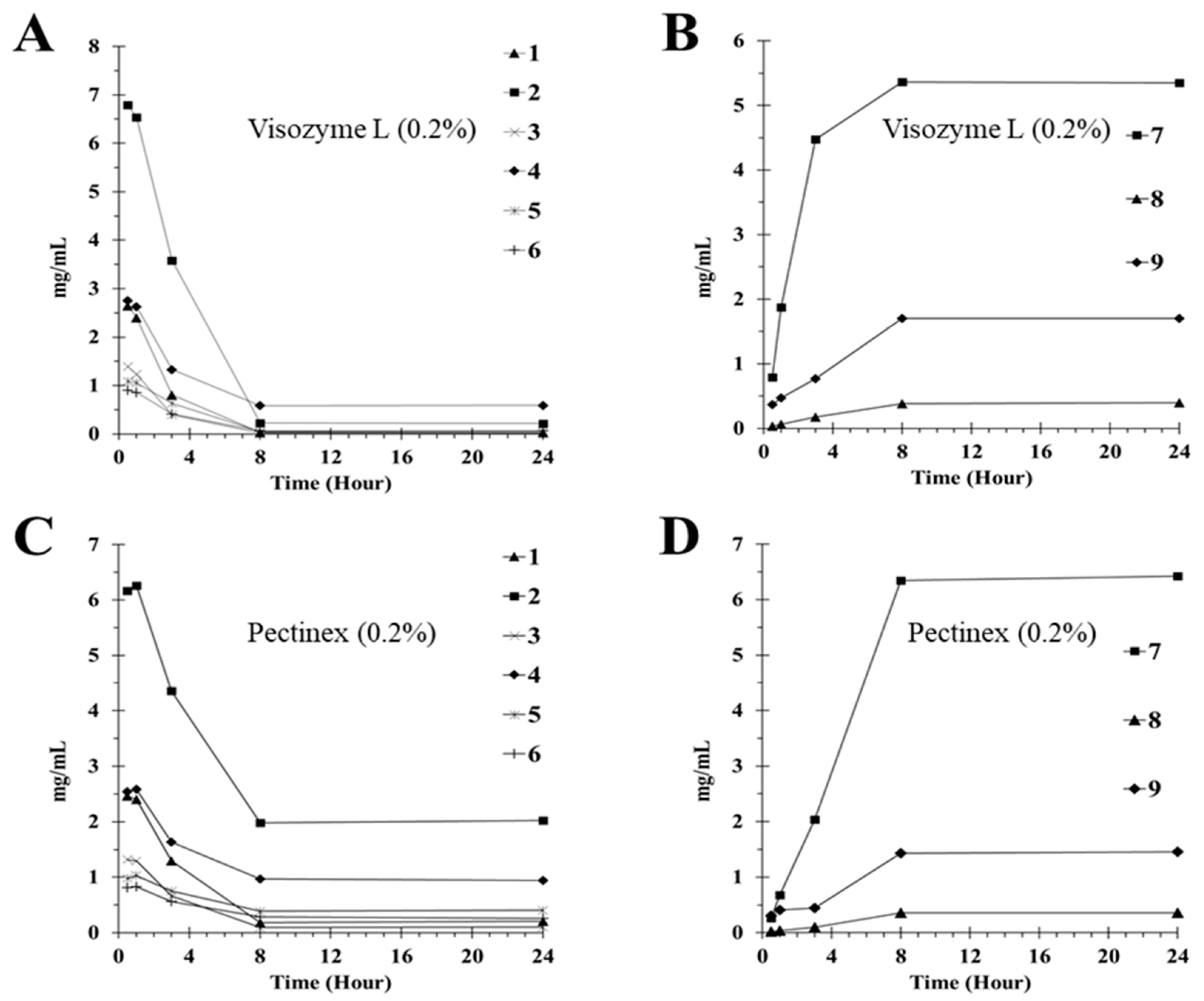
2.4. Deglycosylation of Flavone Glycosides by Enzyme Treatment
2.5. Anti-Inflammatory Activities of the Samples
3. Materials and Methods
3.1. General Procedures
3.2. Sample Preparation
3.3. Separation and Isolation Procedures
3.4. Preparation of Standards and Samples for HPLC Analysis
3.5. Validation of the Developed Analytical Method
3.6. Cell Culture and Reagents
3.7. Reverse Transcription and Real-Time Polymerase Chain Reaction
3.8. Quantitation of TNF-α and IL-2 in Cell Culture Supernatants Using ELISA
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molldrem, K.L.; Li, J.; Simon, P.W.; Tanumihardjo, S.A. Lutein and beta-carotene from lutein-containing yellow carrots are bioavailable in humans. Am. J. Clin. Nutr. 2004, 80, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Mohan, L.; Bharadvaja, N. Disease Prevention and Treatment Using beta-Carotene: The Ultimate Provitamin A. Rev. Bras. Farmacogn. 2022, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Ben-Fadhel, Y.; Maherani, B.; Aragones, M.; Lacroix, M. Antimicrobial Properties of Encapsulated Antimicrobial Natural Plant Products for Ready-to-Eat Carrots. Foods 2019, 8, 535. [Google Scholar] [CrossRef] [PubMed]
- Sodimbaku, V.; Pujari, L.; Mullangi, R.; Marri, S. Carrot (Daucus carota L.): Nephroprotective against gentamicin-induced nephrotoxicity in rats. Indian J. Pharmacol. 2016, 48, 122–127. [Google Scholar] [CrossRef]
- Shebaby, W.N.; El-Sibai, M.; Smith, K.B.; Karam, M.C.; Mroueh, M.; Daher, C.F. The antioxidant and anticancer effects of wild carrot oil extract. Phytother. Res. 2013, 27, 737–744. [Google Scholar] [CrossRef]
- Ma, Z.G.; Kong, X.P.; Liu, L.J.; Ou, C.G.; Sun, T.T.; Zhao, Z.W.; Miao, Z.J.; Rong, J.; Zhuang, F.Y. The unique origin of orange carrot cultivars in China. Euphytica 2016, 212, 37–49. [Google Scholar] [CrossRef]
- Kainulainen, P.; Nissinen, A.; Piirainen, A.; Tiilikkala, K.; Holopainen, J.K. Essential oil composition in leaves of carrot varieties and preference of specialist and generalist sucking insect herbivores. Agric. For. Entomol. 2002, 4, 211–216. [Google Scholar] [CrossRef]
- Srednicka-Tober, D.; Kopczynska, K.; Goralska-Walczak, R.; Hallmann, E.; Baranski, M.; Marszalek, K.; Kazimierczak, R. Are Organic Certified Carrots Richer in Health-Promoting Phenolics and Carotenoids than the Conventionally Grown Ones? Molecules 2022, 27, 4184. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hamauzu, Y. Phenolic compounds and their antioxidant properties in different tissues of carrots (Daucus carota L.). J. Food Agric. Environ. 2004, 2, 95–100. [Google Scholar]
