
Comparative G-Protein-Coupled Estrogen Receptor (GPER) Systems in Diabetic and Cancer Conditions: A Review

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
Methodology
2. GPER System in DM
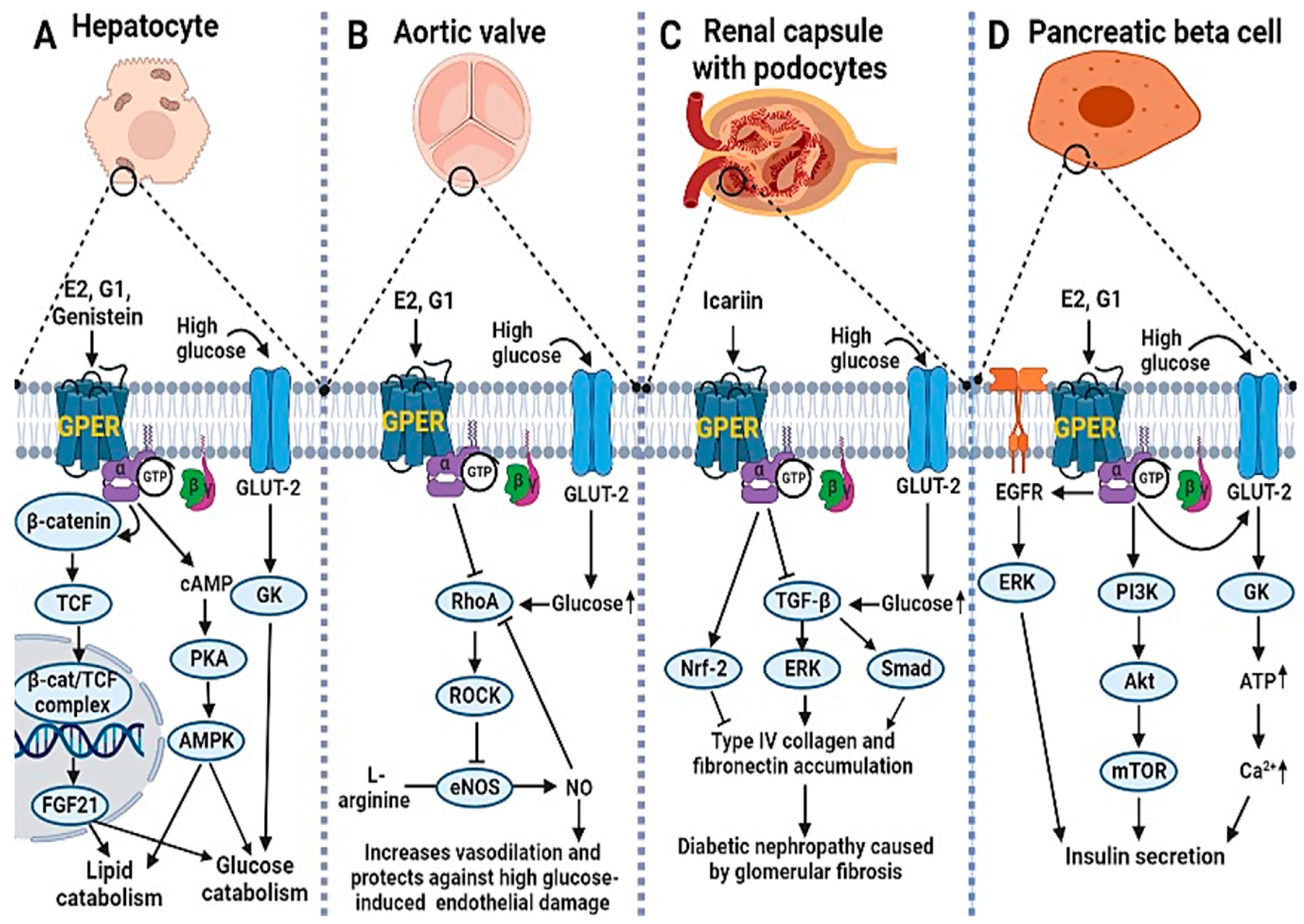
2.1. GPER Deficiency and Signaling in the Pathogenesis of DM and Its Complications
2.2. GPER Signaling in Diabetic Complications: The Protective Roles of Estrogens and Their Mimetics
2.3. Molecular Mechanism of GPER Signaling in DM: The Potential Role of Epigenetic Modifications
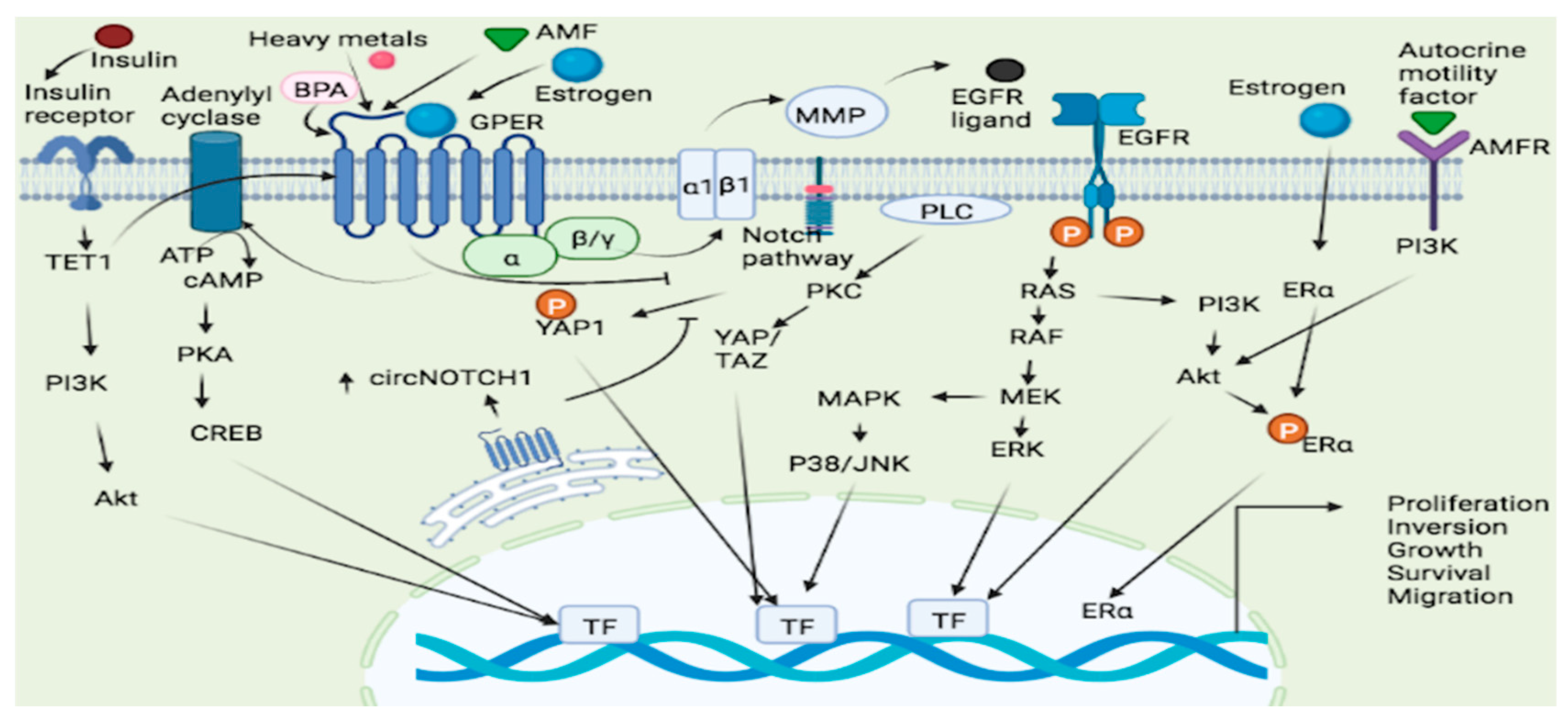
3. GPER System in Various Malignancies
3.1. GPER in Testicular Germ Cell Cancer
3.2. GPER in Breast Cancer
3.2.1. GPER Localization and Predisposition to Breast Cancer Subtype
3.2.2. GPER Target Genes in the Progression of Breast Cancer Malignancies
3.3. GPER in Lung Cancer
3.4. GPER in Gastric Cancer
3.5. GPER in Colon/Colorectal Cancer
3.6. GPER Signaling in Renal, Liver, and Pancreatic Cancer
3.6.1. Renal Cancer
3.6.2. Liver Cancer
3.6.3. Pancreatic Cancer
3.7. GPER in Endometrial Cancer
3.8. GPER in Melanoma Skin Cancer
3.9. GPER in Cervical Cancer

{kind=link}
{kind=link}
| Study-Driven Aim | Methodology | Findings | Conclusion | Reference |
|---|---|---|---|---|
| Evaluate the mutual regulations of estradiol, and prolactin with HBV E6 and E7 oncogenes as well as the expression and localization of the ERα, GPER/GPR30, and PRLR receptors. | qPCR, Western blot, and immunofluorescence | (a) The hormones induced E6 and E7 expressions. (b) E6 and E7 increased the expressions of the receptors (c) Localization: PRLR (cytoplasm) while ERα, ERβ, and GPER (nucleus). | Mutual regulation exists between the hormones and the oncogenes which leads to an increase in the hormonal receptors and hence could influence CC carcinogenesis. | [192] |
| Investigate the expressionand distribution of ER, GPER, androgen receptor (AR), progesterone receptor (PR)A, PRB and connective tissue growth factor (CTGF) in the vaginal wall in CC women treated with radiotherapy. | qPCR and immunohistochemistry | (a) lower expression of ERα in CC survivors compared to the normal women (b) ERα, and AR protein expression in the vaginalMucosa significantly reduced, while GPER expression was not affected in CC survivors (compared to normal women) after radiation. | The expression of ERα, and AR but not GPER reduced following radiation in the CC survivors. | [194] |
| Determine GPER immunopositivity in a cohort of CC patients and investigate its association with other pathological parameters of CC. | Immunohistochemistry | (a) GPER was detected in the tissues of the majority of the CC patients. (b) Cytoplasmic GPER staining was positively correlated to p16 and p53 (non-mutant) and no association was observed between the receptor and HPV E6 and E7. | The study revealed that GPER correlated with HPV-induced tumor suppressor proteins p16 and p53. | [195] |
| Study the role of LSD1 in cervical cancer patients as well as determine its correlation with GPER/GPR30 receptor | Immunochemistry | (a) A disadvantaged 10-year survival was observed in patients that showed strong LSD1 expressions. (b) A negative correlation between LSD1 and GPER/GPR30 was observed. | Epigenetic changes that could be mediated through the LSD1 and the GPER/GPR30 could serve as an important marker in understanding the expression pattern of the LSD1. | [196] |
| To study the effects and related mechanisms of MEHP –induced proliferation of CC | Western blotting, Knockdown techniques, and immunofluorescence | (a) MEHP induced the proliferation of the HeLa and SiHa cells as well as increased the phosphorylation and nuclear localization of Akt in the CC cells (b) GPER knockdown reversed the EHP-induced cell proliferation o | Activation of the GPER/Akt system via the action of MEHP could influence the progression of CC. | [197] |
| To investigate the possible role of GPER in WRO and FRO TC cell lines | Western blotting, MTT, Transwell, ELISA, BrdU incorporation assays | (a) GPER was expressed in the TC cell lines. (b) GPER stimulation was reported to be the initial step for the activation of ERK and AKT pathways, nuclear translocation of NF-kB, and subsequent activation of downstream genes. | GPER signaling pathway (in addition to other related pathways) plays an important role in the progression of TC. | [198] |
| To access the gene and protein expressions of GPER in PTC and non-malignant thyroid tissues. | qPCR, In silico, and immunohistochemistry | (a) low expression of GPER in PTC which could be associated with BRAF mutation | GPER has a role in PTC development and progression. | [199] |
3.10. GPER in Thyroid Cancer
3.11. GPER in Ovarian Carcinoma
3.12. GPER in Prostate Cancer
4. GPER System in Cardiovascular and Other Pathological Conditions
4.1. Cardiovascular Diseases
4.2. Reproductive Disorders
4.3. Nervous Disorders
4.4. Immune-Related Diseases
5. Comparative Perspectives of the Dysregulated GPER System as a Therapeutic Target in DM and Malignancy
6. The Sex-Dependent Function of GPER
7. Pharmacological Interventions on GPER System
7.1. Diabetes Mellitus
7.2. Malignancies
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
List of Abbreviations
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| E2 | 17β-estradiol |
| G1 | A synthetic and selective GPER agonist |
| ATF-4 | Activating transcription factor 4 |
| AMPK | Adenosine 5′-monophosphate-activated protein kinase |
| ATP | Adenosine triphosphate |
| Akt | Akt strain transforming protein/protein kinase B |
| ABCG2 | ATP binding cassette subfamily G member 2 |
| AMF | Autocrine Motility Factor |
| Bcl-2 | B-cell lymphoma 2 |
| BPA | Bisphenol A |
| CHOP | C/EBP Homologous Protein |
| CREB | cAMP response element binding protein |
| CAFs | Carcinoma-associated fibroblast |
| c-FOS | Cellular oncogene |
| CC | Cervical Cancer |
| CTGF | Connective Tissue Growth Factor |
| cAMP | Cyclic adenosine monophosphate |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CYP19A1 | Cytochrome P450 Family 19 Subfamily A Member 1 |
| DGK | Diacylglycerol Kinase |
| DIO | Diet-induced obese |
| DOX | Doxorubicin |
| EGR-1 | Early growth response protein 1 |
| EDCs | Endocrine disruptors |
| eNOS | Endothelial nitric oxide synthase |
| EGFR | Epidermal growth factor receptor |
| ERα | Estrogen receptor α |
| ERβ | Estrogen receptor β |
| ECM | Extracellular Matrix |
| ERK | Extracellular signal-Regulated Kinase |
| FASN | Fatty acid synthase |
| FGF21 | Fibroblast growth factor 21 |
| FAK | Focal adhesion kinase |
| FSHR | Follicle Stimulating Hormone |
| GPER | G-protein-coupled estrogen receptor |
| GC | Gastric cancer |
| GDM | Gestational diabetes mellitus |
| GK | Glucokinase |
| GRP78 | Glucose regulated protein-78 |
| GLUT-2 | Glucose transporter 2 |
| HOTAIR | HOX antisense intergenic RNA |
| HLA | Human Leukocyte Antigen |
| HPV | Human papillomavirus |
| OHT | Hydroxytamoxifen |
| HIF-1 α | Hypoxia-inducible factor 1-alpha |
| LH/FSH | Luteinizing hormone/ follicle stimulating hormone |
| LSD1 | Lysine Specific Demethylase 1 |
| mTOR | Mammalian target of rapamycin |
| MMPs | Matrix Metalloproteinases |
| MEK, | Mitogen-activated protein kinase kinase |
| miRNA | Micro Ribonucleic Acid, microRNAs |
| MAPK | Mitogen-activated protein kinase |
| MEHP | Mono-2- Ethylhexyl phthalate |
| NHERF1 | Na+/H+ exchanger regulatory factor 1 |
| Notch 1 | Neurogenic locus notch homolog protein 1 |
| NOS | Nitric oxide synthase |
| NO | Nitric oxide |
| NSCLC | Non-Small Cell Lung Cancer |
| STX | Novel tamoxifen analogue |
| Nrf-2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | Nuclear Factor kappa- B |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| PIT | Pancreatic islet transplant |
| TAZ | PDZ-binding domain |
| PLC | Phospholipase C |
| PI3K | Phosphatidyl inositol 3-kinase |
| PERK | PKR-like endoplasmic reticulum kinase |
| PKA | Protein kinase A |
| QKI | Quaking homolog/KH domain RNA binding |
| RAF | Rapidly Accelerated Fibrosarcoma |
| RhoA | Ras homolog family member A |
| RAS, | Rat sarcoma |
| ROCK | Rho-associated protein kinase |
| RUNX1 | Runt-related transcription factor 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| SNP | Single-Nucleotide Polymorphism |
| SRC | SRC Proto-Oncogene |
| Smad | Suppressor of mother against decapentaplegi |
| TGCC | Testicular germ cell cancer |
| TET1 | Tetmethylcytosine dioxygenase 1 |
| TC | Thyroid cancer |
| TCF7L2 | Transcription factor 7-like 2 |
| TG | Triglyceride |
| TNBC | Triple-negative breast cancer |
| T1D | Type 1 Diabetic |
| T2DM | Type 2 Diabetes Mellitus |
| VEGF-A | Vascular Endothelial Growth Factor A |
| VEGF | Vascular endothelial growth factor |
| β-cat | β-catenin |
