3.2. General Procedure for the Synthesis of Mannich Products 3a–k by Enantioselective Mannich Reaction of N-Boc Ketimines with β-Diketones
To a mixture of N-Boc ketimine 1a–g (0.1 mmol), catalyst C4 (0.002 mmol, 0.02 equiv) in 1.0 mL of toluene, β-diketone 2a–c (0.11 mmol, 1.1 equiv) was added at room temperature, and the reaction mixture was stirred in a Wheaton vial. The progress of the reaction was monitored by TLC analysis. After the completion of the reaction, the solvent was removed under reduced pressure. The crude reaction mixture was purified by flash column chromatography to afford the corresponding product 3a–k. The enantiomeric excess was determined by chiral-phase HPLC analysis using mixtures of hexane/isopropanol as eluent.
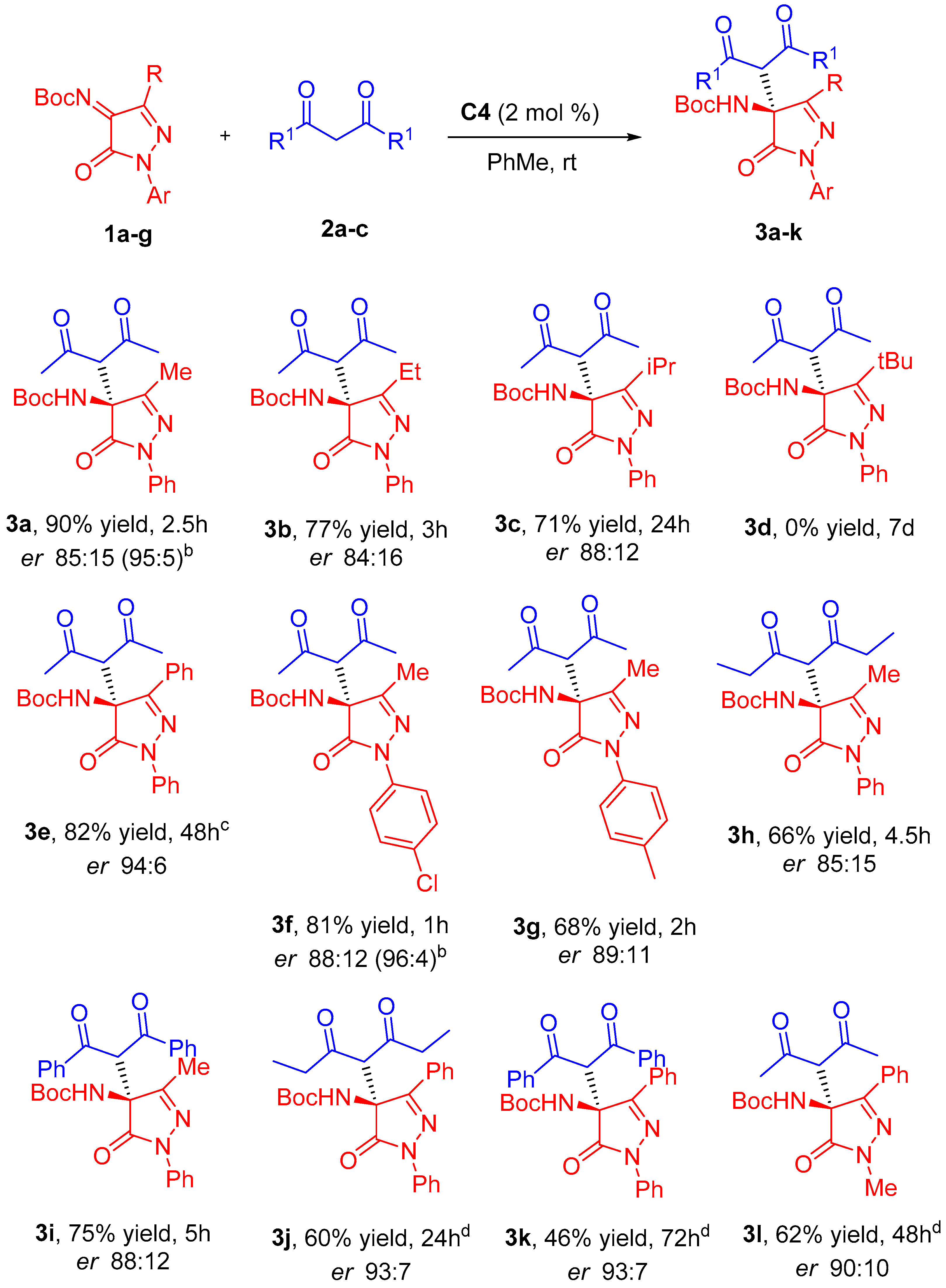
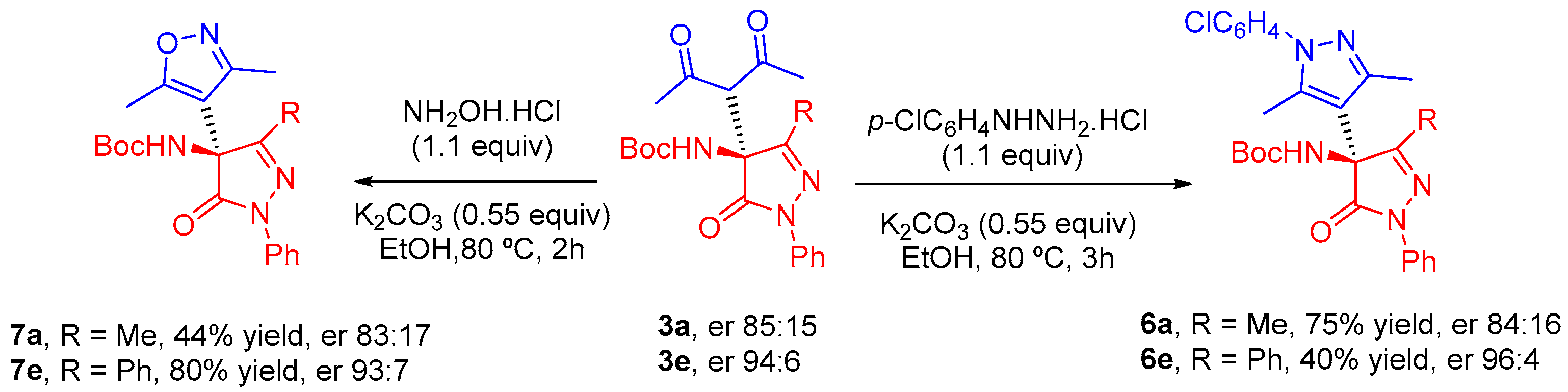
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-3-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3a). Product 3a was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 3:1 as eluent afforded compound 3a as a colorless solid (35 mg, 0.09 mmol, 90% yield); Mp 140–141 °C (hexane-ethyl acetate); [α]D25 = +18.9 (c = 0.9, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.87 (dd, J = 8.6, 1.2 Hz, 2H, Har), 7.39 (dd, J = 8.6, 7.4 Hz, 2H, Har), 7.19 (tt, J = 7.4, 1.3 Hz, 1H, Har), 6.38 (br s, 1H, NH), 4.08 (s, 1H, CH), 2.31 (s, 3H, CH3CO), 2.30 (s, 3H, CH3CO), 2.08 (s, 3H, CH3), 1.36 (s, 9H, C(CH3)3) ppm; 13C NMR (126 MHz, CDCl3): δ 200.4 (CO), 169.9 (CON), 137.7 (Car), 128.9 (CHar), 125.4 (CHar), 118.9 (CHar), 77.3 (C(CH3)3), 66.9 (CH), 66.7 (CNHBoc), 32.1 (CH3CO), 31.9 (CH3CO), 28.1 (C(CH3)3), and 14.8 (CH3) ppm. IR (ATR): 3403, 3356, 2974, 2931, 1707, 1596, 1496, 1375, 1254, 1154, 758, 688 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C20H26N3O5 388.1867; Found 388.1868. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 11.5 min, minor enantiomer (R) tR = 26.1 min. (er 85:15).
A sample of 3a (er 85:15) was recrystallized from MeOH to afford 3a as white crystals (quasi-racemic mixture, er 58:42) and enantioenriched 3a from the mother liquors (er 95:5). This last fraction was then used to prepare compound 4a.
