
Glycoconjugation of Quinoline Derivatives Using the C-6 Position in Sugars as a Strategy for Improving the Selectivity and Cytotoxicity of Functionalized Compounds
 , , , and
, , , and 
Abstract
:1. Introduction
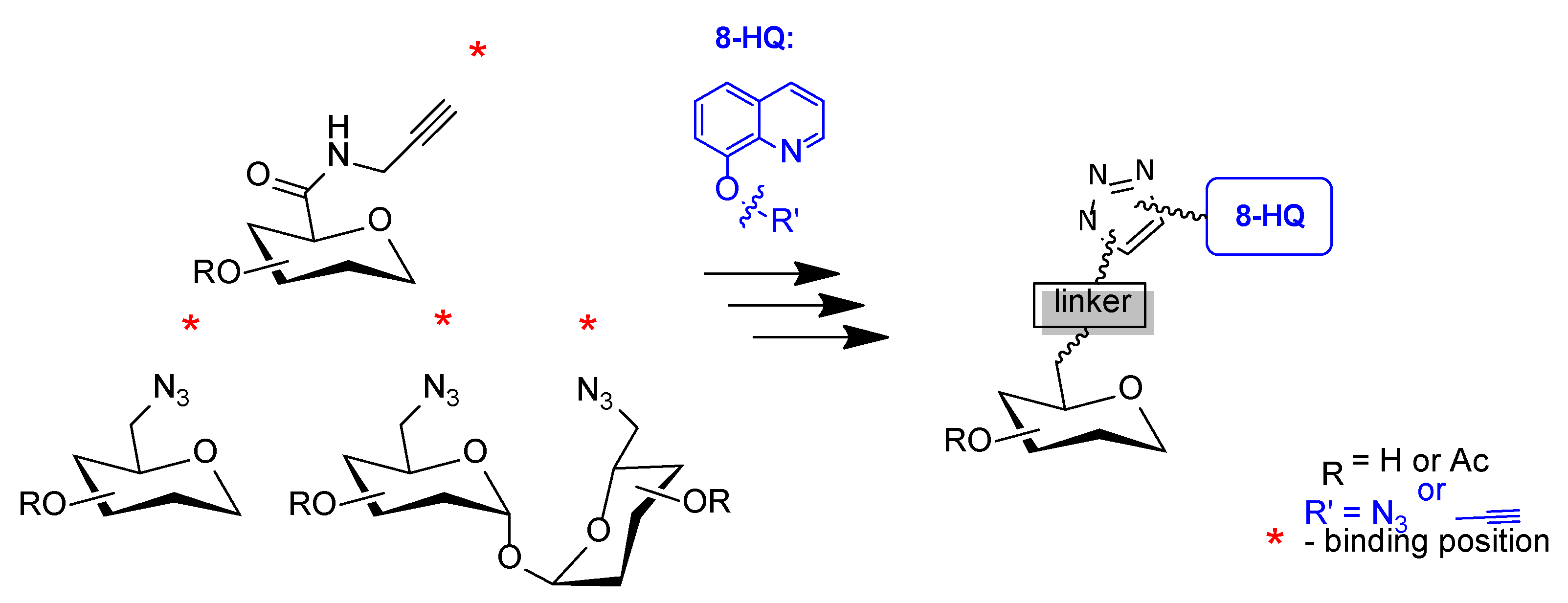