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2021, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes-Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathmann, W.; Kostev, K. Association of glucose-lowering drugs with incident stroke and transient ischaemic attacks in primary care patients with type 2 diabetes: Disease analyzer database. Acta Diabetol. 2022, 59, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.J.; Li, L.; Kim, J. Racial and ethnic disparities in gastric cancer outcomes: More important than surgical technique? World J. Gastroenterol. WJG 2014, 20, 11546. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Casadei-Gardini, A.; Ulivi, P.; Arechederra, M.; Berasain, C.; Lollini, P.; Fernández-Barrena, M.G.; Avila, M.A. Epigenetic mechanisms in gastric cancer: Potential new therapeutic opportunities. Int. J. Mol. Sci. 2020, 21, 5500. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef]
- Tian, S.; Zhan, N.; Li, R.; Dong, W. Downregulation of G Protein-Coupled Estrogen Receptor (GPER) is Associated with Reduced Prognosis in Patients with Gastric Cancer. J. Pharmacol. Exp. Ther. 2019, 25, 3115–3126. [Google Scholar] [CrossRef]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front. Endocrinol. 2020, 11, 563165. [Google Scholar] [CrossRef]
- Liang, S.; Chen, Z.; Jiang, G.; Zhou, Y.; Liu, Q.; Su, Q.; Wei, W.; Du, J.; Wang, H. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-κB/IL-6 signals. Cancer Lett. 2017, 386, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.; Wong, J.M.L.; Sim, Y.J.; Wong, S.S.; Elhassan, S.A.M.; Tan, S.H.; Lim, J.P.L.; Tay, N.W.R.; Annan, N.C.; Bhattamisra, S.K.; et al. Type 1 and 2 diabetes mellitus: A review on current treatment approach and gene therapy as potential intervention. Diabetes Metab. Syndr. 2019, 13, 364–372. [Google Scholar] [CrossRef] [PubMed]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Prossnitz, E.R. Targeting the G protein-coupled estrogen receptor (GPER) in obesity and diabetes. Endocr. Metab. Sci. 2021, 2, 100080. [Google Scholar] [CrossRef]
- Barton, M.; Prossnitz, E.R. Early life stress determines insulin signalling in adulthood. J. Physiol. 2020, 598, 427–428. [Google Scholar] [CrossRef]
- Barton, M.; Prossnitz, E.R. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol. Metab. 2015, 26, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Mauvais-Jarvis, F.; Prossnitz, E.R. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J. Steroid Biochem. Mol. Biol. 2018, 176, 31–37. [Google Scholar] [CrossRef]
- Kooptiwut, S.; Mahawong, P.; Hanchang, W.; Semprasert, N.; Kaewin, S.; Limjindaporn, T.; Yenchitsomanus, P.-T. Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic β-cell death. J. Steroid Biochem. Mol. Biol. 2013, 139, 25–32. [Google Scholar] [CrossRef]
- Liu, S.; Le May, C.; Wong, W.P.; Ward, R.D.; Clegg, D.J.; Marcelli, M.; Korach, K.S.; Mauvais-Jarvis, F. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 2009, 58, 2292–2302. [Google Scholar] [CrossRef]
- Liu, S.; Mauvais-Jarvis, F. Rapid, nongenomic estrogen actions protect pancreatic islet survival. Islets 2009, 1, 273–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Pan, Z.; Yang, X. Key genes and co-expression network analysis in the livers of type 2 diabetes patients. J. Diabetes Investig. 2018, 10, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Prossnitz, E.R. G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. Diabetes Obes. 2017, 1043, 427–453. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E.R. Mechanisms of Estradiol-Induced Insulin Secretion by the G Protein-Coupled Estrogen Receptor GPR30/GPER in Pancreatic β-Cells. Endocrinology 2011, 152, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J.; et al. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci. Transl. Med. 2020, 12, 528. [Google Scholar] [CrossRef] [PubMed]
- Williams, G. Aromatase up-regulation, insulin and raised intracellular oestrogens in men, induce adiposity, metabolic syndrome and prostate disease, via aberrant ER-α and GPER signalling. Mol. Cell. Endocrinol. 2012, 351, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Babiloni-Chust, I.; Dos Santos, R.S.; Medina-Gali, R.M.; Perez-Serna, A.A.; Encinar, J.-A.; Martinez-Pinna, J.; Gustafsson, J.-A.; Marroqui, L.; Nadal, A. G protein-coupled estrogen receptor activation by bisphenol-A disrupts the protection from apoptosis conferred by the estrogen receptors ERα and ERβ in pancreatic beta cells. Environ. Int. 2022, 164, 107250. [Google Scholar] [CrossRef]
- Nadal, A.; Fuentes, E.; Ripoll, C.; Villar-Pazos, S.; Castellano-Muñoz, M.; Soriano, S.; Martinez-Pinna, J.; Quesada, I.; Alonso-Magdalena, P. Extranuclear-initiated estrogenic actions of endocrine disrupting chemicals: Is there toxicology beyond paracelsus? J. Steroid Biochem. Mol. Biol. 2018, 176, 16–22. [Google Scholar] [CrossRef]
- Meyer, M.R.; Barton, M. GPER blockers as Nox downregulators: A new drug class to target chronic non-communicable diseases. J. Steroid Biochem. Mol. Biol. 2018, 176, 82–87. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Hathaway, H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015, 153, 114–126. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Hu, C.; Brigman, J.L.; Zhu, G.; Hathaway, H.J.; Prossnitz, E.R. GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State. Endocrinology 2013, 154, 4136–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuasa, T.; Takata, Y.; Aki, N.; Kunimi, K.; Satoh, M.; Nii, M.; Izumi, Y.; Otoda, T.; Hashida, S.; Osawa, H.; et al. Insulin receptor cleavage induced by estrogen impairs insulin signaling. BMJ Open Diabetes Res. Care 2021, 9, e002467. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.S.; Cau, S.; Silva, M.; Manzato, C.P.; Mestriner, F.L.A.C.; Matsumoto, T.; Carneiro, F.; Tostes, R.C. Diabetes impairs the vascular effects of aldosterone mediated by G protein-coupled estrogen receptor activation. Front. Pharmacol. 2015, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, J.; Veit, M.; Zingg, S.; Di Santo, S.; Traupe, T.; Yang, Z.; Völzmann, J.; Dubey, R.K.; Christen, S.; Baumgartner, I. Estrogen Receptor-α But Not -β or GPER Inhibits High Glucose-Induced Human VSMC Proliferation: Potential Role of ROS and ERK. J. Clin. Endocrinol. Metab. 2011, 96, 220–228. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Cirillo, F.; Vivacqua, A.; Malaguarnera, R.; Belfiore, A.; Maggiolini, M. Novel Aspects Concerning the Functional Cross-Talk between the Insulin/IGF-I System and Estrogen Signaling in Cancer Cells. Front. Endocrinol. 2015, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Gigoux, V.; Fourmy, D. Acting on Hormone Receptors with Minimal Side Effect on Cell Proliferation: A Timely Challenge Illustrated with GLP-1R and GPER. Front. Endocrinol. 2013, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Kilic, G.; Meyers, M.S.; Navarro, G.; Wang, Y.; Oberholzer, J.; Mauvais-Jarvis, F. Oestrogens improve human pancreatic islet transplantation in a mouse model of insulin deficient diabetes. Diabetologia 2012, 56, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Bian, C.; Bai, B.; Gao, Q.; Li, S.; Zhao, Y. 17β-Estradiol Regulates Glucose Metabolism and Insulin Secretion in Rat Islet β Cells Through GPER and Akt/mTOR/GLUT2 Pathway. Front. Endocrinol. 2019, 10, 531. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, Y.; Wang, H.; Jiang, Z.; Ma, H. Genistein accelerates glucose catabolism via activation the GPER-mediated cAMP/PKA-AMPK signaling pathway in broiler chickens. Life Sci. 2022, 303, 120676. [Google Scholar] [CrossRef]
- Badakhshi, Y.; Shao, W.; Liu, D.; Tian, L.; Pang, J.; Gu, J.; Hu, J.; Jin, T. Estrogen-Wnt signaling cascade regulates expression of hepatic fibroblast growth factor 21. Am. J. Physiol. Metab. 2021, 321, E292–E304. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cheng, L.; Liang, H.; Duan, W.; Hu, J.; Zhi, W.; Yang, J.; Liu, Z.; Zhao, M.; Liu, J. GPER inhibits diabetes-mediated RhoA activation to prevent vascular endothelial dysfunction. Eur. J. Cell Biol. 2016, 95, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Ding, X.S.; Li, H.M.; Zhang, C. Icariin attenuates high glucose-induced type IV collagen and fibronectin accumulation in glomerular mesangial cells by inhibiting transforming growth factor-β production and signalling through G protein-coupled oestrogen receptor 1. Clin. Exp. Pharmacol. Physiol. 2013, 40, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Ye, W.; Li, S.; Wang, H.; Ding, X. Icariin modulates mitochondrial function and apoptosis in high glucose-induced glomerular podocytes through G protein-coupled estrogen receptors. Mol. Cell. Endocrinol. 2018, 473, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zheng, X.; Pan, Z.; Yao, W.; Gao, X.; Wang, X.; Ding, X. Icariin Prevents Extracellular Matrix Accumulation and Ameliorates Experimental Diabetic Kidney Disease by Inhibiting Oxidative Stress via GPER Mediated p62-Dependent Keap1 Degradation and Nrf2 Activation. Front. Cell Dev. Biol. 2020, 8, 559. [Google Scholar] [CrossRef]