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-3-ethyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3b). Product 3b was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 3:1 as eluent afforded compound 3b as a colorless solid (31 mg, 0.077 mmol, 77% yield). [α]D25 = +10.7 (c = 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.89 (dd, J = 8.7, 1.2 Hz, 2H, Har), 7.38 (dd, J = 8.7, 7.4 Hz, 2H, Har), 7.18 (tt, J = 7.4, 1.2 Hz, 1H, Har), 6.40 (br s, 1H, NH), 4.05 (s, 1H, CH), 2.41 (m, 1H, CHHCH3), 2.34 (m, 1H, CHHCH3), 2.30 (s, 3H, CH3CO), 2.29 (s, 3H, CH3CO), 1.34 (s, 9H, C(CH3)3), 1.27 (t, J = 7.3 Hz, 3H, CH3CH2) ppm. 13C NMR (126 MHz, CDCl3): δ 200.7 (CO), 170.1 (CON), 137.9 (Car), 128.8 (CHar), 125.3 (CHar), 118.9 (CHar), 77.3 (C(CH3)3), 67.1 (CH), 66.9 (CNHBoc), 32.1 (CH3CO), 31.9 (CH3CO), 28.1 (C(CH3)3), 22.2 (CH2CH3), 9.6 (CH3CH2) ppm. IR (ATR): 3388, 2985, 2942, 1707, 1596, 1493, 1453, 1351, 1279, 1152, 1054, 761, 692 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C21H28N3O5 402.2023; Found 402.2029. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 10.0 min, minor enantiomer (R) tR = 20.0 min. (er 84:16).
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-3-isopropyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3c). Product 3c was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 3c as a colorless solid (30 mg, 0.071 mmol, 71% yield). [α]D25 = +42.5 (c = 0.8, MeOH). 1H NMR (500 MHz, CDCl3): δ 7.89 (d, J = 8.1 Hz, 2H, Har), 7.38 (dd, J = 8.7, 7.4 Hz, 2H, Har), 7.17 (tt, J = 7.4, 1.2 Hz, 1H, Har), 6.49 (br s, 1H, NH), 4.04 (s, 1H, CH), 2.65 (sept, J = 6.9 Hz, 1H, CH(CH3)2), 2.29 (s, 3H, CH3CO), 2.28 (s, 3H, CH3CO), 1.37 (s, 9H, C(CH3)3), 1.27 (d, J = 6.8 Hz, 6H, (CH3)2CH) 1.24 (d, J = 7.0 Hz, 6H, (CH3)2CH) ppm. 13C NMR (126 MHz, CDCl3): δ 201.1 (CO), 169.8 (CON), 138.0 (Car), 128.8 (CHar), 125.2 (CHar), 119.1 (CHar), 77.3 (C(CH3)3), 67.4 (CH(COCH3)2), 67.0 (CNHBoc), 32.1 (CH3CO), 31.7 (CH3CO), 28.8 (CH(CH3)2), 28.1 (C(CH3)3), 20.3 ((CH3)2CH) ppm. IR (ATR): 3413, 2975, 2935, 1710, 1598, 1493, 1457, 1359, 1283, 1156, 1083, 1054, 754, 688 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C22H29N3NaO5 438.1999; Found 438.1999. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 95:5, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 18.6 min, minor enantiomer (R) tR = 27.0 min. (er 88:12).