- Tian, C.L.; Liu, X.; Chang, Y.; Wang, R.X.; Lv, T.M.; Cui, C.C.; Liu, M.C. Investigation of the anti-inflammatory and antioxidant activities of luteolin, kaempferol, apigenin and quercetin. S. Afr. J. Bot. 2021, 137, 257–264. [Google Scholar] [CrossRef]
- Jiang, Z.B.; Wang, W.J.; Xu, C.; Xie, Y.J.; Wang, X.R.; Zhang, Y.Z.; Huang, J.M.; Huang, M.; Xie, C.; Liu, P.; et al. Luteolin and its derivative apigenin suppress the inducible PD-L1 expression to improve anti-tumor immunity in KRAS-mutant lung cancer. Cancer Lett. 2021, 515, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Dahuja, A.; Sachdev, A.; Tomar, M.; Lorenzo, J.M.; Dhumal, S.; Chandran, D.; Varghese, E.; Saha, S.; Sairam, K.V.S.S.; et al. Optimization of the use of cellulolytic enzyme preparation for the extraction of health promoting anthocyanins from black carrot using response surface methodology. LWT—Food Sci. Technol. 2022, 163, 113528. [Google Scholar] [CrossRef]
- Zhu, X.F.; Zhang, H.X.; Lo, R. Phenolic compounds from the leaf extract of artichoke (Cynara scolymus L.) and their antimicrobial activities. J. Agr. Food Chem. 2004, 52, 7272–7278. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.C.; Pai, Y.F.; Tsai, T.H. Isolation of Luteolin and Luteolin-7-O-glucoside from Dendranthema morifolium Ramat Tzvel and Their Pharmacokinetics in Rats. J. Agr. Food Chem. 2015, 63, 7700–7706. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Mohammed, A.; Dampuri, G.; Ghanta, R.G.; Raghavan, S.C. Phytochemical and Antimicrobial Studies of Methyl Angolensate and Luteolin-7-O-glucoside Isolated from Callus Cultures of Soymida febrifuga. Int. J. Biomed. Sci. 2007, 3, 269–278. [Google Scholar]
- Wang, M.F.; Simon, J.E.; Aviles, I.F.; He, K.; Zheng, Q.Y.; Tadmor, Y. Analysis of antioxidative phenolic compounds in artichoke (Cynara scolymus L.). J. Agr. Food Chem. 2003, 51, 601–608. [Google Scholar] [CrossRef]
- Plazonic, A.; Bucar, F.; Males, Z.; Mornar, A.; Nigovic, B.; Kujundzic, N. Identification and Quantification of Flavonoids and Phenolic Acids in Burr Parsley (Caucalis platycarpos L.), Using High-Performance Liquid Chromatography with Diode Array Detection and Electrospray Ionization Mass Spectrometry. Molecules 2009, 14, 2466–2490. [Google Scholar] [CrossRef]