- Taheri, M.; Shoorei, H.; Dinger, M.E.; Ghafouri-Fard, S. Perspectives on the Role of Non-Coding RNAs in the Regulation of Expression and Function of the Estrogen Receptor. Cancers 2020, 12, 2162. [Google Scholar] [CrossRef]
- Vidal-Gómez, X.; Pérez-Cremades, D.; Mompeon, A.; Dantas, A.P.; Novella, S.; Hermenegildo, C. MicroRNA as Crucial Regulators of Gene Expression in Estradiol-Treated Human Endothelial Cells. Cell. Physiol. Biochem. 2018, 45, 1878–1892. [Google Scholar] [CrossRef]
- Ma, Y.; Xia, W.; Wang, D.Q.; Wan, Y.J.; Xu, B.; Chen, X.; Li, Y.Y.; Xu, S.Q. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia 2013, 56, 2059–2067. [Google Scholar] [CrossRef]
- Treviño, L.S.; Dong, J.; Kaushal, A.; Katz, T.A.; Jangid, R.K.; Robertson, M.J.; Grimm, S.L.; Ambati, C.S.R.; Putluri, V.; Cox, A.R.; et al. Epigenome environment interactions accelerate epigenomic aging and unlock metabolically restricted epigenetic reprogramming in adulthood. Nat. Commun. 2020, 11, 2316. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Sabir, S.; Rehman, K. Bisphenol A-induced metabolic disorders: From exposure to mechanism of action. Environ. Toxicol. Pharmacol. 2020, 77, 103373. [Google Scholar] [CrossRef]
- Hagobian, T.A.; Bird, A.; Stanelle, S.; Williams, D.; Schaffner, A.; Phelan, S. Pilot Study on the Effect of Orally Administered Bisphenol A on Glucose and Insulin Response in Nonobese Adults. J. Endocr. Soc. 2019, 3, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Pjanic, M. The role of polycarbonate monomer bisphenol-A in insulin resistance. PeerJ 2017, 5, e3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Ding, D.; Wang, T.; Liu, Q.; Lin, Y. MiR-338 controls BPA-triggered pancreatic islet insulin secretory dysfunction from compensation to decompensation by targeting Pdx-1. FASEB J. 2017, 31, 5184–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knabl, J.; Hiden, U.; Hüttenbrenner, R.; Riedel, C.; Hutter, S.; Kirn, V.; Günthner-Biller, M.; Desoye, G.; Kainer, F.; Jeschke, U. GDM Alters Expression of Placental Estrogen Receptor α in a Cell Type and Gender-Specific Manner. Reprod. Sci. 2015, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Zhou, X.; Hu, W.; Chu, Z.; Ruan, Q.; Zhang, H.; Li, M.; Zhang, H.; Huang, X.; Yao, P. Pterostilbene accelerates wound healing by modulating diabetes-induced estrogen receptor β suppression in hematopoietic stem cells. Burn. Trauma 2021, 9, tkaa045. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.R.; Liu, D. Anti-diabetic functions of soy isoflavone genistein: Mechanisms underlying its effects on pancreatic β-cell function. Food Funct. 2012, 4, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Gan, M.; Shen, L.; Wang, S.; Guo, Z.; Zheng, T.; Tan, Y.; Fan, Y.; Liu, L.; Chen, L.; Jiang, A.; et al. Genistein inhibits high fat diet-induced obesity through miR-222 by targeting BTG2 and adipor1. Food Funct. 2020, 11, 2418–2426. [Google Scholar] [CrossRef]
- Rago, V.; Romeo, F.; Giordano, F.; Maggiolini, M.; Carpino, A. Identification of the estrogen receptor GPER in neo-plastic and non-neoplastic human testes. Reprod. Biol. Endocrinol. 2011, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, N.; Paul-Bellon, R.; Camparo, P.; Michiels, J.-F.; Chevallier, D.; Fénichel, P. Genetic Variants of GPER/GPR30, a Novel Estrogen-Related G Protein Receptor, Are Associated with Human Seminoma. Int. J. Mol. Sci. 2014, 15, 1574–1589. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, N.; Bouskine, A.; Fenichel, P. Bisphenol A promotes testicular seminoma cell proliferation through GPER/GPR30. Int. J. Cancer 2011, 130, 241–242. [Google Scholar] [CrossRef]
- Bouskine, A.; Nebout, M.; Brücker-Davis, F.; Benahmed, M.; Fenichel, P. Low Doses of Bisphenol A Promote Human Seminoma Cell Proliferation by Activating PKA and PKG via a Membrane G-Protein–Coupled Estrogen Receptor. Environ. Health Perspect. 2009, 117, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallacides, A.; Chesnel, A.; Ajj, H.; Chillet, M.; Flament, S.; Dumond, H. Estrogens promote proliferation of the seminoma-like TCam-2 cell line through a GPER-dependent ERα36 induction. Mol. Cell. Endocrinol. 2012, 350, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; Passaro, C.; Gigantino, V.; Perdonà, S.; Franco, R.; Portella, G.; Chieffi, S.; Chieffi, P. High Levels of Gpr30 Protein in Human Testicular Carcinoma In Situ and Seminomas Correlate with Low Levels of Estrogen Receptor-Beta and Indicate a Switch in Estrogen Responsiveness. J. Cell. Physiol. 2014, 230, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Boscia, F.; Gigantino, V.; Marra, L.; Esposito, F.; Ferrara, D.; Pariante, P.; Botti, G.; Caraglia, M.; Minucci, S.; et al. GPR30 is overexpressed in post-puberal testicular germ cell tumors. Cancer Biol. Ther. 2011, 11, 609–613. [Google Scholar] [CrossRef] [Green Version]
- Bandini, E.; Fanini, F. MicroRNAs and Androgen Receptor: Emerging Players in Breast Cancer. Front. Genet. 2019, 10, 203. [Google Scholar] [CrossRef]
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell. Endocrinol. 2020, 506, 110762. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Santolla, M.F.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Galli, G.R.; Talia, M.; Muglia, L.; Pellegrino, M.; Nohata, N.; et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.-H.; Chu, N.-M.; Lin, Y.-F.; Kao, S.-H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2018, 174, 121–127. [Google Scholar] [CrossRef]
- Weißenborn, C.; Ignatov, T.; Ochel, H.-J.; Costa, S.D.; Zenclussen, A.; Ignatova, Z.; Ignatov, A. GPER functions as a tumor suppressor in triple-negative breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 713–723. [Google Scholar] [CrossRef]
- Castillo-Sanchez, R.; Ramirez-Ricardo, J.; Martinez-Baeza, E.; Cortes-Reynosa, P.; Candanedo-Gonzales, F.; Gomez, R.; Salazar, E.P. Bisphenol A induces focal adhesions assembly and activation of FAK, Src and ERK2 via GPER in MDA-MB-231 breast cancer cells. Toxicol. In Vitro 2020, 66, 104871. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, Z.; Meng, R.; Tao, T.; Wang, Q.; Zhao, C.; Liu, H.; Song, R.; Zheng, J.; Qin, Q.; et al. NHERF1 inhibits proliferation of triple-negative breast cancer cells by suppressing GPER signaling. Oncol. Rep. 2017, 38, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgert, R.; Emons, G.; Gründker, C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol. Rep. 2016, 37, 1212–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Li, J.; Pan, F.; Zhang, B.; Yao, Y. The activation of GPER inhibits cells proliferation, invasion and EMT of triple-negative breast cancer via CD151/miR-199a-3p bio-axis. AM. J. Transl Res. 2020, 12, 32. [Google Scholar] [PubMed]
- Chen, Z.-J.; Wei, W.; Jiang, G.-M.; Liu, H.; Yang, X.; Wu, Y.-M.; Liu, H.; Wong, C.K.; Du, J.; Wang, H.-S. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol. Oncol. 2016, 10, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.G.; Lebot, M.N.; Sukkarn, B.; Ball, G.; Green, A.R.; Rakha, E.A.; Ellis, I.O.; Storr, S.J. Low expression of G protein-coupled oestrogen receptor 1 (GPER) is associated with adverse survival of breast cancer patients. Oncotarget 2018, 9, 25946–25956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, W.K.; Dennis, M.K.; Hu, C.; Dai, D.; Arterburn, J.B.; Smith, H.O.; Hathaway, H.J.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor-Selective Ligands Modulate Endometrial Tumor Growth. Obstet. Gynecol. Int. 2013, 2013, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöström, M.; Hartman, L.; Grabau, R.; Fornander, T.; Malmström, P.; Nordenskjöld, B.; Sgroi, D.C.; Skoog, L.; Stål, O.; Leeb-Lundberg, L.M.F.; et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res. Treat. 2014, 145, 61–71. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Meisel, A.; Varga, Z.; Fink, D.; Imesch, P. The G Protein-Coupled Estrogen Receptor (GPER) Is Expressed in Two Different Subcellular Localizations Reflecting Distinct Tumor Properties in Breast Cancer. PLoS ONE 2014, 9, e83296. [Google Scholar] [CrossRef] [Green Version]
- Molina, L.; Figueroa, C.D.; Bhoola, K.D.; Ehrenfeld, P. GPER-1/GPR30 a novel estrogen receptor sited in the cell membrane: Therapeutic coupling to breast cancer. Expert Opin. Ther. Targets 2017, 21, 755–766. [Google Scholar] [CrossRef]
- Tutzauer, J.; Sjöström, M.; Bendahl, P.-O.; Rydén, L.; Fernö, M.; Leeb-Lundberg, L.M.F.; Alkner, S. Plasma membrane expression of G protein-coupled estrogen receptor (GPER)/G protein-coupled receptor 30 (GPR30) is associated with worse outcome in metachronous contralateral breast cancer. PLoS ONE 2020, 15, e0231786. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, M.; Vivacqua, A.; Perrotta, I.; Pisano, A.; Aquila, S.; Abonante, S.; Gasperi-Campani, A.; Pezzi, V.; Maggiolini, M. The nuclear localization signal is required for nuclear GPER translocation and function in breast Cancer-Associated Fibroblasts (CAFs). Mol. Cell. Endocrinol. 2013, 376, 23–32. [Google Scholar] [CrossRef]