2. Results and Discussion
2.1. Synthesis
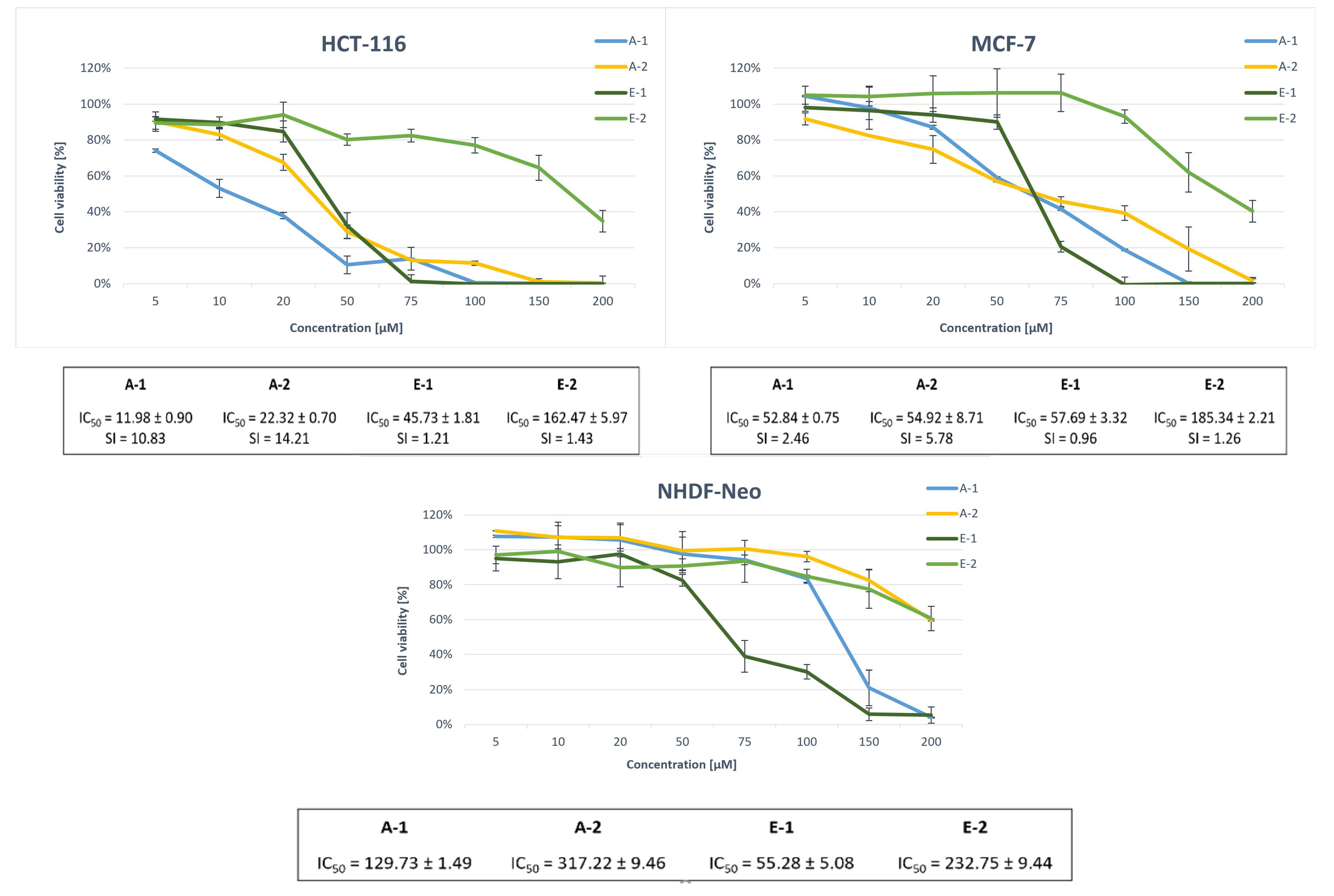
2.2. Biological Evaluation