- Kumar, K.S.; Sabu, V.; Sindhu, G.; Rauf, A.A.; Helen, A. Isolation, identification and characterization of apigenin from Justicia gendarussa and its anti-inflammatory activity. Int. Immunopharmacol. 2018, 59, 157–167. [Google Scholar] [CrossRef]
- Nickavar, B.; Rezaee, J.; Nickavar, A. Effect-Directed Analysis for the Antioxidant Compound in Salvia verticillata. Iran J. Pharm. Res. 2016, 15, 241–246. [Google Scholar]
- Svehlikova, V.; Bennett, R.N.; Mellon, F.A.; Needs, P.W.; Piacente, S.; Kroon, P.A.; Bao, Y.P. Isolation, identification and stability of acylated derivatives of apigenin 7-O-glucoside from chamomile (Chamomilla recutita [L.] Rauschert). Phytochemistry 2004, 65, 2323–2332. [Google Scholar] [CrossRef]
- Angelini, L.G.G.; Tavarini, S.; Ascrizzi, R.; Flamini, G.; Vannacci, G.; Sarrocco, S. Composition and antifungal activity of the essential oils hydrodistilled from three accessions of pastinocello carrot (Daucus carota L. ssp. major). Ind. Crop Prod. 2022, 181, 114797. [Google Scholar] [CrossRef]
- Leiss, K.A.; Cristofori, G.; van Steenis, R.; Verpoorte, R.; Klinkhamer, P.G.L. An eco-metabolomic study of host plant resistance to Western flower thrips in cultivated, biofortified and wild carrots. Phytochemistry 2013, 93, 63–70. [Google Scholar] [CrossRef]
- Koutouan, C.; Le Clerc, V.; Baltenweck, R.; Claudel, P.; Halter, D.; Hugueney, P.; Hamama, L.; Suel, A.; Huet, S.; Merlet, M.H.B.; et al. Link between carrot leaf secondary metabolites and resistance to Alternaria dauci. Sci. Rep. 2018, 8, 13746. [Google Scholar] [CrossRef]
- Lin, L.Z.; Lu, S.M.; Harnly, J.M. Detection and quantification of glycosylated flavonoid malonates in celery, Chinese celery, and celery seed by LC-DAD-ESI/MS. J. Agr. Food Chem. 2007, 55, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; He, W.N.; Xiang, C.; Han, J.; Wu, L.J.; Guo, D.A.; Ye, M. Qualitative and Quantitative Analyses of Flavonoids in Spirodela polyrrhiza by High-performance Liquid Chromatography Coupled with Mass Spectrometry. Phytochem. Anal. 2011, 22, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.F.; Li, Y.Y.; Jia, Y.S.; Wang, Z.J.; Rong, R.; Bao, J.; Zhao, M.Q.; Fu, Z.H.; Ge, G.T. Comparative Analysis of Major Flavonoids among Parts of Lactuca indica during Different Growth Periods. Molecules 2021, 26, 7445. [Google Scholar] [CrossRef]