- Pupo, M.; Bodmer, A.; Berto, M.; Maggiolini, M.; Dietrich, P.-Y.; Picard, D. A genetic polymorphism repurposes the G-protein coupled and membrane-associated estrogen receptor GPER to a transcription factor-like molecule promoting paracrine signaling between stroma and breast carcinoma cells. Oncotarget 2017, 8, 46728–46744. [Google Scholar] [CrossRef] [Green Version]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef] [Green Version]
- Pietras, R.J.; Márquez-Garbán, D.C. Membrane-Associated Estrogen Receptor Signaling Pathways in Human Cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [Green Version]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef]
- Wang, D.; Hu, L.; Zhang, G.; Zhang, L.; Chen, C. G protein-coupled receptor 30 in tumor development. Endocrine 2010, 38, 29–37. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Lappano, R.; Santolla, M.F.; Pupo, M.; Sinicropi, M.S.; Caruso, A.; Rosano, C.; Maggiolini, M. MIBE acts as antagonist ligand of both estrogen receptor α and GPER in breast cancer cells. Breast Cancer Res. 2012, 14, 1–13. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor re-ceptor through release of HB-EGF. J. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Quinn, J.A.; Graeber, C.T.; Frackelton, A.R.; Kim, M. Schwarzbauer JE, Filardo EJ. Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. J. Mol. Endocrinol. 2009, 23, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 2011, 159, 212–218. [Google Scholar] [CrossRef]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- Whyte, J.; Bergin, O.; Bianchi, A.; McNally, S.; Martin, F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009, 11, 209. [Google Scholar] [CrossRef]
- Li, Y.; Birnbaumer, L.; Teng, C.T. Regulation of ERRα gene expression by estrogen receptor agonists and antagonists in SKBR3 breast cancer cells: Differential molecular mechanisms mediated by G protein-coupled receptor GPR30/GPER-1. J. Mol. Endocrinol. 2010, 24, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Madeo, A.; Maggiolini, M. Nuclear alternate estrogen receptor GPR30 mediates 17β-estradiol–induced gene expression and migration in breast cancer–associated fibroblasts. Cancer Res. 2010, 70, 6036–6046. [Google Scholar] [CrossRef] [Green Version]
- Vivacqua, A.; Romeo, E.; De Marco, P.; De Francesco, E.M.; Abonante, S.; Maggiolini, M. GPER mediates the Egr-1 expression induced by 17β-estradiol and 4-hydroxitamoxifen in breast and endometrial cancer cells. Breast Cancer Res. Treat. 2011, 133, 1025–1035. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal Adhesion Kinase Fine Tunes Multifaced Signals toward Breast Cancer Progression. Cancers 2021, 13, 645. [Google Scholar] [CrossRef]
- Castillo, S.R.; Gomez, R.; Perez Salazar, E. Bisphenol A induces migration through a GPER-, FAK-, Src-, and ERK2-dependent pathway in MDA-MB-231 breast cancer cells. Chem. Res. Toxicol. 2016, 29, 285–295. [Google Scholar] [CrossRef]
- Tsai, C.-L.; Wu, H.-M.; Lin, C.-Y.; Lin, Y.-J.; Chao, A.; Wang, T.-H.; Hsueh, S.; Lai, C.-H.; Wang, H.-S. Estradiol and Tamoxifen Induce Cell Migration through GPR30 and Activation of Focal Adhesion Kinase (FAK) in Endometrial Cancers with Low or without Nuclear Estrogen Receptor α (ERα). PLoS ONE 2013, 8, e72999. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Luo, H.; Yang, G.; Yu, T.; Luo, S.; Wu, C.; Sun, Y.; Liu, M.; Tu, G. GPER-mediated proliferation and estradiol production in breast cancer-associated fibroblasts. Endocr.-Relat. Cancer 2014, 21, 355–369. [Google Scholar] [CrossRef] [Green Version]
- De Marco, P.; Bartella, V.; Vivacqua, A.; Lappano, R.; Santolla, M.F.; Morcavallo, A.; Pezzi, V.; Belfiore, A.; Maggiolini, M. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene. 2013, 32, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Duursen, M.B.D.; Nijmeijer, S.; Morree, E.D.; Jong, P.C.D.; Van den Berg, M. Genistein induces breast cancer-associated aromatase and stimulates estrogen-dependent tumor cell growth in in vitro breast cancer model. Toxicology 2011, 289, 67–73. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, M.; Yang, L.; Tu, G.; Zhu, Q.; Chen, M.; Cheng, H.; Luo, H.; Fu, W.; Li, Z.; et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: A new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res. 2015, 17, 1–18. [Google Scholar]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1α (HIF-1α) in breast cancer cells and cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.W.; Maggiolini, M.; et al. Tamoxifen through GPER upregulates aromatase expression: A novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 2014, 146, 273–285. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; He, H.; Chen, Q. Estradiol induces HOTAIR levels via GPER-mediated miR-148a inhibition in breast cancer. J. Transl. Med. 2015, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Lappano, R.; De Marco, P.; Pupo, M.; Vivacqua, A.; Sisci, D.; Abonante, S.; Iacopetta, D.; Cappello, A.R.; Dolce, V.; et al. G Protein-coupled Estrogen Receptor Mediates the Up-regulation of Fatty Acid Synthase Induced by 17β-Estradiol in Cancer Cells and Cancer-associated Fibroblasts. J. Biol. Chem. 2012, 287, 43234–43245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivacqua, A.; Sebastiani, A.; Miglietta, A.M.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Santolla, M.F.; Lappano, R.; Giordano, F.; et al. miR-338-3p Is Regulated by Estrogens through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts (CAFs). Cells 2018, 7, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Greve, B.; Pavao, M.S.; Kiesel, L.; Ibrahim, S.A.; Götte, M. Syndecan-1 modulates β-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. FEBS J. 2013, 280, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of Triple-Negative Breast Cancer Cells Relies upon Coordinate Autocrine Expression of the Proinflammatory Cytokines IL-6 and IL-8Tandem Expression of IL-6 and IL-8 Are Critical for TNBCs. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Wang, S.; Wang, Z.; Feng, X.; Liu, P.; Lv, X.; Li, F.; Yu, F.; Sun, Y.; Yuan, H.; et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J. Clin. Investig. 2019, 125, 2123–2135. [Google Scholar] [CrossRef]
- Kim, K.M.; Jung, J. Upregulation of G protein-coupled estrogen receptor by Chrysin-nanoparticles inhibits tumor proliferation and metastasis in triple negative breast Cancer Xenograft model. Front. Endocrinol. 2020, 11, 560605. [Google Scholar] [CrossRef]
- Benson, C.S.; Babu, S.D.; Radhakrishna, S.; Selvamurugan, N.; Sankar, B.R. Expression of matrix metalloproteinases in human breast cancer tissues. Dis. Markers 2013, 34, 395–405. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000, 16, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, R.; Efroni, S. MicroRNA regulation of molecular pathways as a generic mechanism and as a core disease phenotype. Oncotarget 2015, 6, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bade, B.C.; Cruz, C.S.D. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jala, V.R.; Radde, B.N.; Haribabu, B.; Klinge, C.M. Enhanced expression of G-protein coupled estrogen receptor (GPER/GPR30) in lung cancer. BMC Cancer 2012, 12, 624. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, C.; Zhou, L.; Huang, J. G protein-coupled oestrogen receptor promotes cell growth of non-small cell lung cancer cells via YAP1/QKI/circNOTCH1/m6A methylated NOTCH1 signalling. J. Cell. Mol. Med. 2020, 25, 284–296. [Google Scholar] [CrossRef]
- Huff, M.O.; Todd, S.L.; Smith, A.L.; Elpers, J.T.; Smith, A.P.; Murphy, R.D.; Bleser-Shartzer, A.S.; Hoerter, J.E.; Radde, B.N.; Klinge, C.M. Arsenite and Cadmium Activate MAPK/ERK via Membrane Estrogen Receptors and G-Protein Coupled Estrogen Receptor Signaling in Human Lung Adenocarcinoma Cells. Toxicol. Sci. 2016, 152, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.-S.; Chen, H.-Q.; Chen, Y.-S.; Qiu, K.-F.; Zheng, X.-B.; Li, G.-C.; Yang, H.-D.; Wen, C.-J. Bisphenol A stimulates human lung cancer cell migration via upregulation of matrix metalloproteinases by GPER/EGFR/ERK1/2 signal pathway. Biomed. Pharmacother. 2014, 68, 1037–1043. [Google Scholar] [CrossRef]
- Sheng, Z.-G.; Huang, W.; Liu, Y.-X.; Zhu, B.-Z. Bisphenol A at a low concentration boosts mouse spermatogonial cell proliferation by inducing the G protein-coupled receptor 30 expression. Toxicol. Appl. Pharmacol. 2013, 267, 88–94. [Google Scholar] [CrossRef]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.; Ladeira, C.; Zeferino, S.; Dias, A.; Faria, I.; Cristovam, E.; Gomes, M.; Ribeiro, E. Cytotoxic and genotoxic effects of environmental relevant concentrations of bisphenol A and interactions with doxorubicin. Mutat. Res. Toxicol. Environ. Mutagen. 2018, 838, 28–36. [Google Scholar] [CrossRef]