2.2.1. Antiproliferative Studies
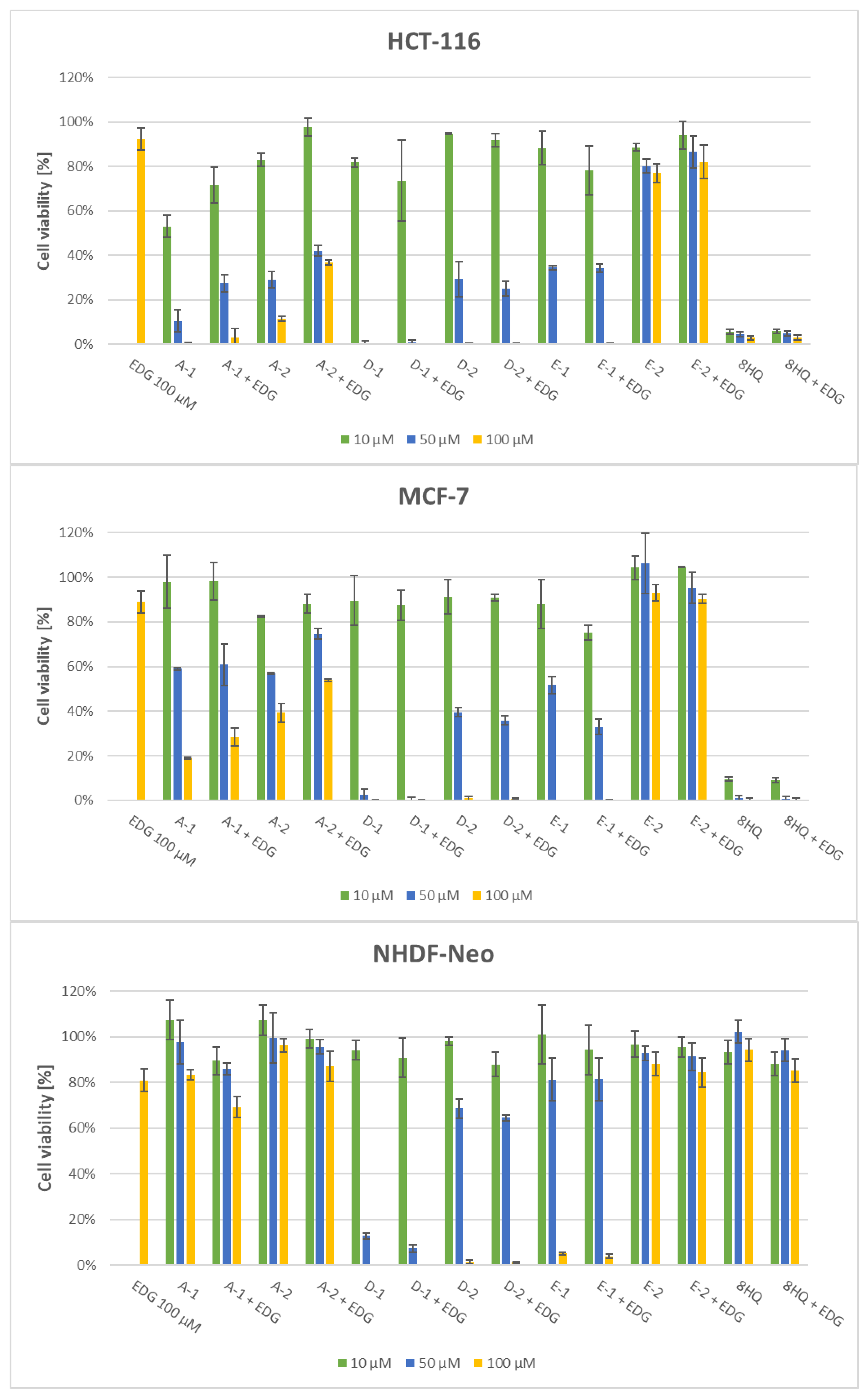
2.2.2. Effect of the GLUT1 Inhibitor