- Wang, Y.G.; Fu, J.X.; Zhang, C.; Zhao, H.B. HPLC-DAD-ESI-MS Analysis of Flavonoids from Leaves of Different Cultivars of Sweet Osmanthus. Molecules 2016, 21, 1224. [Google Scholar] [CrossRef]
- Sharma, K.D.; Karki, S.; Thakur, N.S.; Attri, S. Chemical composition, functional properties and processing of carrot-a review. J. Food Sci. Tech. Mys. 2012, 49, 22–32. [Google Scholar] [CrossRef]
- Teubert, H.; Wunscher, G.; Herrmann, K. Flavonols and flavones of vegetables. VIII. Flavones of carrot leaves (author’s transl). Z. Für Lebensm. Unters. Und Forsch. 1977, 165, 147–150. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Naayer, M.; van Beek, T.A.; Capelle, A.; Haaksman, I.K.; van Doren, H.A.; de Groot, A.E. Chemical and enzymatic hydrolysis of anthraquinone glycosides from madder roots. Phytochem. Anal. 2003, 14, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Caro, G.; Ordonez-Diaz, J.L.; de Santiago, E.; Moreno-Ortega, A.; Caceres-Jimenez, S.; Sanchez-Parra, M.; Roldan-Guerra, F.J.; Ortiz-Somovilla, V.; Moreno-Rojas, J.M. Antioxidant Activity and Bio-Accessibility of Polyphenols in Black Carrot (Daucus carota L. ssp. sativus var. atrorubens Alef.) and Two Derived Products during Simulated Gastrointestinal Digestion and Colonic Fermentation. Foods 2021, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Mizui, M. Natural and modified IL-2 for the treatment of cancer and autoimmune diseases. Clin. Immunol. 2019, 206, 63–70. [Google Scholar] [CrossRef]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Gravallese, E.M. Immune-mediated inflammatory disease therapeutics: Past, present and future. Nat. Rev. Immunol. 2021, 21, 680–686. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Grootaert, C.; Capanoglu, E.; Ozkan, C.; Smagghe, G.; Raes, K.; Van Camp, J. Anti-inflammatory potential of black carrot (Daucus carota L.) polyphenols in a co-culture model of intestinal Caco-2 and endothelial EA.hy926 cells. Mol. Nutr. Food Res. 2017, 61, 1600455. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Content (n = 5) | |
|---|---|---|
| DCL | DCR | |
| 1 | 2.7378 ± 0.0295 b | N.D. |
| 2 | 6.6595 ± 0.0105 | N.D. |
| 3 | 1.4797 ± 0.0150 | N.D. |
| 4 | 2.7711 ± 0.0148 | 0.0682 ± 0.0009 |
| 5 | 1.0465 ± 0.0110 | N.D. |
| 6 | 0.8607 ± 0.0128 | 1.0062 ± 0.0082 |
| 7 | 0.0593 ± 0.0004 | N.D. |
| 8 | N.D. a | N.D. |
| 9 | 0.2947 ± 0.0019 | N.D. |