- Słowikowski, B.K.; Lianeri, M.; Jagodziński, P.P. Exploring estrogenic activity in lung cancer. Mol. Biol. Rep. 2016, 44, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Konings, G.; Reynaert, N.; Delvoux, B.; Verhamme, F.; Bracke, K.; Brusselle, G.; Romano, A.; Vernooy, J. Increased levels of enzymes involved in local estradiol synthesis in chronic obstructive pulmonary disease. Mol. Cell. Endocrinol. 2017, 443, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-H.; Chu, N.-M.; Kao, S.-H. Estrogen, Estrogen Receptor and Lung Cancer. Int. J. Mol. Sci. 2017, 18, 1713. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liao, Y.; Fan, S.; Fu, X.; Xiong, J.; Zhou, S.; Zou, M.; Wang, J. G-Protein-Coupled Estrogen Receptor Antagonist G15 Decreases Estrogen-Induced Development of Non-Small Cell Lung Cancer. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2019, 27, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zali, H.; Rezaei-Tavirani, M.; Azodi, M. Gastric cancer: Prevention, risk factors and treatment. Gastroenterol. Hepatol. Bed Bench 2011, 4, 175–185. [Google Scholar]
- Agudo, A.; Cayssials, V.; Bonet, C.; Tjønneland, A.; Overvad, K.; Boutron-Ruault, M.-C.; Affret, A.; Fagherazzi, G.; Katzke, V.; Schübel, R.; et al. Inflammatory potential of the diet and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Am. J. Clin. Nutr. 2018, 107, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Kim, T.W.; Park, G.L.; Hwang, Y.S.; Cho, H.J.; Kim, J.-T.; Lee, J.-T.K.; Amp, H.G. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic effect via ER stress signaling in gastric cancer. BMB Rep. 2019, 52, 647–652. [Google Scholar] [CrossRef] [Green Version]
- Xu, E.; Xia, X.; Jiang, C.; Li, Z.; Yang, Z.; Zheng, C.; Wang, X.; Du, S.; Miao, J.; Wang, F.; et al. GPER1 Silencing Suppresses the Proliferation, Migration, and Invasion of Gastric Cancer Cells by Inhibiting PI3K/AKT–Mediated EMT. Front. Cell Dev. Biol. 2020, 8, 591239. [Google Scholar] [CrossRef]
- Ulhaq, Z.S.; Soraya, G.V.; Milliana, A.; Tse, W.K.F. Association between GPER gene polymorphisms and GPER expression levels with cancer predisposition and progression. Heliyon 2021, 7, e06428. [Google Scholar] [CrossRef]
- Paschke, S.; Jafarov, S.; Staib, L.; Kreuser, E.-D.; Maulbecker-Armstrong, C.; Roitman, M.; Holm, T.; Harris, C.C.; Link, K.-H.; Kornmann, M. Are Colon and Rectal Cancer Two Different Tumor Entities? A Proposal to Abandon the Term Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 2577. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K. A Review of Colorectal Cancer in Terms of Epidemiology, Risk Factors, Development, Symptoms and Diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V. Colorectal Cancer: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2021, 9, 1858. [Google Scholar] [CrossRef] [PubMed]
- Accarie, A.; Toth, J.; Wauters, L.; Farré, R.; Tack, J.; Vanuytsel, T. Estrogens Play a Critical Role in Stress-Related Gastrointestinal Dysfunction in a Spontaneous Model of Disorders of Gut–Brain Interaction. Cells 2022, 11, 1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gong, X.; Yang, X.; Shang, X.; Du, Q.; Liao, Q.; Xie, R.; Chen, Y.; Xu, J. The roles of estrogen and estrogen receptors in gastrointestinal disease (Review). Oncol. Lett. 2019, 18, 5673–5680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielińska, M.; Fichna, J.; Bashashati, M.; Habibi, S.; Sibaev, A.; Timmermans, J.-P.; Storr, M. G protein-coupled estrogen receptor and estrogen receptor ligands regulate colonic motility and visceral pain. Neurogastroenterol. Motil. 2017, 29, e13025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Jiang, F.; Jiang, Z.; Liu, C.; Li, L.; Luo, Y.; Lu, R.; Mu, Y.; Xue, B. G protein-coupled estrogen receptor is involved in modulating colonic motor function via nitric oxide release in C57BL/6 female mice. Neurogastroenterol. Motil. 2015, 28, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; Liu, H.; Du, J.; Wang, H. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bubb, M.; Beyer, A.-S.L.; Dasgupta, P.; Kaemmerer, D.; Sänger, J.; Evert, K.; Wirtz, R.M.; Schulz, S.; Lupp, A. Assessment of G Protein-Coupled Oestrogen Receptor Expression in Normal and Neoplastic Human Tissues Using a Novel Rabbit Monoclonal Antibody. Int. J. Mol. Sci. 2022, 23, 5191. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Morelos-Garnica, L.; García-Vázquez, J.; Bello, M.; Padilla-Martínez, I.; Fragoso-Vázquez, M.; González, A.D.; De Pedro, N.; Gómez-Vidal, J.; Mendoza-Figueroa, H.; et al. Modifications on the Tetrahydroquinoline Scaffold Targeting a Phenylalanine Cluster on GPER as Antiproliferative Compounds against Renal, Liver and Pancreatic Cancer Cells. Pharmaceuticals 2021, 14, 49. [Google Scholar] [CrossRef]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.-Z.; Yan, R.-L.; Huang, J.-W.; Li, F.-L.; Zhong, Y.-X.; Chen, Y.; Liu, F.-N.; Hu, B.; Huang, S.-B.; Yin, L.-H. Activation of G protein coupled estrogen receptor (GPER) promotes the migration of renal cell carcinoma via the PI3K/AKT/MMP-9 signals. Cell Adhes. Migr. 2018, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D.; Ding, Q.; Hussain, Y.; Limbird, L.E.; Pickering, J.G.; Gros, R. Aldosterone mediates metastatic spread of renal cancer via the G protein-coupled estrogen receptor (GPER). FASEB J. 2016, 30, 2086–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.-A.; Xiong, J.; Fu, Q.; Dong, Y.; Liu, M.; Peng, M.; Jin, W.; Zhou, L.; Xu, X.; Huang, X.; et al. GPER-Induced ERK Signaling Decreases Cell Viability of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 638171. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Shi, H. Estradiol and Estrogen Receptor Agonists Oppose Oncogenic Actions of Leptin in HepG2 Cells. PLoS ONE 2016, 11, e0151455. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Chen, W.; Wen, L.; Zhang, J.; Zhang, Q.; Yang, J.; Liu, H.; Chen, B.W.; Zhou, Y.; Feng, X.; et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett. 2016, 382, 195–202. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Rice, A.; Thorpe, S.D.; Robinson, B.; Yeldag, G.; Lee, D.A.; Ghemtio, L.; Rombouts, K.; Hernández, A.E.D.R. Tamoxifen mechanically deactivates hepatic stellate cells via the G protein-coupled estrogen receptor. Oncogene 2018, 38, 2910–2922. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Radde, B.N.; Litchfield, L.M.; Ivanova, M.M.; Prough, R.A.; Clark, B.J.; Doll, M.A.; Hein, D.W.; Klinge, C.M. Dehydroepiandrosterone Activation of G-protein-coupled Estrogen Receptor Rapidly Stimulates MicroRNA-21 Transcription in Human Hepatocellular Carcinoma Cells. J. Biol. Chem. 2015, 290, 15799–15811. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.; Cortes, E.; Lachowski, D.; Oertle, P.; Matellan, C.; Thorpe, S.D.; Ghose, R.; Wang, H.; Lee, D.A.; Plodinec, M.; et al. GPER Activation Inhibits Cancer Cell Mechanotransduction and Basement Membrane Invasion via RhoA. Cancers 2020, 12, 289. [Google Scholar] [CrossRef] [Green Version]
- Natale, C.A.; Li, J.; Pitarresi, J.R.; Norgard, R.J.; Dentchev, T.; Capell, B.C.; Seykora, J.T.; Stanger, B.Z.; Ridky, T.W. Pharmacologic Activation of the G Protein–Coupled Estrogen Receptor Inhibits Pancreatic Ductal Adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 868–880.e1. [Google Scholar] [CrossRef]
- Pein, M.; Oskarsson, T. Tamoxifen calms down the distressed PDAC stroma. EMBO Rep. 2018, 20, e47334. [Google Scholar] [CrossRef] [PubMed]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.; Lieberthal, T.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF -1A and reduces cancer cell survival. EMBO Rep. 2019, 20, e46557. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jia, Y.; Bian, Y.; Tong, H.; Qu, J.; Wang, K.; Wan, X.-P. Autocrine motility factor promotes endometrial cancer progression by targeting GPER-1. Cell Commun. Signal. 2019, 17, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filigheddu, N.; Sampietro, S.; Chianale, F.; Porporato, P.E.; Gaggianesi, M.; Gregnanin, I.; Rainero, E.; Ferrara, M.; Perego, B.; Riboni, F.; et al. Diacylglycerol kinase α mediates 17-β-estradiol-induced proliferation, motility, and anchorage-independent growth of Hec-1A endometrial cancer cell line through the G protein-coupled estrogen receptor GPR30. Cell. Signal. 2011, 23, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Huang, Z.-Y.; Xu, X.-L.; Li, J.; Fu, X.-W.; Deng, S.-L. Estrogen Receptor Function: Impact on the Human Endometrium. Front. Endocrinol. 2022, 13, 827724. [Google Scholar] [CrossRef]
- Skrzypczak, M.; Schüler, S.; Lattrich, C.; Ignatov, A.; Ortmann, O.; Treeck, O. G protein-coupled estrogen receptor (GPER) expression in endometrial adenocarcinoma and effect of agonist G-1 on growth of endometrial adenocarcinoma cell lines. Steroids 2013, 78, 1087–1091. [Google Scholar] [CrossRef]