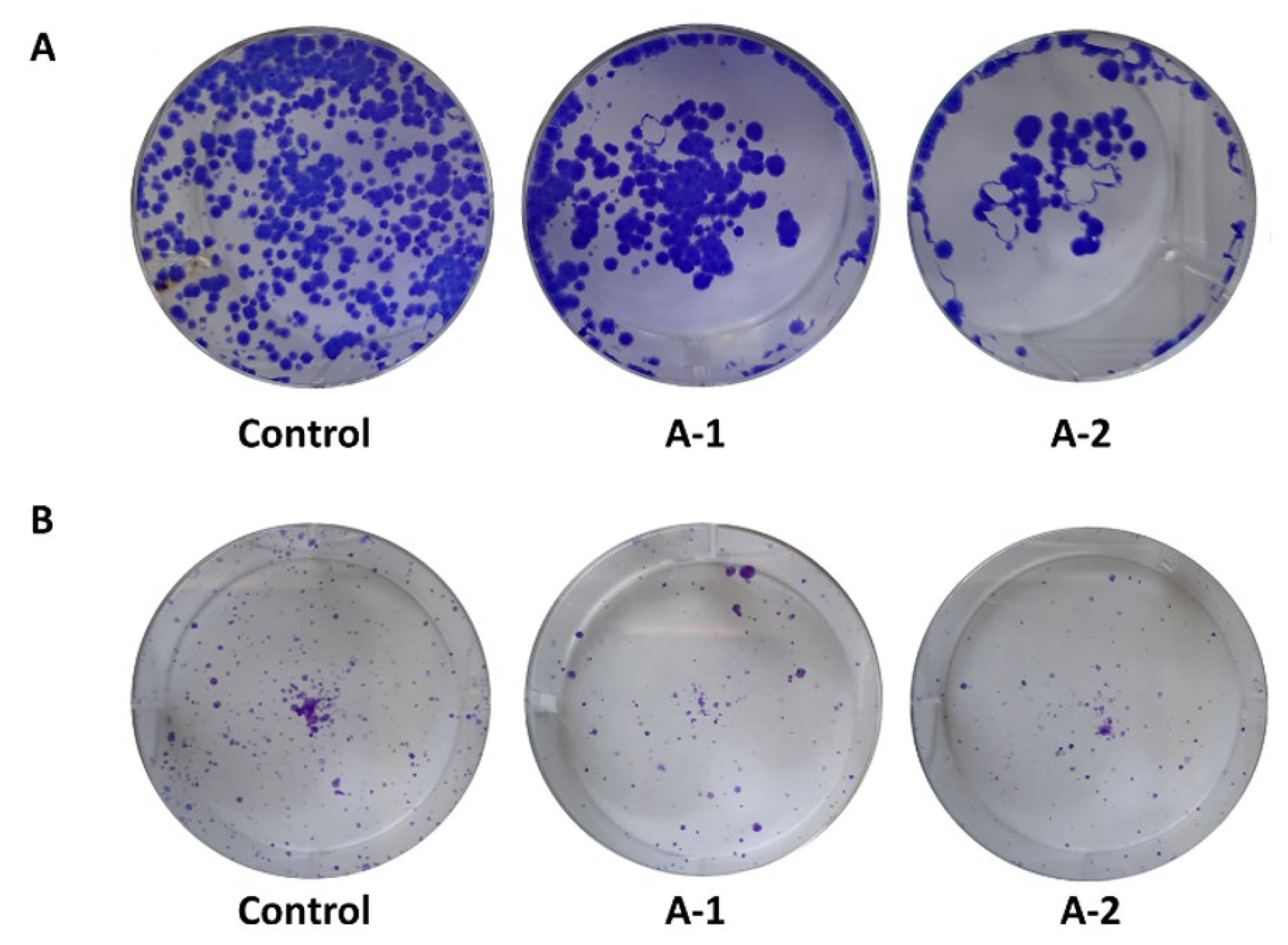
2.2.3. Clonogenic Assay
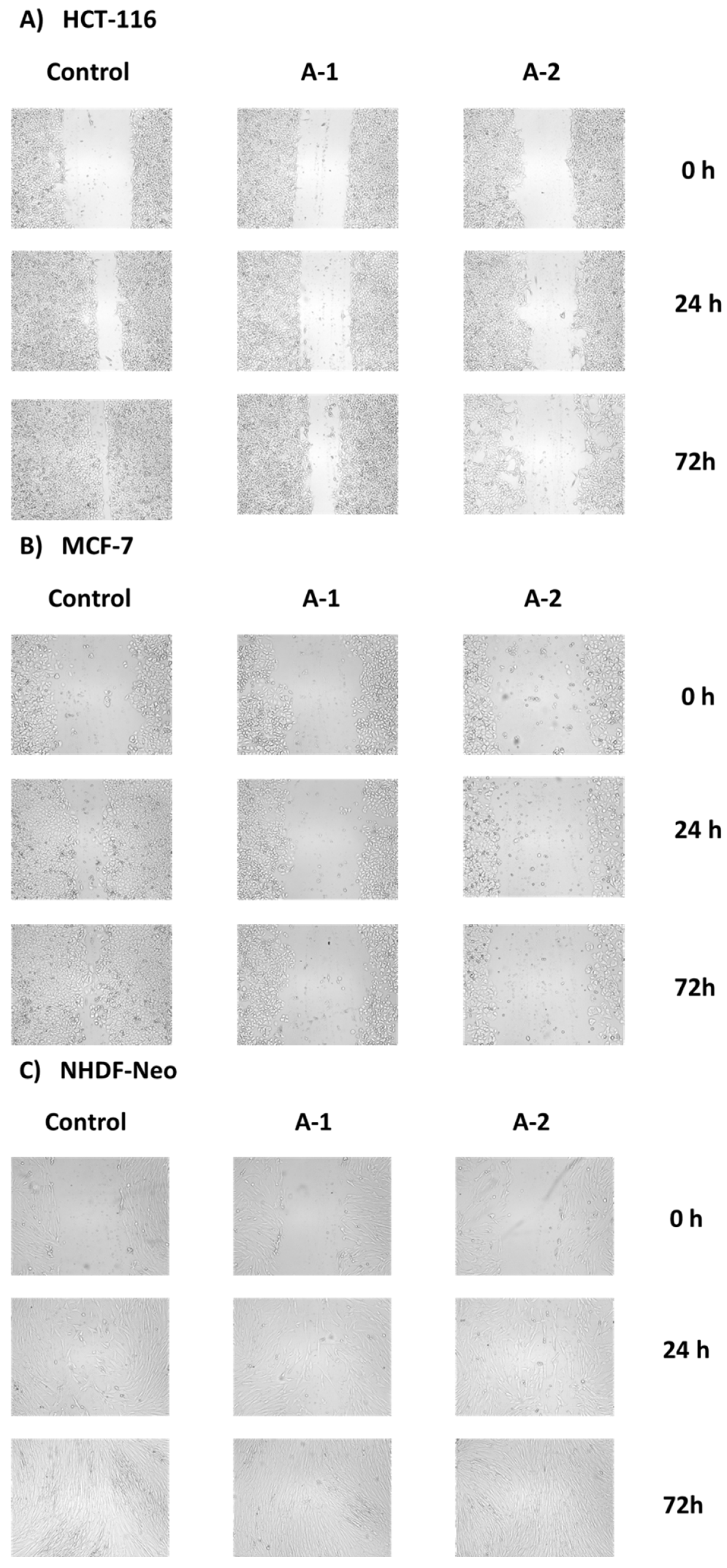
2.2.4. Wound Healing Assay