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3e). Product 3e was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (3.2 mg, 0.005 mmol, 0.05 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 3e as a colorless solid (37 mg, 0.082 mmol, 82% yield). [α]D25 = +56.3 (c = 0.6, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.98 (dd, J = 8.4, 1.2 Hz, 2H, Har), 7.90 (dd, J = 7.8, 2.0 Hz, 2H, Har), 7.43 (m, 5H, Har), 7.23 (tt, J = 7.4, 1.0 Hz, 1H, Har), 6.82 (br s, 1H, NH), 3.97 (s, 1H, CH), 2.11 (s, 3H, CH3CO), 2.03 (s, 3H, CH3CO), 1.31 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 202.2 (CO), 200.5 (CO), 170.0 (CON), 137.9 (Car), 130.8 (CHar), 129.0 (CHar), 128.9 (CHar), 126.7 (CHar), 125.6 (CHar), 119.2 (CHar), 77.3 (C(CH3)3), 66.3 (CH), 66.2 (CNHBoc), 32.3 (CH3CO), 32.2 (CH3CO), 28.1 (C(CH3)3) ppm. IR (ATR): 3379, 2982, 1714, 1597, 1490, 1358, 1255, 1156, 751, 689 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C25H27N3NaO5 472.1843; Found 472.1842. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 12.7 min, minor enantiomer (R) tR = 29.1 min. (er 94:6).
tert-Butyl (S)-(1-(4-chlorophenyl)-4-(2,4-dioxopentan-3-yl)-3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3f). Product 3f was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 3:1 as eluent afforded compound 3f as a colorless solid (34 mg, 0.081 mmol, 81% yield). Mp 170–171 °C (hexane-ethyl acetate). [α]D25 = +17.9 (c = 0.6, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.84 (d, J = 8.9 Hz, 2H, Har), 7.34 (d, J = 8.9 Hz, 2H, Har), 6.36 (br s, 1H, NH), 4.05 (s, 1H, CH), 2.30 (s, 3H, CH3CO), 2.29 (s, 3H, CH3CO), 2.07 (s, 3H, CH3), 1.35 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 200.4 (CO), 169.9 (CON), 158.1 (CO2tBu), 153.5 (CCH3), 136.3 (Car), 130.4 (Car), 128.9 (CHar), 119.9 (CHar), 81.7 (C(CH3)3), 66.9 (CH), 66.6 (CNHBoc), 32.1 (CH3CO), 31.9 (CH3CO), 28.1 (C(CH3)3), 14.7 (CH3) ppm. IR (ATR): 3419, 2975, 2905, 1714, 1494, 1461, 1365, 1251, 1152, 836, 810 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. ForC20H25ClN3O5 422.1477; Found 422.1487. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 10.0 min, minor enantiomer (R) tR = 31.8 min. (er 88:12). A sample of 3f (er 88:12) was recrystallized from hexane-ethyl acetate to afford 3f as white crystals (quasi-racemic mixture, er 58:42) and enantioenriched 3f from the mother liquors (er 96:4). This last fraction was then used to prepare compound 4f.
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-3-methyl-5-oxo-1-(p-tolyl)-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3g). Product 3g was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 3:1 as eluent afforded compound 3g as a colorless solid (27 mg, 0.068 mmol, 68% yield). Mp 150–151 °C (hexane-ethyl acetate). [α]D25 = +19.0 (c = 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.73 (d, J = 8.4 Hz, 2H, Har), 7.18 (d, J = 8.4 Hz, 2H, Har), 6.35 (br s, 1H, NH), 4.07 (s, 1H, CH), 2.33 (s, 3H, CH3C6H4), 2.30 (s, 3H, CH3CO), 2.29 (s, 3H, CH3CO), 2.07 (s, 3H, CH3), 1.35 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 200.4 (CO), 169.7 (CON), 135.3 (Car), 135.1 (Car), 129.4 (CHar), 119.0 (CHar), 77.3 (C(CH3)3), 67.0 (CH), 66.7 (CNHBoc), 32.1 (CH3CO), 31.9 (CH3CO), 28.1 (C(CH3)3), 21.0 (CH3C6H4), 14.8 (CH3) ppm. IR (ATR): 3423, 2978, 2923, 1714, 1703, 1512, 1472, 1369, 1255, 1156, 1056, 814 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C21H28N3O5 402.2023; Found 402.2043. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 11.5 min, minor enantiomer (R) tR = 44.7 min. (er 89:11).
tert-Butyl (S)-(4-(3,5-dioxoheptan-4-yl)-3-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3h). Product 3h was obtained according to general procedure using heptane-3,5-dione (15 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 3h as a colorless solid (27 mg, 0.066 mmol, 66% yield). Mp 119–120 °C (hexane-ethyl acetate). [α]D25 = +12.9 (c = 0.6, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.85 (dd, J = 8.7, 1.2 Hz, 2H, Har), 7.37 (dd, J = 8.6, 7.4 Hz, 2H, Har), 7.17 (tt, J = 7.4, 1,2 Hz, 1H, Har), 6.55 (br s, 1H, NH), 4.03 (s, 1H, CH), 2.60 (m, 2H, CHHCH3), 2.54 (m, 2H, CHHCH3), 2.05 (s, 3H, CH3), 1.36 (s, 9H, C(CH3)3), 1.08 (t, J = 7.1 Hz, 3H, CH3CH2), 0.99 (t, J = 7.1 Hz, 3H, CH3CH2) ppm. 13C NMR (126 MHz, CDCl3): δ 202.7 (CO), 170.1 (CON), 137.8 (Car), 128.8 (CHar), 125.3 (CHar), 118.9 (CHar), 77.3 (C(CH3)3), 67.0 (CH), 65.1 (CNHBoc), 38.6 (CH2CO), 38.4 (CH2CO), 28.1 (C(CH3)3), 14.8 (CH3), 7.4 (CH3CH2), 7.3 (CH3CH2) ppm. IR (ATR): 3339, 2978, 2942, 1714, 1597, 1497, 1369, 1270, 1152, 1104, 759, 689 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C22H30N3O5 416.2180; Found 416.2189. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 9.2 min, minor enantiomer (R) tR = 23.8 min. (er 85:15).