- Lv, Q.-Y.; Xie, B.-Y.; Yang, B.-Y.; Ning, C.-C.; Shan, W.-W.; Gu, C.; Luo, X.-Z.; Chen, X.-J.; Zhang, Z.-B.; Feng, Y.-J. Increased TET1 Expression in Inflammatory Microenvironment of Hyperinsulinemia Enhances the Response of Endometrial Cancer to Estrogen by Epigenetic Modulation of GPER. J. Cancer 2017, 8, 894–902. [Google Scholar] [CrossRef]
- Krakstad, C.; Trovik, J.; Wik, E.; Engelsen, I.B.; Werner, H.M.J.; Birkeland, E.; Raeder, M.B.; Øyan, A.M.; Stefansson, I.M.; Kalland, K.H.; et al. Loss of GPER identifies new targets for therapy among a subgroup of ERα-positive endometrial cancer patients with poor outcome. Br. J. Cancer 2012, 106, 1682–1688. [Google Scholar] [CrossRef]
- Deng, J.; Wang, W.; Yu, G.; Ma, X. MicroRNA-195 inhibits epithelial-mesenchymal transition by targeting G protein-coupled estrogen receptor 1 in endometrial carcinoma. Mol. Med. Rep. 2019, 20, 4023–4032. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.C.; Suzawa, M.; Blind, R.D.; Tobias, S.C.; Bulun, S.E.; Scanlan, T.S.; Ingraham, H.A. Stimulating the GPR30 Estrogen Receptor with a Novel Tamoxifen Analogue Activates SF-1 and Promotes Endometrial Cell Proliferation. Cancer Res. 2009, 69, 5415–5423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, Y.; Lan, L.; Liu, R.; Wu, Y.; Qu, Q.; Wen, K. Tamoxifen has a proliferative effect in endometrial carcinoma mediated via the GPER/EGFR/ERK/cyclin D1 pathway: A retrospective study and an in vitro study. Mol. Cell. Endocrinol. 2016, 437, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.-Y.; Lv, Q.-Y.; Ning, C.-C.; Yang, B.-Y.; Shan, W.-W.; Cheng, Y.-L.; Gu, C.; Luo, X.-Z.; Zhang, Z.-B.; Chen, X.-J.; et al. TET1-GPER-PI3K/AKT pathway is involved in insulin-driven endometrial cancer cell proliferation. Biochem. Biophys. Res. Commun. 2017, 482, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Hevir-Kene, N.; Rižner, T.L. The endometrial cancer cell lines Ishikawa and HEC-1A, and the control cell line HIEEC, differ in expression of estrogen biosynthetic and metabolic genes, and in androstenedione and estrone-sulfate metabolism. Chem. Interact. 2015, 234, 309–319. [Google Scholar] [CrossRef]
- Isola, A.L.; Eddy, K.; Chen, S. Biology, Therapy and Implications of Tumor Exosomes in the Progression of Melanoma. Cancers 2016, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Melanoma Skin Cancer Statistics. Available online: https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html (accessed on 17 September 2022).
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2021, 10, 626129. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Prim. 2015, 1, 15003. [Google Scholar] [CrossRef]
- Bannister-Tyrrell, M.; Patterson, J.A.; Ford, J.B.; Morris, J.M.; Nicholl, M.C.; Roberts, C.L. Variation in hospital caesarean section rates for preterm births. Aust. N. Z. J. Obstet. Gynaecol. 2015, 55, 350–356. [Google Scholar] [CrossRef] [Green Version]
- Gandini, S.; Iodice, S.; Koomen, E.; Di Pietro, A.; Sera, F.; Caini, S. Hormonal and reproductive factors in relation to melanoma in women: Current review and meta-analysis. Eur. J. Cancer 2011, 47, 2607–2617. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R. Identification of a Gene (GPR30) with Homology to the G-Protein-Coupled Receptor Superfamily Associated with Estrogen Receptor Expression in Breast Cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F.; George, S.R. Discovery of Three Novel G-Protein-Coupled Receptor Genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Duperret, E.K.; Zhang, J.; Sadeghi, R.; Dahal, A.; O’Brien, K.T.; Cookson, R.; Winkler, J.D.; Ridky, T.W. Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors. Elife 2016, 5, e15104. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. Elife 2018, 7, e31770. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Xie, H.-F.; Tang, Y.; Lin, S.-Q.; Li, J.-M.; Sun, S.-N.; Hu, X.-L.; Huang, Y.-X.; Shi, W.; Jian, D. G protein-coupled estrogen receptor enhances melanogenesis via cAMP-protein kinase (PKA) by upregulating microphthalmia-related transcription factor-tyrosinase in melanoma. J. Steroid Biochem. Mol. Biol. 2017, 165, 236–246. [Google Scholar] [CrossRef]
- Ribeiro, M.P.; Santos, A.E.; Custódio, J.B. The activation of the G protein-coupled estrogen receptor (GPER) inhibits the proliferation of mouse melanoma K1735-M2 cells. Chem. Interact. 2017, 277, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Lilyquist, J.; White, K.A.M.; Lee, R.J.; Philips, G.K.; Hughes, C.R.; Torres, S.M. Quantitative Analysis of Immunohistochemistry in Melanoma Tumors. Medicine 2017, 96, e6432. [Google Scholar] [CrossRef]
- Spałkowska, M.; Dyduch, G.; Broniatowska, E.; Damiani, G.; Wojas-Pelc, A. Molecular Proof of a Clinical Concept: Expression of Estrogen Alpha-, Beta-Receptors and G Protein-Coupled Estrogen Receptor 1 (GPER) in Histologically Assessed Common Nevi, Dysplastic Nevi and Melanomas. Medicina 2021, 57, 1228. [Google Scholar] [CrossRef]
- Kombe, A.J.K.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.; Zhou, Y.; Jin, T. Epidemiology and burden of human papillomavirus and related dis-eases, molecular pathogenesis, and vaccine evaluation. Front. Public Health 2021, 8, 552028. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-López, I.G.; Arellano, A.R.; Jave-Suárez, M.L.F.; Hernández-Silva, C.D.; García-Chagollan, M.; Hernández-Bello, J.; Lopez-Pulido, E.I.; Macias-Barragan, J.; Montoya-Buelna, M.; Muñoz-Valle, J.F.; et al. Interaction between 17β-estradiol, prolactin and human papillomavirus induce E6/E7 transcript and modulate the expression and localization of hormonal receptors. Cancer Cell Int. 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofsjö, A.; Bohm-Starke, N.; Bergmark, K.; Masironi, B.; Sahlin, L. Sex steroid hormone receptor expression in the vaginal wall in cervical cancer survivors after radiotherapy. Acta Oncol. 2019, 58, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Friese, K.; Kost, B.; Vattai, A.; Marmé, F.; Kuhn, C.; Mahner, S.; Dannecker, C.; Jeschke, U.; Heublein, S. The G protein-coupled estrogen receptor (GPER/GPR30) may serve as a prog-nostic marker in early-stage cervical cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 13–19. [Google Scholar] [CrossRef]
- Beilner, D.; Kuhn, C.; Kost, B.P.; Jückstock, J.; Mayr, D.; Schmoeckel, E.; Dannecker, C.; Mahner, S.; Jeschke, U.; Heidegger, H.H. Lysine-specific histone demethylase 1A (LSD1) in cervical cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2843–2850. [Google Scholar] [CrossRef]
- Yang, W.; Tan, W.; Zheng, J.; Zhang, B.; Li, H.; Li, X. MEHP promotes the proliferation of cervical cancer via GPER mediated activation of Akt. Eur. J. Pharmacol. 2018, 824, 11–16. [Google Scholar] [CrossRef]
- Zhu, H.; Lv, Z.; An, C.; Shi, M.; Pan, W.; Zhou, L.; Yang, W.; Yang, M. Onco-lncRNA HOTAIR and its functional genetic variants in papillary thyroid carcinoma. Scientific Reports. 2016, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bertoni, A.P.S.; Manfroi, P.D.A.; Tomedi, J.; Assis-Brasil, B.M.; Souza, E.L.D.S.; Furlanetto, T.W. The gene ex-pression of GPER1 is low in fresh samples of papillary thyroid carcinoma (PTC), and in silico analysis. Mol. Cell. Endocrinol. 2021, 535, 111397. [Google Scholar] [CrossRef]
- Manfroi, P.D.A.; Bertoni, A.P.S.; Furlanetto, T.W. GPER1 in the thyroid: A systematic review. Life Sci. 2020, 241, 117112. [Google Scholar] [CrossRef]
- Prat, J. New insights into ovarian cancer pathology. Ann. Oncol. 2012, 23, x111–x117. [Google Scholar] [CrossRef]
- Matz, M.; Coleman, M.P.; Sant, M.; Chirlaque, M.D.; Visser, O.; Gore, M.; Allemani, C.; Bouzbid, S.; Hamdi-Chérif, M.; Zaidi, Z.; et al. The histology of ovarian cancer: Worldwide distribution and implications for international survival comparisons (CONCORD-2). Gynecol. Oncol. 2016, 144, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.T.; Kim, H.; Yun, J.; Chung, J.S.; Kwon, S.-M. Promising Therapeutic Strategies for Mesenchymal Stem Cell-Based Cardiovascular Regeneration: From Cell Priming to Tissue Engineering. Stem Cells Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Douglass, J.; Habibi, M. Microbiota and Estrogen Metabolism. Compr. Gut Microbiota 2022, 27–34. [Google Scholar]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Mungenast, F.; Thalhammer, T. Estrogen Biosynthesis and Action in Ovarian Cancer. Front. Endocrinol. 2014, 5, 192. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, S.; Terai, Y.; Kawaguchi, H.; Takai, M.; Yoo, S.; Tanaka, Y.; Tanaka, T.; Tsunetoh, S.; Sasaki, H.; Kanemura, M.; et al. GPR30 regulates the EGFR-Akt cascade and predicts lower survival in patients with ovarian cancer. J. Ovarian Res. 2012, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Casarini, L.; Lazzaretti, C.; Paradiso, E.; Limoncella, S.; Riccetti, L.; Sperduti, S.; Melli, B.; Marcozzi, S.; Anzivino, C.; Sayers, N.S.; et al. Membrane estrogen receptor (GPER) and follicle-stimulating hor-mone receptor (FSHR) heteromeric complexes promote human ovarian follicle survival. Iscience 2020, 23, 101812. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, H.; Wen, H.; Jiang, X.; Cao, X.; Zhang, G.; Liu, G. The novel estrogen receptor GPER regulates the migration and invasion of ovarian cancer cells. Mol. Cell. Biochem. 2013, 378, 1–7. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, X.; Zhao, Y.; Wen, H.; Liu, G. Role of GPER on proliferation, migration and invasion in ligand-independent manner in human ovarian cancer cell line SKOV3. Cell Biochem. Funct. 2015, 33, 552–559. [Google Scholar] [CrossRef]