| Compound | tR (min) | Linear Range (mg/mL) | Equation (Linear Model) a | r2 b |
|---|---|---|---|---|
| 1 | 13.7 | 0.0002–0.2 | y = 12,536,160.4409x − 3665.4389 | 0.9998 |
| 2 | 14.5 | 0.0002–0.2 | y = 29,391,463.6658x − 10,361.4359 | 0.9998 |
| 3 | 16.4 | 0.0002–0.2 | y = 10,086,940.3468x − 2969.9097 | 0.9998 |
| 4 | 17.2 | 0.0002–0.2 | y = 12,664,623.4452x − 3672.0329 | 0.9998 |
| 5 | 17.9 | 0.0002–0.2 | y = 30,244,089.0439x − 8460.5572 | 0.9998 |
| 6 | 20.1 | 0.0002–0.2 | y = 20,910,201.0414x − 6370.9364 | 0.9998 |
| 7 | 27.3 | 0.0002–0.2 | y = 31,733,472.2675x − 10,337.5869 | 0.9998 |
| 8 | 30.5 | 0.0002–0.2 | y = 52,480,014.7485x − 16,388.1711 | 0.9998 |
| 9 | – | 0.0002–0.2 | y = 18,491,893.6013x − 5697.0590 | 0.9998 |
| Compound | Spiked (mg/mL) | Inter-Day | Intra-Day | ||
|---|---|---|---|---|---|
| Recovery (%, n = 4) | RSD a (%) | Recovery (%, n = 4) | RSD (%) | ||
| 1 | 1.2 | 95.15 ± 1.25 | 1.31 | 97.10 ± 0.28 | 0.29 |
| 2.4 | 100.73 ± 0.89 | 0.89 | 101.70 ± 0.19 | 0.18 | |
| 3.8 | 97.44 ± 0.33 | 0.33 | 97.26 ± 0.26 | 0.27 | |
| 2 | 1.2 | 103.10 ± 1.95 | 1.89 | 96.38 ± 0.30 | 0.31 |
| 2.4 | 102.71 ± 1.19 | 1.16 | 102.51 ± 1.22 | 1.19 | |
| 3.8 | 94.89 ± 0.45 | 0.48 | 95.44 ± 0.72 | 0.76 | |
| 3 | 1.2 | 101.55 ± 1.54 | 1.51 | 102.77 ± 0.81 | 0.79 |
| 2.4 | 100.67 ± 0.75 | 0.75 | 101.41 ± 0.82 | 0.81 | |
| 3.8 | 101.16 ± 0.43 | 0.42 | 100.97 ± 0.39 | 0.39 | |
| 4 | 1.2 | 97.51 ± 1.72 | 1.76 | 99.17 ± 1.59 | 1.60 |
| 2.4 | 96.94 ± 1.82 | 1.88 | 98.98 ± 1.42 | 1.44 | |
| 3.8 | 97.24 ± 0.55 | 0.56 | 96.42 ± 0.92 | 0.96 | |
| 5 | 1.2 | 103.72 ± 0.72 | 0.70 | 104.03 ± 1.10 | 1.06 |
| 2.4 | 100.57 ± 1.27 | 1.26 | 101.23 ± 0.47 | 0.46 | |
| 3.8 | 102.35 ± 0.48 | 0.47 | 102.62 ± 0.58 | 0.56 | |
| 6 | 1.2 | 104.74 ± 1.60 | 1.52 | 102.52 ± 0.40 | 0.39 |
| 2.4 | 101.60 ± 1.53 | 1.51 | 102.41 ± 1.43 | 1.40 | |
| 3.8 | 102.36 ± 1.13 | 1.11 | 101.05 ± 1.42 | 1.14 | |
| 7 | 1.2 | 102.33 ± 0.98 | 0.96 | 101.51 ± 0.45 | 0.45 |
| 2.4 | 100.75 ± 0.87 | 0.86 | 100.95 ± 0.22 | 0.22 | |
| 3.8 | 102.70 ± 0.23 | 0.23 | 102.48 ± 0.41 | 0.40 | |
| 8 | 1.2 | 105.59 ± 0.47 | 0.45 | 105.97 ± 0.27 | 0.26 |
| 2.4 | 102.74 ± 0.18 | 0.18 | 102.89 ± 0.17 | 0.17 | |
| 3.8 | 103.86 ± 0.09 | 0.09 | 103.86 ± 0.16 | 0.15 | |
| 9 | 1.2 | 105.77 ± 1.66 | 1.57 | 102.43 ± 1.16 | 1.13 |
| 2.4 | 101.15 ± 0.48 | 0.48 | 100.97 ± 0.84 | 0.83 | |
| 3.8 | 103.64 ± 0.61 | 0.58 | 104.51 ± 0.51 | 0.49 | |
| Enzyme | Compound | Compound Contents (mg/g) | ||||