tert-Butyl (S)-(4-(1,3-dioxo-1,3-diphenylpropan-2-yl)-3-methyl-5-oxo-1-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3i). Product 3i was obtained according to general procedure using 1,3-diphenylpropane-1,3-dione (25 mg, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (1.3 mg, 0.002 mmol, 0.02 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 3i as a colorless solid (38 mg, 0.075 mmol, 75% yield). Mp 201-202 °C (hexane-ethyl acetate). [α]D25 = -68.6 (c = 0.7, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.89 (dd, J = 11.9, 8.6 Hz, 4H, Har), 7.51 (m, 4H, Har), 7.39 (m, 4H, Har), 7.22 (dd, J = 8.6, 7.2 Hz, 2H, Har), 7.07 (td, J = 7.4, 1.3 Hz, 1H, Har), 6.61 (br s, 1H, NH), 5.91 (s, 1H, CH), 2.22 (s, 3H, CH3), 1.36 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 191.1 (CO), 170.1 (CON), 137.5 (Car), 136.1 (Car), 135.2 (Car), 134.5 (CHar), 134.2 (CHar), 129.1 (CHar), 129.1 (CHar), 128.7 (CHar), 128.5 (CHar), 128.5 (CHar), 125.0 (CHar), 118.6 (CHar), 77.3 (C(CH3)3), 67.8 (CNHBoc), 56.6 (CH), 28.1 (C(CH3)3), 15.8 (CH3) ppm. IR (ATR): 3276, 3147, 2986, 1729, 1700, 1593, 1490, 1446, 1365, 1270, 1214, 1152, 770, 751, 696, 682 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C30H30N3O5 512.2180; Found 512.2214. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 14.2 min, minor enantiomer (R) tR = 35.8 min. (er 88:12).
tert-Butyl (S)-(4-(3,5-dioxoheptan-4-yl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3j). Product 3j was obtained according to general procedure using heptane-3,5-dione (15 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (6.3 mg, 0.01 mmol, 0.1 equiv). Chromatography on a silica gel using hexane/EtOAc = 8:1 as eluent afforded compound 3j as a colorless solid (29 mg, 0.06 mmol, 60% yield). [α]D25 = +48.4 (c = 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.98 (dd, J = 8.6, 1.3 Hz, 2H, Har), 7.88 (dd, J = 7.3, 2.5 Hz, 2H, Har), 7.43 (m, 5H, Har), 7.23 (m, 1H, Har), 7.12 (br s, 1H, NH), 3.94 (s, 1H, CH), 2.59 (dq, J = 20.6, 7.1 Hz, 1H, CHHCH3), 2.38 (dq, J = 19.0, 7.0 Hz, 1H, CHHCH3), 2.19 (dq, J = 19.2, 7.1 Hz, 1H, CHHCH3), 2.03 (dq, J = 19.4, 7.0 Hz, 1H, CHHCH3), 1.33 (s, 9H, C(CH3)3), 0.90 (t, J = 7.1 Hz, 3H, CH3), 0.79 (t, J = 7.0 Hz, 3H, CH3) ppm. 13C NMR (126 MHz, CDCl3): δ 204.0 (CO), 202.7 (CO), 170.2 (CON), 137.9 (Car), 130.7 (CHar), 128.9 (CHar), 126.8 (CHar), 125.5 (CHar), 119.2 (CHar), 77.3 (C(CH3)3), 66.7 (CNHBoc), 39.0 (2 CH2), 28.1 (C(CH3)3), 7.2 (CH3), 6.9 (CH3) ppm. IR (ATR): 3369, 2979, 2939, 1731, 1698, 1599, 1489, 1397, 1283, 1158, 1114, 1015, 758, 736, 689 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C27H32N3O5 478.2336; Found 478.2345. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 85:15, 0.7 mL/min, λ = 254 nm, major enantiomer (S) tR = 8.9 min, minor enantiomer (R) tR = 22.3 min. (er 93:7).
tert-Butyl (S)-(4-(1,3-dioxo-1,3-diphenylpropan-2-yl)-5-oxo-1,3-diphenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3k). Product 3k was obtained according to general procedure using 1,3-diphenylpropane-1,3-dione (25 mg, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (6.3 mg, 0.01 mmol, 0.1 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as an eluent afforded compound 3k as a colorless solid (26 mg, 0.046 mmol, 46% yield). Mp 189–190 °C (hexane-EtOAc). [α]D25 = +41.7 (c = 0.34, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.88 (ddd, J = 8.6, 7.6, 1.3 Hz, 4H, Har), 7.75 (d, J = 7.2 Hz, 2H, Har), 7.58 (m, 1H, Har), 7.49 (dd, J = 8.5, 1.3 Hz, 2H, Har), 7.40 (m, 5H, Har), 7.21 (m, 6H, Har), 5.80 (s, 1H, CH), 1.39 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 190.4 (CO), 170.3 (CON), 137.9 (Car), 137.0 (Car), 136.6 (Car), 134.1 (CHar), 133.8 (CHar), 130.4 (CHar), 129.1 (CHar), 128.9 (CHar), 128.6 (CHar), 128.4 (CHar), 128.2 (CHar), 128.1 (CHar), 127.5 (CHar), 125.4 (CHar), 118.9 (CHar), 77.3 (C(CH3)3), 68.6 (CNHBoc), 51.9 (CH), 28.2 ((CH3)3C) ppm. IR (ATR): 3420, 3068, 2979, 2928, 1709, 1695, 1595, 1482, 1280, 1258, 1159, 971, 758, 685 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C35H32N3O5 574.2336; Found 574.2342. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 80:20, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 10.9 min, minor enantiomer (R) tR = 23.8 min. (er 93:7).