- Ignatov, T.; Modl, S.; Thulig, M.; Weißenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J. Ovarian Res. 2013, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis. 2013, 4, e869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüler-Toprak, S.; Skrzypczak, M.; Ignatov, T.; Ignatov, A.; Ortmann, O.; Treeck, O. G protein-coupled estrogen receptor 1 (GPER-1) and agonist G-1 inhibit growth of ovarian cancer cells by activation of anti-tumoral tran-scriptome responses: Impact of GPER-1 mRNA on survival. J. Cancer Res. Clin. Oncol. 2020, 146, 3175–3188. [Google Scholar] [CrossRef] [PubMed]
- Fraungruber, P.; Kaltofen, T.; Heublein, S.; Kuhn, C.; Mayr, D.; Burges, A.; Mahner, S.; Rathert, P.; Jeschke, U.; Trillsch, F. G protein-coupled estrogen receptor correlates with Dkk2 expres-sion and has prognostic impact in ovarian cancer patients. Front. Endocrinol. 2021, 12, 564002. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.X.; Xiong, W.; Wang, M.L.; Yang, J.; Shi, H.J.; Chen, H.Q.; Niu, G. Nuclear G protein-coupled oestrogen receptor (GPR30) predicts poor survival in patients with ovarian cancer. J. Int. Med. Res. 2018, 46, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Kolkova, Z.; Casslén, V.; Henic, E.; Ahmadi, S.; Ehinger, A.; Jirström, K.; Casslén, B. The G protein-coupled estrogen receptor 1 (GPER/GPR30) does not predict survival in patients with ovarian cancer. J. Ovarian Res. 2012, 5, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-protein-coupled estrogen receptor (GPER) regulates trimethylation of histone h3 at lysine 4 and represses migration and proliferation of ovarian cancer cells in vitro. Cells 2021, 10, 619. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [Google Scholar] [CrossRef]
- Thompson-Elliott, B.; Johnson, R.; Khan, S.A. Alterations in TGFβ signaling during prostate cancer progression. Am. J. Clin. Exp. Urol. 2021, 9, 318. [Google Scholar]
- Lappano, R.; Marco, P.D.; Francesco, E.M.D.; Chimento, A.; Pezzi, V.; Maggiolini, M. Cross-talk between GPER and growth factor signaling. J. Steroid Biochem. Mol. Biol. 2013, 137, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.L.; Eckhart, A.D. Regulation of GPCR signaling in hypertension. Biochim. Biophys. Acta. Mol. Basis. Dis. 2010, 1802, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, H.; Nakatani, T. Involvement of estrogen receptors in prostatic diseases. Int. J. Urol. 2012, 19, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H.; Fixemer, T.; Hunsicker, I.; Remberger, K. Estrogen Receptor Expression in Prostate Cancer and Premalignant Prostatic Lesions. Am. J. Pathol. 1999, 155, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellem, S.J.; Risbridger, G.P. Treating prostate cancer: A rationale for targeting local oestrogens. Nat. Cancer 2007, 7, 621–627. [Google Scholar] [CrossRef]
- Ramírez-De-Arellano, A.; Pereira-Suárez, A.L.; Rico-Fuentes, C.; López-Pulido, E.I.; Villegas-Pineda, J.C.; Sierra-Diaz, E. Distribution and Effects of Estrogen Receptors in Prostate Cancer: Associated Molecular Mechanisms. Front. Endocrinol. 2022, 12, 811578. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Kanda, N.; Watanabe, S. 17β-Estradiol Inhibits Oxidative Stress-Induced Apoptosis in Keratinocytes by Promoting Bcl-2 Expression. J. Investig. Dermatol. 2003, 121, 1500–1509. [Google Scholar] [CrossRef] [Green Version]
- Kanda, N.; Watanabe, S. 17β-Estradiol Stimulates the Growth of Human Keratinocytes by Inducing Cyclin D2 Expression. J. Investig. Dermatol. 2004, 123, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Jacquot, Y.; Kampa, M.; Lindsey, S.H. Editorial: GPER and Human Pathologies. Front. Endocrinol. 2021, 12, 794332. [Google Scholar] [CrossRef] [PubMed]
- Barrett-Connor, E. Sex Differences in Coronary Heart Disease. Circulation 1997, 95, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, A.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Fredette, N.C.; Daniel, C.; Sharma, G.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. Obligatory role for GPER in cardiovascular aging and disease. Sci. Signal. 2016, 9, ra105. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Oparil, S. Hypertension in Women. Hypertension 2017, 70, 19–26. [Google Scholar] [CrossRef]
- Yoon, S.S.S.; Carroll, M.D.; Fryar, C.D. Hypertension Prevalence and Control Among Adults: United States, 2011−2014. NCHS 2015, 220, 1–8. [Google Scholar]
- Aryan, L.; Younessi, D.; Zargari, M.; Banerjee, S.; Agopian, J.; Rahman, S.; Borna, R.; Ruffenach, G.; Umar, S.; Eghbali, M. The Role of Estrogen Receptors in Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4314. [Google Scholar] [CrossRef]
- Zhu, Y.; Bian, Z.; Lu, P.; Karas, R.H.; Bao, L.; Cox, D.; Hodgin, J.; Shaul, P.W.; Thorén, P.; Smithies, O.; et al. Abnormal Vascular Function and Hypertension in Mice Deficient in Estrogen Receptor β. Science 2002, 295, 505–508. [Google Scholar] [CrossRef]
- Haas, E.; Bhattacharya, I.; Brailoiu, E.; Damjanović, M.; Brailoiu, G.C.; Gao, X.; Mueller-Guerre, L.; Marjon, N.A.; Gut, A.; Minotti, R.; et al. Regulatory Role of G Protein–Coupled Estrogen Receptor for Vascular Function and Obesity. Circ. Res. 2009, 104, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Francesco, E.M.D.; Angelone, T.; Pasqua, T.; Pupo, M.; Cerra, M.C.; Maggiolini, M. GPER mediates cardiotropic effects in spontaneously hypertensive rat hearts. PLoS ONE 2013, 8, e69322. [Google Scholar] [CrossRef] [Green Version]
- Rocca, C.; Femminò, S.; Aquila, G.; Granieri, M.C.; De Francesco, E.M.; Pasqua, T.; Rigiracciolo, D.C.; Fortini, F.; Cerra, M.C.; Maggiolini, M.; et al. Notch1 mediates preconditioning protection induced by gper in normotensive and hypertensive female rat hearts. Front. Physiol. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Debortoli, A.R.; Rouver, W.D.N.; Delgado, N.T.B.; Mengal, V.; Claudio, E.R.G.; Pernomian, L.; Bendhack, L.M.; Moysés, M.R.; Santos, R.L.d. GPER modulates tone and coronary vascular reactivity in male and female rats. J. Mol. Endocrinol. 2017, 59, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rižner, T.L. Estrogen metabolism and action in endometriosis. Mol. Cell. Endocrinol. 2009, 307, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yuan, X.; Zhang, Y. The co-expression of GPER and Gankyrin in ovarian endometriosis and its correlation with the rASRM stages. Arch. Gynecol. Obstet. 2015, 293, 133–141. [Google Scholar] [CrossRef]
- Jiang, X.; Ye, X.; Ma, J.; Li, W.; Wu, R.; Jun, L. G protein-coupled estrogen receptor 1 (GPER 1) mediates estrogen-induced, proliferation of leiomyoma cells. Gynecol. Endocrinol. 2015, 31, 894–898. [Google Scholar] [CrossRef]
- Kasap, B.; Turhan, N.O.; Edgunlu, T.; Duran, M.; Akbaba, E.; Oner, G. G-protein-coupled estrogen receptor-30 gene polymorphisms are associated with uterine leiomyoma risk. Bosn. J. Basic Med. Sci. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Broughton, B.R.; Brait, V.H.; Kim, H.A.; Lee, S.; Chu, H.X.; Gardiner-Mann, C.V.; Guida, E.; Evans, M.A.; Miller, A.A.; Arumugam, T.; et al. Sex-Dependent Effects of G Protein–Coupled Estrogen Receptor Activity on Outcome After Ischemic Stroke. Stroke 2014, 45, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Bourque, M.; Poirier, A.-A.; Aubé, B.; Morissette, M.; Di Paolo, T.; Soulet, D. GPER1-mediated immunomodulation and neuroprotection in the myenteric plexus of a mouse model of Parkinson’s disease. Neurobiol. Dis. 2015, 82, 99–113. [Google Scholar] [CrossRef]
- Callier, S.; Le Saux, M.; Lhiaubet, A.-M.; Di Paolo, T.; Rostène, W.; Pelaprat, D. Evaluation of the protective effect of oestradiol against toxicity induced by 6-hydroxydopamine and 1-methyl-4-phenylpyridinium ion (MPP+) towards dopaminergic mesencephalic neurones in primary culture. J. Neurochem. 2002, 80, 307–316. [Google Scholar] [CrossRef]
- Mendes-Oliveira, J.; Campos, F.L.; Videira, R.A.; Baltazar, G. GPER activation is effective in protecting against inflammation-induced nigral dopaminergic loss and motor function impairment. Brain Behav. Immun. 2017, 64, 296–307. [Google Scholar] [CrossRef]