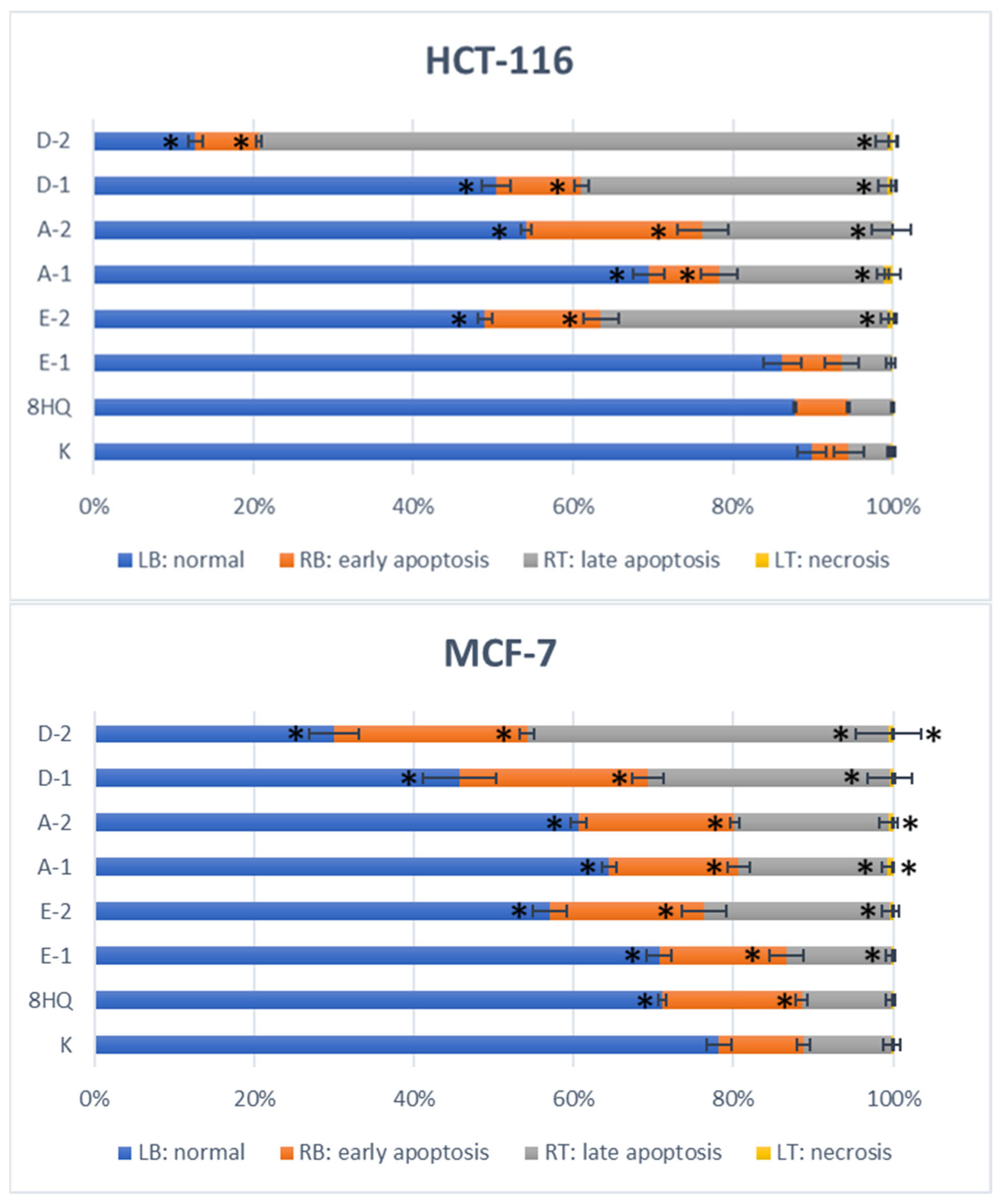
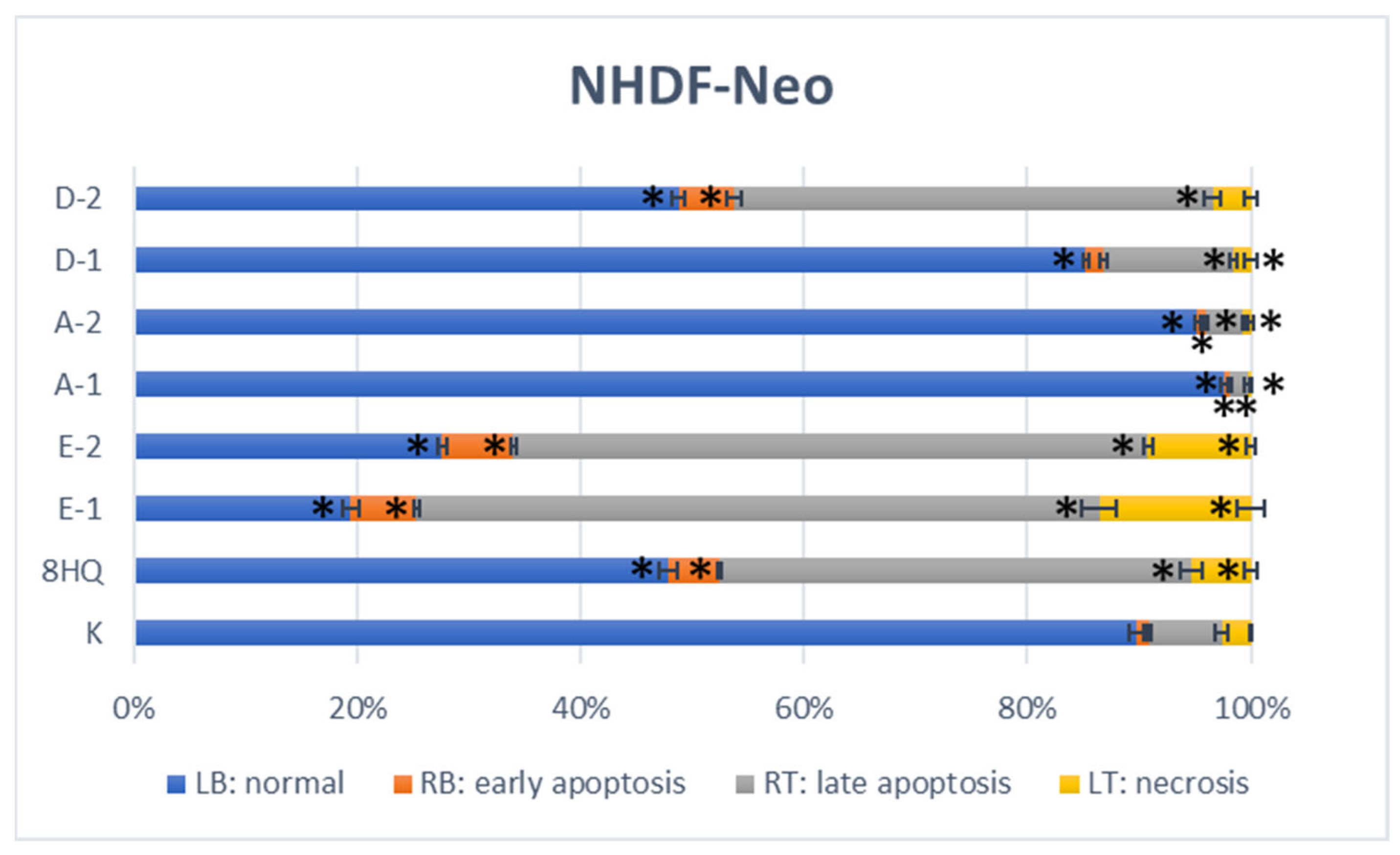
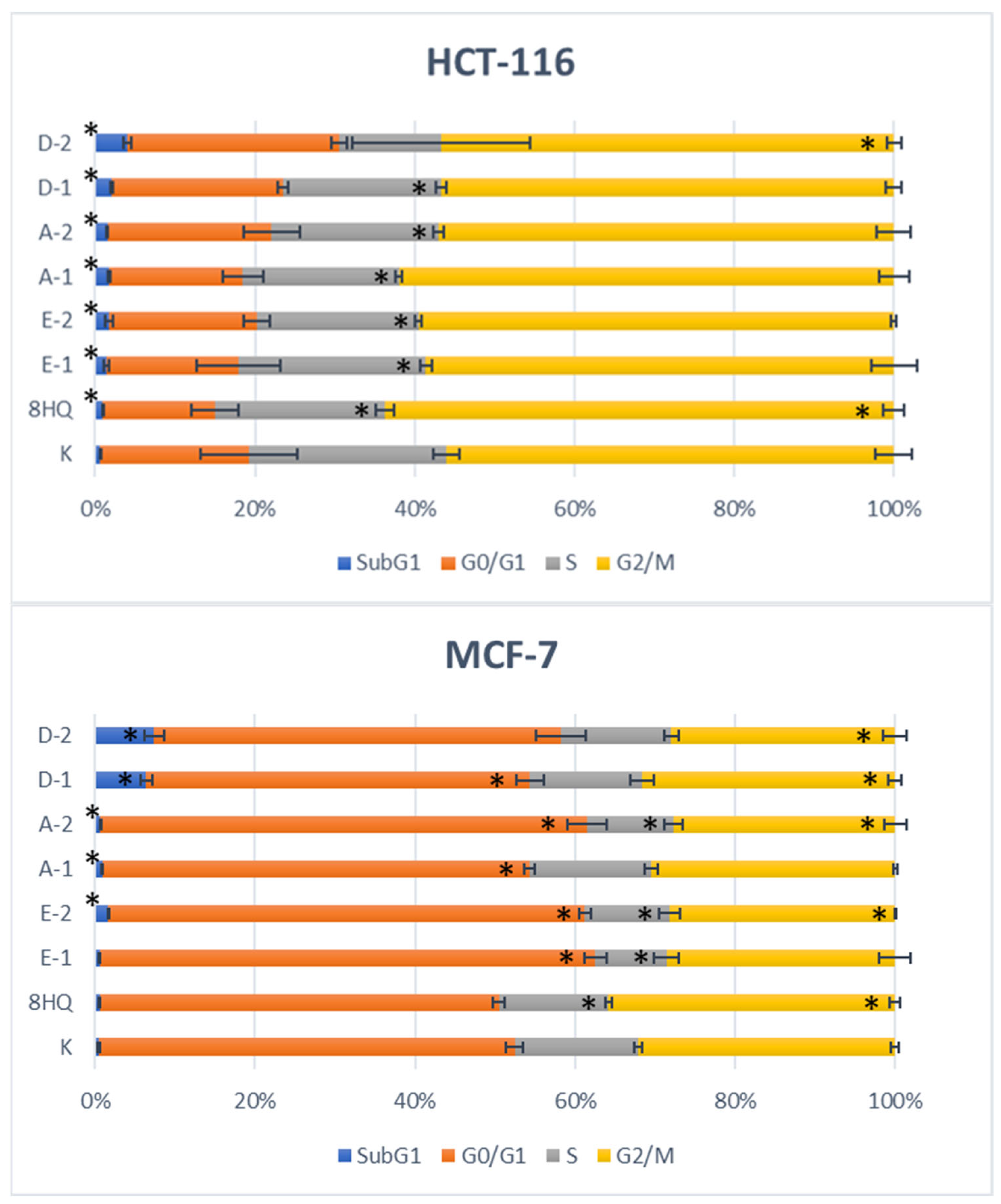