tert-Butyl (S)-(4-(2,4-dioxopentan-3-yl)-1-methyl-5-oxo-3-phenyl-4,5-dihydro-1H-pyrazol-4-yl)carbamate (3l). Product 3l was obtained according to general procedure using pentane-2,4-dione (11 µL, 0.11 mmol, 1.1 equiv) as β-diketone and catalyst C4 (6.4 mg, 0.01 mmol, 0.1 equiv). Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 3l as a colorless solid (24 mg, 0.062 mmol, 62% yield). [α]D25 = +46.0 (c = 0.3, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.77 (m, 2H, Har), 7.40 (m, 3H, Har), 6.76 (br s, 1H, NH), 3.86 (s, 1H, CH), 3.45 (s, 1H, CH3N), 2.07 (s, 3H, CH3CO), 2.03 (s, 3H, CH3CO), 1.36 (br s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 202.4 (CO), 200.5 (CO), 171.4 (CON), 133.9 (Car), 130.4 (CHar), 128.9 (CHar), 126.4 (CHar), 77.2 (C(CH3)3), 66.0 (CH), 64.9 (CNHBoc), 32.2 (CH3N), 32.2 (CH3CO), 28.1 (C(CH3)3) ppm. IR (ATR): 3375, 2978, 2926, 1703, 1483, 1351, 1252, 1157, 765, 695 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C20H25N3NaO5 410.1686; Found 410.1686. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 95:5, 0.8 mL/min, λ = 254 nm, major enantiomer (S) tR = 31.6 min, minor enantiomer (R) tR = 43.1 min. (er 90:10).
3.3. General Procedure for the Synthesis of Pyrazole Derivatives 4a–j by Reaction of Adducts 3a–j with Hydrazine Hydrate
To a solution of adduct 3 (0.1 mmol) in 1.0 mL of methanol, hydrated hydrazine (12 µL, 0.2 mmol, 2 equiv) was added at 0 °C, and the reaction mixture was then stirred at rt. The progress of the reaction was monitored by TLC analysis. After the completion of the reaction, the solvent was removed under reduced pressure. The crude reaction mixture was purified by flash column chromatography to afford the corresponding product 4. The enantiomeric excess was determined by chiral-phase HPLC analysis using mixtures of hexane/isopropanol as eluent.
tert-Butyl (S)-(3,3′,5-trimethyl-5′-oxo-1′-phenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4a). Product 4a was obtained from 3a according to general procedure. Chromatography on a silica gel using EtOAc as eluent afforded compound 4a as a colorless solid (31 mg, 0.081 mmol, 81% yield). Mp 220–222 °C (hexane-ethyl acetate). [α]D25 = +96.5 (c = 0.5, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J = 8.0, 0.8 Hz, 2H, Har), 7.39 (t, J = 7.8 Hz, 2H, Har), 7.18 (tt, J = 7.4, 1.2 Hz, 1H, Har), 5.92 (br s, 1H, NH), 2.28 (s, 6H, CH3), 2.11 (s, 3H, CH3), 1.37 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 172.0 (CON), 160.5 (CO2tBu), 154.1 (CCH3), 142.3 (CCH3), 138.0 (Car), 128.9 (CHar), 125.0 (CHar), 118.6 (CHar), 107.5 (C4pyrazole), 77.3 (C(CH3)3), 65.6 (CNHBoc), 28.1 (C(CH3)3), 14.2 (CH3), 12.9 (CH3) ppm. IR (ATR): 3247, 2978, 2934, 1711, 1692, 1593, 1501, 1365, 1251, 1156, 759, 693 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C20H25N5NaO3 406.1850; Found 406.1860. Chiral HPLC analysis: Lux Amylose-2 column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, minor enantiomer (R) tR = 19.9 min, major enantiomer (S) tR = 29.1 min. (er 94:6).
tert-Butyl (S)-(3′-ethyl-3,5-dimethyl-5′-oxo-1′-phenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4b). Product 4b was obtained from 3b according to general procedure. Chromatography on a silica gel using EtOAc as eluent afforded compound 4b as a colorless solid (27 mg, 0.068 mmol, 68% yield). [α]D25 = +62.8 (c = 0.14, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.97 (dd, J = 8.7, 1.2 Hz, 2H, Har), 7.40 (dd, J = 8.7, 7.4 Hz, 2H, Har), 7.18 (tt, J = 7.4, 1.2 Hz, 1H, Har), 5.71 (br s, 1H, NH), 2.50 (dq, J = 17.6, 7.4 Hz, 1H, CHHCH3), 2.37 (m, 1H, CHHCH3), 2.28 (s, 6H, CH3), 1.36 (s, 9H, C(CH3)3), 1.30 (t, J = 7.0 Hz, 3H, CH3CH2) ppm. 13C NMR (126 MHz, CDCl3): 172.1 (CON), 164.0 (CO2tBu), 154.0 (CEt), 142.3 (CCH3), 138.2 (Car), 128.8 (CHar), 124.9 (CHar), 118.5 (CHar), 108.0 (C4pyrazole), 77.2 (C(CH3)3), 65.6 (CNHBoc), 28.1 (C(CH3)3), 21.5 (CH2CH3), 12.9 (CH3), 9.1 (CH3CH2) ppm. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C21H28N5O3 398.2187; Found 398.216. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 25.8 min, minor enantiomer (R) tR = 31.8 min. (er 85:15).