- Guan, J.; Yang, B.; Fan, Y.; Zhang, J. GPER Agonist G1 Attenuates Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson Disease. Neuroimmunomodulation 2017, 24, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Olsen, N.J.; Kovacs, W.J. Gonadal Steroids and Immunity. Endocr. Rev. 1996, 17, 369–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chunhe, C.; Dehghani, B.; Magrisso, I.J.; Rick, E.A.; Bonhomme, E.; Cody, D.B.; Elenich, L.A.; Subramanian, S.; Murphy, S.J.; Kelly, M.J.; et al. GPR30 Contributes to Estrogen-Induced Thymic Atrophy. Mol. Endocrinol. 2008, 22, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notas, G.; Kampa, M.; Castanas, E. G Protein-coupled estrogen receptor in immune cells and its role in immune-related diseases. Front. Endocrinol. 2020, 11, 579420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, P.; Deng, Y.; Ma, Z.; Guo, H.; Guo, H.; Hou, Y.; Wang, S.; Zou, W.; Sun, Y.; et al. The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J. Neuroinflam. 2018, 15, 206. [Google Scholar] [CrossRef]
- Blasko, E.; Haskell, C.A.; Leung, S.; Gualtieri, G.; Halks-Miller, M.; Mahmoudi, M.; Dennis, M.; Prossnitz, E.; Karpus, W.J.; Horuk, R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 2009, 214, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Triplett, K.D.; Pokhrel, S.; Castleman, M.J.; Daly, S.M.; Elmore, B.O.; Joyner, J.A.; Sharma, G.; Herbert, G.; Campen, M.J.; Hathaway, H.J.; et al. GPER activation protects against epithelial barrier disruption by Staphylococcus aureus α-toxin. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1870–1882. [Google Scholar] [CrossRef]
- Yang, H.; Wang, C.; Liao, H.; Wang, Q. Activation of GPER by E2 promotes proliferation, invasion and migration of breast cancer cells by regulating the miR-124/CD151 pathway. Oncol. Lett. 2021, 21, 432. [Google Scholar] [CrossRef]
- Choi, I.; Gudas, L.J.; Katzenellenbogen, B.S. Regulation of keratin 19 gene expression by estrogen in human breast cancer cells and identification of the estrogen responsive gene region. Mol. Cell Endocrinol. 2000, 164, 225–237. [Google Scholar] [CrossRef]


| S/No | Breast Cancer Phenotype | Experimental Model | Pattern of GPER Expression Observed | Observations | Reference |
|---|---|---|---|---|---|
| 1 | ER-positive breast cancer | MCF-7 cells and ER-positive breast cancer patients | Elevated | Increased tamoxifen resistance and metastasis | [66] |
| 2 | ER-positive breast cancer | MCF-7 cells | Elevated expression using the G-1 agonist | Reduced cell proliferation, cell cycle arrest at M-phase and enhanced apoptosis | [70] |
| 3 | ER-negative breast cancer | MDA-MB-231 cells | Elevated expression in the presence of bisphenol A (ligand) | Increased invasiveness and metastasis | [71] |
| 4 | ER-negative breast cancer | MDA-MB-231 cells | Inhibition via interaction with Na+/H+ exchanger regulatory factor 1 | Inhibited ERK1/2 and suppressed proliferation of the TNBC cells | [72] |
| 5 | ER-negative breast cancer | HCC70 and HCC1806 cells | Downregulated via treatment with gefitinib | Suppressed proliferation of the TNBC cells | [73] |
| 6 | ER-negative breast cancer | MDA-MB-231 xenograft tumors | Elevated via treatment with G1 agonist | Inhibited angiogenesis and metastasis | [11] |
| 7 | ER negative breast cancer | MDA-MB-231 MDA-MB-468 | Elevated via treatment with G1 agonist | Inhibited cell growth, induction of cell cycle arrest, increased apoptosis | [70] |
| 8 | ER negative breast cancer | TNBC patients in China | Increased expression | Reduced lymph node metastasis | [75] |
| 9 | Primary invasive breast cancer patients | Cohort of breast cancer patients in the UK | Low expression | Aggressive disease progression and adverse survival | [76] |
| S/N | Gene | Gene Product | Mechanism/Function | References |
|---|---|---|---|---|
| 1. | β1-integrin | β1-integrin | Tumor progression inflammation, proliferation, adhesion, invasion, and tumor survival | [107] |
| 2. | c-FOS | c-FOS | Cell proliferation and migration | [67,107] |
| 3. | EGR-1 | EGR-1 | Cell proliferation and migration | [67,107] |
| 4. | CTGF | CTGF | Cell proliferation and migration | [94,108] |
| 5. | CYP19A1 | Aromatase | Proliferation and cell-cycle progression | [104,109] |
| 6. | FAK | FAK | Differentiation, proliferation, apoptosis, migration, invasion, survival, and angiogenesis | [101] |
| 7. | ERK2 | ERK1/2 | Cell migration, proliferation, angiogenesis, and invasion | [67,101] |
| 8. | Src | Src | Cell cycle progression, growth, survival and migration | [101] |
| 9. | HIF1α | HIF1α | Endothelial tube formation | [110] |
| 10. | VEGF | VEGF | Endothelial tube formation | [110] |
| 11. | Endothelial marker (CD34) | Endothelial marker (CD34) | Endothelial tube formation | [110] |
| 12. | HOTAIR | HOTAIR | Unknown | [111] |
| 13. | FASN | FASN | Energy production, cell proliferation, aggressiveness, and metastasis | [112] |
| 14. | miR-338-3p | miR-338-3p | suppress the growth and invasion of SkBr3 cancer cells and CAFs | [113] |
| 15. | miR144 | miR144 | increase cell cycle progression | [113] |
| 16. | onco-suppressor Runx1 | onco-suppressor Runx1 | increase cell cycle progression | [113] |
| Nature of GPER Alteration in Diabetic Conditions | Consequences of GPER Alteration in Diabetic Conditions | Nature of GPERAlteration in Malignancies | Consequences of Alteration in Malignancy |
|---|---|---|---|
| (a) Differential GPER gene expression in the livers of T2D patients [22] (b) Upregulated GPER gene and protein expression in mice model of obesity and T2D [34] (c) Upregulated GPER protein expression in a T2D rat model following E2 treatment [39] | (a) Could be implicated in hepatic insulin resistance [22] (b) Loss of aldosterone-mediated protection against diabetes-induced endothelial damage [34] (c) Increases GLUT-2 protein content and insulin secretion in islet β-cells [39] | (a) GPER is overexpressed at mRNA and protein levels in seminomas [58,59] (aii) SNPs at GPER gene promoter in seminomas [59] (b) GPER is overexpressed at protein levels in ovarian cancer [208] (c) Downregulated GPER protein expression in ovarian cancer compared to benign and low-malignant ovarian tumors Tanja Ignatov et al. [75] (di) GPER expression is upregulated in TNBC [67,71,75] (dii) GPER gene promoter is hypermethylated in MCF-7 (ER-positive breast cancer cells) [70] (e) GPER gene expression is upregulated in NSCLC [124] (fi) GPER mRNA and protein levels are downregulated in gastric cancer [9] (fii) SNPs along the GPER gene sequence of gastric cancer patients [139] (g) Insulin induces the upregulation of GPER mRNA and protein levels in insulin-resistant endometrial cancer cells by upregulating TET-1 [168] (h) GPER protein is differentially expressed in different melanocytic lesions [189] (I) Upregulated LSD1 protein expression correlates negatively with GPER protein expression in cervical cancer patients [196] | (a) Stimulation of seminoma cell proliferation by activating ERK1/2 and protein kinase A [58,59] ERβ downregulation causing a switch in estrogen responsiveness [63] (aii) Correlate positively with GPER upregulation in seminomas [59] (b) Causes poor progression-free survival in ovarian cancer patients [208] (c) GPER could be a tumor suppressor in ovarian cancer Tanja Ignatov et al. [75] (di) Contributes to TNBC invasiveness, metastasis, and recurrence by activating FAK and STAT3, which alter the expression of target genes such as ERK267,72; Reduce lymph node metastasis in TNBC patients [75] (dii) Reduced GPER gene expression and its tumor suppressive role in the studied cell lines [70] (e) Promotes NSCLC cell proliferation [124](fi) Predicts poor prognosis in gastric cancer [9] (fii) Correlate with gastric cancer propensity, advancement, and severity, particularly among the Asian population [139] (g) UpregulatedGPER predicts poor prognosis and stimulates endometrial cancer cells proliferation [168] (h) GPER could be a prognostic biomarker in melanoma [189] (I) Disadvantaged 10-year overall survival in the studied patients [196] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, A.; Forcados, G.E.; Yusuf, A.P.; Abubakar, M.B.; Sadiq, I.Z.; Elhussin, I.; Siddique, M.A.T.; Aminu, S.; Suleiman, R.B.; Abubakar, Y.S.; et al. Comparative G-Protein-Coupled Estrogen Receptor (GPER) Systems in Diabetic and Cancer Conditions: A Review. Molecules 2022, 27, 8943. https://doi.org/10.3390/molecules27248943
Muhammad A, Forcados GE, Yusuf AP, Abubakar MB, Sadiq IZ, Elhussin I, Siddique MAT, Aminu S, Suleiman RB, Abubakar YS, et al. Comparative G-Protein-Coupled Estrogen Receptor (GPER) Systems in Diabetic and Cancer Conditions: A Review. Molecules. 2022; 27(24):8943. https://doi.org/10.3390/molecules27248943
Chicago/Turabian StyleMuhammad, Aliyu, Gilead Ebiegberi Forcados, Abdurrahman Pharmacy Yusuf, Murtala Bello Abubakar, Idris Zubairu Sadiq, Isra Elhussin, Md. Abu Talha Siddique, Suleiman Aminu, Rabiatu Bako Suleiman, Yakubu Saddeeq Abubakar, and et al. 2022. "Comparative G-Protein-Coupled Estrogen Receptor (GPER) Systems in Diabetic and Cancer Conditions: A Review" Molecules 27, no. 24: 8943. https://doi.org/10.3390/molecules27248943