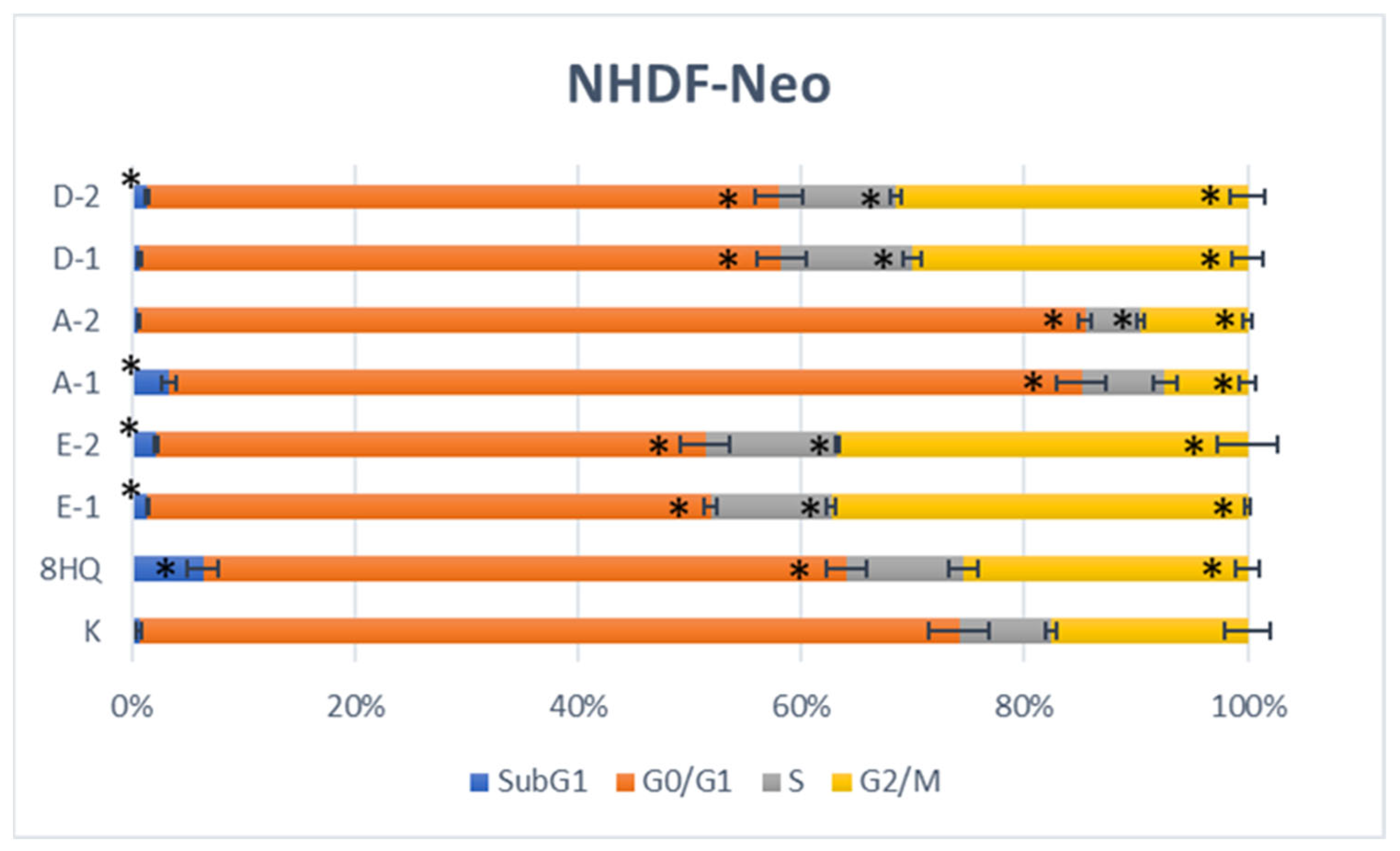
2.2.5. Apoptosis and Cell Cycle Analyses by Flow Cytometry