tert-Butyl (S)-(3′-isopropyl-3,5-dimethyl-5′-oxo-1′-phenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4c). Product 4c was obtained from 3c according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 2:1 as an eluent afforded compound 4c as a colorless solid (32 mg, 0.078 mmol, 78% yield). [α]D25 = +139.8 (c = 0.46, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.99 (d, J = 7.1 Hz, 2H, Har), 7.40 (dd, J = 8.5, 7.4 Hz, 2H, Har), 7.18 (t, J = 7.4 Hz, 1H, Har), 6.22 (br s, 1H, NH), 2.66 (sept, J = 6.8 Hz, 1H, CH(CH3)2), 2.23 (s, 6H, CH3), 1.36 (s, 9H, C(CH3)3), 1.32 (d, J = 7.0 Hz, 3H, CH3CH), 1.07 (d, J = 6.8 Hz, 3H, CH3CH) ppm. 13C NMR (126 MHz, CDCl3): 172.3 (CON), 167.0 (CO2tBu), 154.4 (CiPr), 141.9 (CCH3), 138.1 (Car), 128.8 (CHar), 125.0 (CHar), 118.6 (CHar), 107.8 (C4pyrazole), 77.2 (C(CH3)3), 66.2 (CNHBoc), 28.2 (CH(CH3)2), 28.1 (C(CH3)3), 21.1 (CH3CH), 20.8 (CH3CH), 12.8 (CH3) ppm. IR (ATR): 3290, 2975, 2931, 1708, 1597, 1494, 1367, 1159, 759, 737, 693 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C22H29N5NaO3 434.2163; Found 434.2162. Chiral HPLC analysis: Lux Amylose-2 column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, minor enantiomer (R) tR = 17.5 min, major enantiomer (S) tR = 23.5 min. (er 90:10).
tert-Butyl (S)-(3,5-dimethyl-5′-oxo-1′,3′-diphenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4e). Product 4e was obtained from 3e according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 1:1 as eluent afforded compound 4e as a colorless solid (36 mg, 0.082 mmol, 82% yield). [α]D25 = −190.0 (c = 0.1, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.13 (br s, 1H, NH), 7.99 (dd, J = 8.7, 1.2 Hz, 2H, Har), 7.85 (d, J = 7.1 Hz, 2H, Har), 7.39 (m, 5H, Har), 7.19 (tt, J = 7.4, 1.2 Hz, 1H, Har), 2.30 (s, 6H, CH3), 1.19 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 171.5 (CON), 167.0 (CO2tBu), 153.7 (CPh), 143.1 (CCH3), 138.3 (Car), 138.2 (Car), 128.9 (CHar), 128.8 (CHar), 126.4 (CHar), 125.1 (CHar), 118.7 (CHar), 108.7 (C4pyrazole), 77.2 (C(CH3)3), 64.1 (CNHBoc), 27.9 (C(CH3)3), 12.8 (CH3) ppm. IR (ATR): 3237, 3123, 3060, 2978, 2931, 1730, 1708, 1594, 1500, 1367, 1159, 759, 737, 689 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C25H28N5O3 446.2187; Found 446.2205. Chiral HPLC analysis: Lux i-Amylose-3 column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 12.8 min, minor enantiomer (R) tR = 32.5 min. (er 94:6).
tert-Butyl (S)-(1′-(4-chlorophenyl)-3,3′,5-trimethyl-5′-oxo-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4f). Product 4f was obtained from enantioenriched 3f according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 1:3 as eluent afforded compound 4f as a colorless solid (25 mg, 0.060 mmol, 60% yield). [α]D25 = +51.0 (c = 0.22, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.91 (d, J = 8.9 Hz, 2H, Har), 7.35 (d, J = 8.9 Hz, 2H, Har), 5.84 (br s, 1H, NH), 2.27 (s, 6H, CH3), 2.10 (s, 3H, CH3), 1.37 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 171.9 (CON), 160.7 (CO2tBu), 154.0 (CMe), 142.3 (CCH3pyrazole), 136.6 (Car), 130.1 (Car), 128.9 (CHar), 119.6 (CHar), 107.4 (C4pyrazole), 77.2 (C(CH3)3), 65.5 (CNHBoc), 28.1 (C(CH3)3), 14.2 (CH3), 12.8 (CH3) ppm. IR (ATR): 3268, 2978, 2931, 1708, 1490, 1361, 1254, 1159, 1093, 1011, 910, 828, 727 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C20H25ClN5O3 418.1640; Found 418.1633. HPLC: Lux i-Amylose-3 column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 26.5 min, minor enantiomer (R) tR = 52.5 min. (er 94:6).
tert-Butyl (S)-(3,3′,5-trimethyl-5′-oxo-1′-(p-tolyl)-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4g). Product 4g was obtained from 3g according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 1:3 as an eluent afforded compound 4g as a colorless solid (25 mg, 0.063 mmol, 63% yield). [α]D25 = +61.2 (c = 0.3, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.80 (d, J = 8.6 Hz, 2H, Har), 7.19 (d, J = 8.6 Hz, 2H, Har), 6.00 (br s, 1H, NH), 2.34 (s, 3H, CH3C4H6), 2.26 (s, 6H, CH3), 2.09 (s, 3H, CH3), 1.37 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 171.8 (CON), 160.5 (CO2tBu), 154.1 (CCH3), 142.3 (CCH3pyrazole), 135.6 (Car), 134.7 (Car), 129.4 (CHar), 118.6 (CHar), 107.6 (C4pyrazole), 77.3 (C(CH3)3), 65.5 (CNHBoc), 28.1 (C(CH3)3), 20.9 (CH3C6H4), 14.1 (CH3), 12.8 (CH3) ppm. IR (ATR): 3268, 2982, 2928, 1705, 1509, 1361, 1250, 1159, 815, 730 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C21H28N5O3 398.2187; Found 398.2183. Chiral HPLC analysis: Chiralpak IA, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 23.5 min, minor enantiomer (S) tR = 47.2 min. (er 88:12).