|---|---|---|---|---|---|---|
| 0.5 h | 1 h | 3 h | 8 h | 24 h | ||
| Vis a | 1 | 2.65 ± 0.01 c | 2.40 ± 0.08 | 0.80 ± 0.07 | 0.03 ± 0.00 | 0.03 ± 0.00 |
| 2 | 6.80 ± 0.02 | 6.53 ± 0.23 | 3.58 ± 0.32 | 0.23 ± 0.00 | 0.22 ± 0.00 | |
| 3 | 1.39 ± 0.01 | 1.23 ± 0.04 | 0.39 ± 0.03 | 0.02 ± 0.00 | 0.02 ± 0.00 | |
| 4 | 2.75 ± 0.01 | 2.63 ± 0.09 | 1.33 ± 0.12 | 0.59 ± 0.01 | 0.59 ± 0.10 | |
| 5 | 1.09 ± 0.01 | 1.05 ± 0.03 | 0.63 ± 0.06 | 0.05 ± 0.00 | 0.06 ± 0.03 | |
| 6 | 0.90 ± 0.01 | 0.86 ± 0.02 | 0.41 ± 0.04 | 0.06 ± 0.00 | 0.07 ± 0.04 | |
| 7 | 0.79 ± 0.01 | 1.88 ± 0.06 | 4.47 ± 0.40 | 5.96 ± 0.12 | 6.34 ± 0.09 | |
| 8 | 0.03 ± 0.00 | 0.07 ± 0.00 | 0.18 ± 0.01 | 0.39 ± 0.01 | 0.40 ± 0.05 | |
| 9 | 0.37 ± 0.00 | 0.48 ± 0.01 | 0.77 ± 0.07 | 1.71 ± 0.04 | 1.72 ± 0.03 | |
| 1 | 2.47 ± 0.14 | 2.40 ± 0.11 | 1.30 ± 0.02 | 0.19 ± 0.01 | 0.21 ± 0.01 | |
| Pec b | 2 | 6.16 ± 0.35 | 6.06 ± 0.26 | 4.53 ±0.04 | 1.98 ± 0.19 | 2.02 ±0.12 |
| 3 | 1.32 ± 0.07 | 1.29 ± 0.05 | 0.67 ± 0.01 | 0.09 ± 0.01 | 0.10 ± 0.01 | |
| 4 | 2.54 ± 0.14 | 2.39 ± 0.10 | 1.63 ± 0.01 | 0.96 ± 0.09 | 0.94 ±0.05 | |
| 5 | 0.98 ± 0.05 | 0.82 ± 0.04 | 0.75 ± 0.01 | 0.39 ± 0.04 | 0.40 ± 0.02 | |
| 6 | 0.81 ± 0.05 | 0.83 ± 0.03 | 0.56 ± 0.00 | 0.29 ± 0.02 | 0.26 ± 0.01 | |
| 7 | 0.26 ±0.02 | 0.68 ± 0.03 | 2.04 ± 0.03 | 5.34 ± 0.63 | 5.43 ±0.09 | |
| 8 | 0.02 ± 0.00 | 0.03 ± 0.00 | 0.10 ± 0.00 | 0.35 ± 0.03 | 0.35 ± 0.01 | |
| 9 | 0.31 ± 0.02 | 0.41 ± 0.02 | 0.44 ± 0.01 | 1.43 ± 0.15 | 1.45 ± 0.09 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.T.; Kim, H.J.; Ryuk, J.A.; Jung, D.H.; Ko, B.S. Efficiency of the Enzymatic Conversion of Flavone Glycosides Isolated from Carrot Leaves and Anti-Inflammatory Effects of Enzyme-Treated Carrot Leaves. Molecules 2023, 28, 4291. https://doi.org/10.3390/molecules28114291
Hwang JT, Kim HJ, Ryuk JA, Jung DH, Ko BS. Efficiency of the Enzymatic Conversion of Flavone Glycosides Isolated from Carrot Leaves and Anti-Inflammatory Effects of Enzyme-Treated Carrot Leaves. Molecules. 2023; 28(11):4291. https://doi.org/10.3390/molecules28114291
Chicago/Turabian StyleHwang, Joo Tae, Hye Jin Kim, Jin Ah Ryuk, Dong Ho Jung, and Byoung Seob Ko. 2023. "Efficiency of the Enzymatic Conversion of Flavone Glycosides Isolated from Carrot Leaves and Anti-Inflammatory Effects of Enzyme-Treated Carrot Leaves" Molecules 28, no. 11: 4291. https://doi.org/10.3390/molecules28114291