tert-Butyl (S)-(3,5-diethyl-5′-oxo-1′,3′-diphenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (4j). Product 4j was obtained from 3j according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 2:1 as eluent afforded compound 4j as a colorless solid (19 mg, 0.040 mmol, 40% yield). [α]D25 = −134.3 (c = 0.2, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.97 (dd, J = 8.6, 1.2 Hz, 2H, Har), 7.82 (d, J = 7.6 Hz, 2H, Har), 7.42 (m, 5H, Har), 7.21 (tt, J = 7.4, 1.2 Hz, 1H, Har), 2.98 (m, 2H, CHHCH3), 2.86 (dq, J = 15.5, 7.6 Hz, CHHCH3), 1.24 (t, J = 7.5, 6H), 1.20 (s, 9H, C(CH3)3 ppm. 13C NMR (126 MHz, CDCl3): δ 166.1 (CON), 148.5 (CPh), 144.0 (CEt), 133.4 (Car), 124.3 (CHar), 124.2 (CHar), 121.7 (CHar), 120.6 (CHar), 114.1 (CHar), 104.4 (C4pyrazole), 72.3 (C(CH3)3), 23.2 (C(CH3)3), 15.1 (CH3CH2), 8.7 (CH3CH2), 7.3 (CH3CH2) ppm. IR (ATR): 3250, 2975, 2928, 1701, 1594, 1490, 1368, 1159, 756, 693 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C27H32N5O3 474.2500; Found 474.2486. Chiral HPLC analysis: Chiralpak IA, hexane/i-PrOH 95:5, 1 mL/min, λ = 254 nm, minor enantiomer (R) tR = 20.1 min, major enantiomer (S) tR = 25.0 min. (er 95:5).
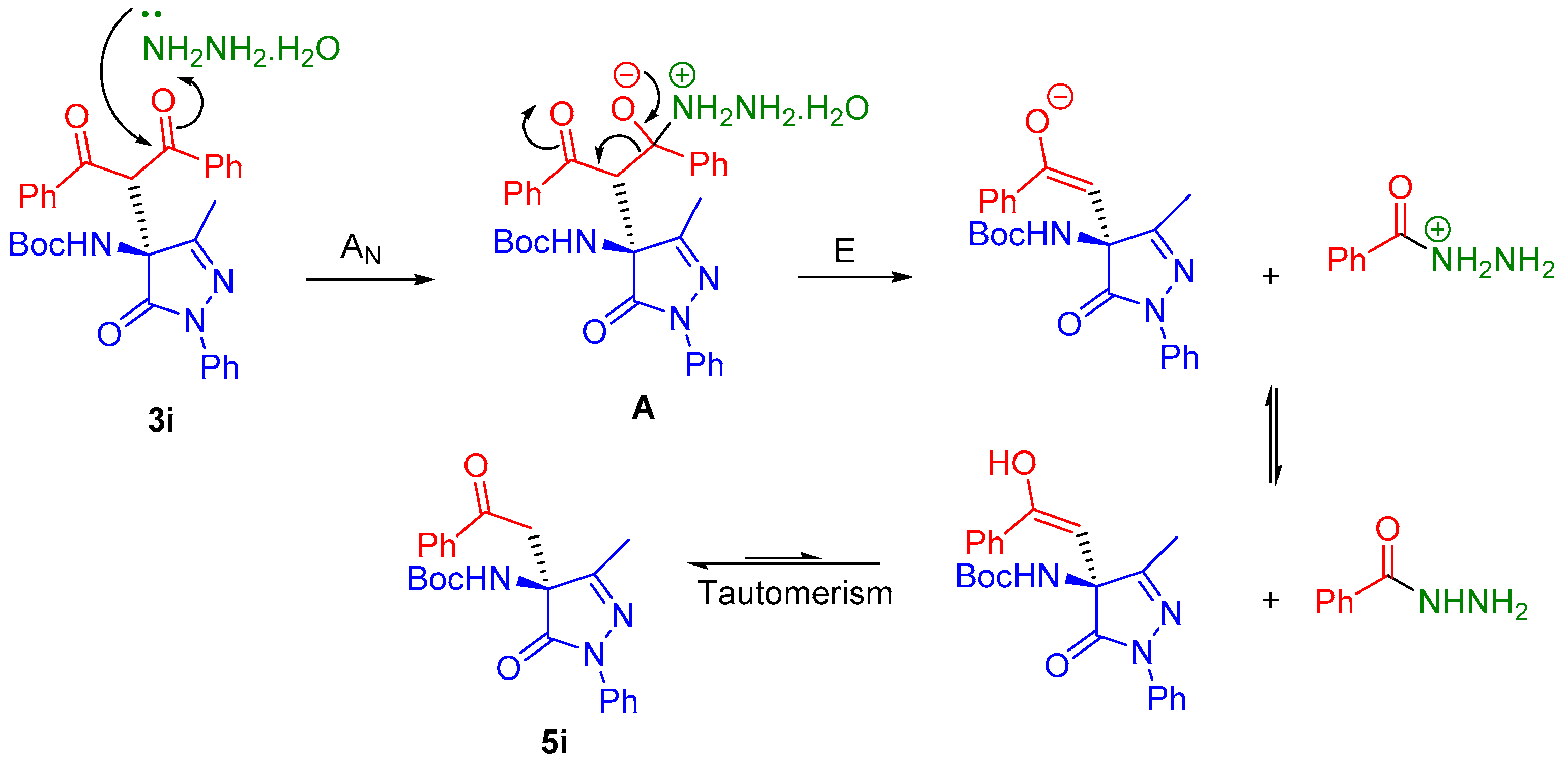
tert-Butyl (
S)-(3-methyl-5-oxo-4-(2-oxo-2-phenylethyl)-1-phenyl-4,5-dihydro-1
H-pyrazol-4-yl)carbamate (
5i) [
19]. Product
5i was obtained from
3i according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 3:1 as eluent afforded compound
5i as a colorless solid (21 mg, 0.052 mmol, 52% yield). [α]
D25 = −17.5 (
c = 0.3, CH
2Cl
2). [Lit. [
19] [α]
D20 = −20.2 (
c = 1, CH
2Cl
2, er 94:6 for (
S) enantiomer)].
1H NMR (400 MHz, DMSO-d
6): δ 7.90 (br s, 1H,
Har), 7.83 (m, 2H,
Har), 7.74 (d, J = 7.8 Hz, 2H,
Har), 7.62 (tt, J = 7.4, 1.3 Hz, 1H,
Har), 7.49 (t, J = 7.8 Hz, 2H,
Har), 7.38 (dd, J = 8.7, 7.4 Hz, 2H,
Har), 7.14 (tt, J = 7.4, 1.3 Hz, 1H,
Har), 3.74 (d, J = 17.2 Hz, 1H, C
HHCOPh), 3.62 (d, J = 17.2 Hz, 1H, C
HHCOPh), 1.99 (s, 3H, C
H3), 1.31 (s, 9H, C(C
H3)
3) ppm.
13C NMR (100 MHz, DMSO-d
6): δ 195 (
CO), 172.3 (
CON), 158.8 (
CO
2tBu), 153.8 (
CCH
3), 138.7 (
Car), 136.2 (
Car), 134.2 (
CHar), 129.3 (
CHar), 129.2 (
CHar), 128.4 (
CHar), 124.7 (
CHar), 118.1 (
CHar), 80.0 (
C(CH
3)
3), 63.6 (
CNHBoc), 42.6 (
CH
2), 28.4 (C(
CH
3)
3), 13.5 (
CH
3) ppm. IR (ATR): 2856, 1714, 1594, 1500, 1364, 1251, 1159, 753, 693 cm
−1. HRMS (ESI-QTOF) m/z: [M+Na]
+ Calcd. For C
35H
25N
3NaO
4 430.1737; Found 430.1759. Chiral HPLC analysis: Chiralpak IA column, hexane/
i-PrOH 80:20, 1 mL/min, λ = 254 nm, minor enantiomer (
R)
tR = 6.9 min, major enantiomer (
S)
tR = 32.4 min. (
er 77:23).
3.4. General Procedure for the Synthesis of Pyrazole Derivatives 6a,e
A solution of 3a,e (0.1 mmol), 4-chlorophenylhydrazine hydrochloride (19 mg, 0.11 mmol, 1.1 equiv) and and K2CO3 (8 mg, 0.055 mmol, 0.55 equiv) in ethanol (1 mL) was heated to 80 °C for 2–3 h. After that, the solvent of reaction mixture was removed under reduced pressure. The crude product was purified by flash chromatography on silica gel to afford 6a,e.
tert-Butyl (S)-(1-(4-chlorophenyl)-3,3′,5-trimethyl-5′-oxo-1′-phenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (6a). Product 6a was obtained from 3a according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 4:1 as an eluent afforded compound 6a as a colorless solid (37 mg, 0.075 mmol, 75% yield). Mp 196–197 °C (hexane-ethyl acetate). [α]D25 = +59.9 (c = 0.7, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.94 (dd, J = 8.7, 1.2 Hz, 2H, Har), 7.40 (m, 4H, Har), 7.28 (d, J = 8.5 Hz, 2H, Har), 7.18 (tt, J = 7.4, 1.2 Hz, 1H, Har), 5.41 (br s, 1H, NH), 2.40 (s, 3H, CH3), 2.33 (s, 3H, CH3), 2.19 (s, 3H, CH3), 1.39 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 171.5 (CON), 159.8 (CO2tBu), 153.7 (CCH3), 146.6 (CCH3), 138.0 (Car), 137.1 (Car), 134.4 (Car), 129.4 (CHar), 128.9 (CHar), 127.1 (CHar), 125.1 (CHar), 118.6 (CHar), 110.0 (C4pyrazole), 79.7 (C(CH3)3), 65.4 (CNHBoc), 28.1 (C(CH3)3), 14.5 (CH3), 14.1 (CH3), 12.3 (CH3) ppm. IR (ATR): 3269, 2982, 2928, 1711, 1598, 1500, 1393, 1364, 1295, 1254, 1163, 1093, 1014, 838, 759, 690, 645 cm−1. HRMS (ESI-QTOF) m/z: [M+H]+ Calcd. For C26H29ClN5O3 494.1953; Found 494.1931. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 45.0 min, minor enantiomer (R) tR = 74.2 min. (er 84:16).
tert-Butyl (S)-(1-(4-chlorophenyl)-3,5-dimethyl-5′-oxo-1′,3′-diphenyl-1′,5′-dihydro-1H,4′H-[4,4′-bipyrazol]-4′-yl)carbamate (6e). Product 6e was obtained from 3e according to general procedure. Chromatography on a silica gel using hexane/EtOAc = 4:1 as eluent afforded compound 6e as a colorless solid (22 mg, 0.040 mmol, 40% yield). [α]D25 = −155.0 (c = 0.4, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.01 (dd, J = 8.8, 1.1 Hz, 2H, Har), 7.91 (d, J = 6.5 Hz, 2H, Har), 7.44 (m, 5H, Har), 7.42 (d, J = 8.7 Hz, 2H, Har), 7.29 (d, J = 8.7 Hz, 2H, Har), 7.20 (tt, J = 7.4, 1.2 Hz, 1H, Har), 5.54 (br s, 1H, NH), 2.41 (s, 3H, CH3), 2.31 (s, 3H, CH3), 1.22 (s, 9H, C(CH3)3) ppm. 13C NMR (126 MHz, CDCl3): δ 171.7 (CON), 153.6 (CCH3), 146.9 (CPh), 138.4 (Car), 137.5 (Car), 134.1 (Car), 130.7 (CCH3), 130.3 (Car), 129.3 (CHar), 128.9 (CHar), 126.9 (CHar), 126.5 (CHar), 125.1 (CHar), 118.9 (CHar), 110.5 (C4pyrazole), 77.2 (C(CH3)3), 64.4 (CNHBoc), 27.9 (C(CH3)3), 14.2 (CH3), 12.6 (CH3) ppm. IR (ATR): 3245, 2975, 2854, 1727, 1701, 1596, 1500, 1362, 1260, 1158, 1092, 1016, 829, 756, 735, 691 cm−1. HRMS (ESI-QTOF) m/z: [M+Na]+ Calcd. For C31H30N5ClNaO3 578.1929; Found 578.1943. Chiral HPLC analysis: Chiralpak AD-H column, hexane/i-PrOH 90:10, 1 mL/min, λ = 254 nm, major enantiomer (S) tR = 10.1 min, minor enantiomer (R) tR = 63.7 min. (er 96:4).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