2.2.6. Intercalation Study
3. Materials and Methods
3.1. General Information
3.2. Chemistry
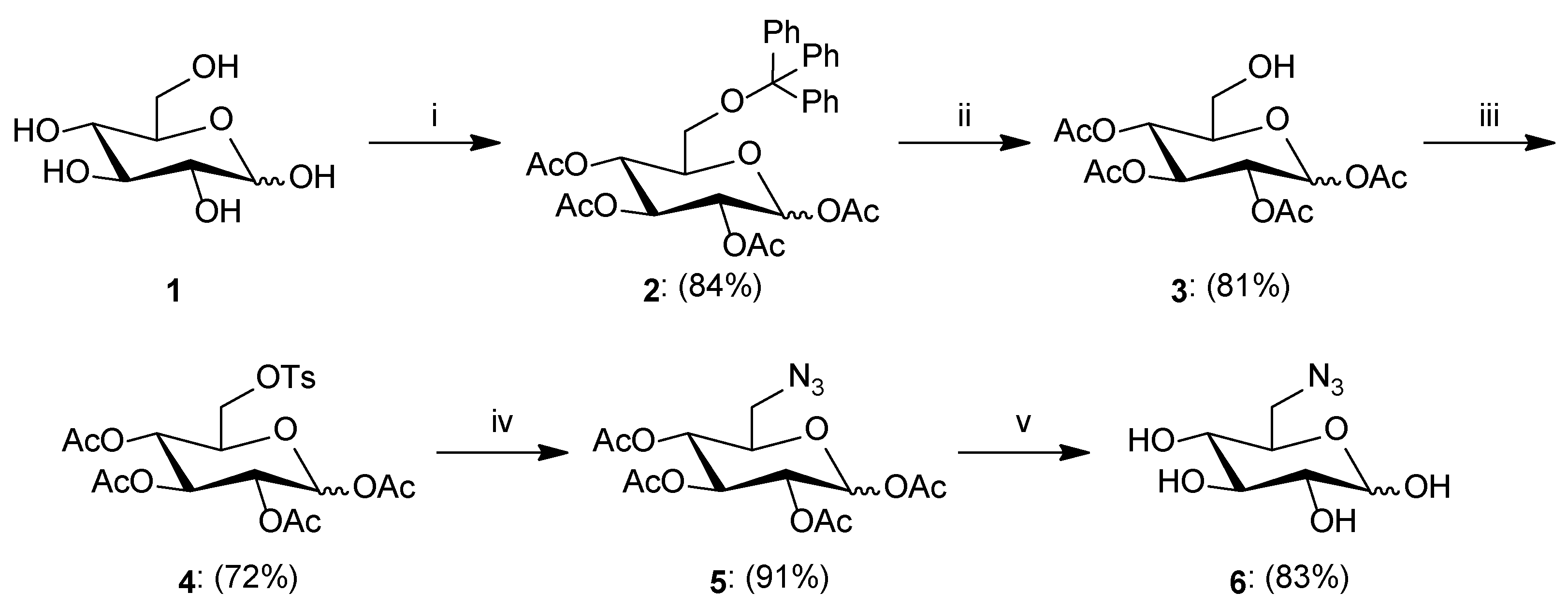
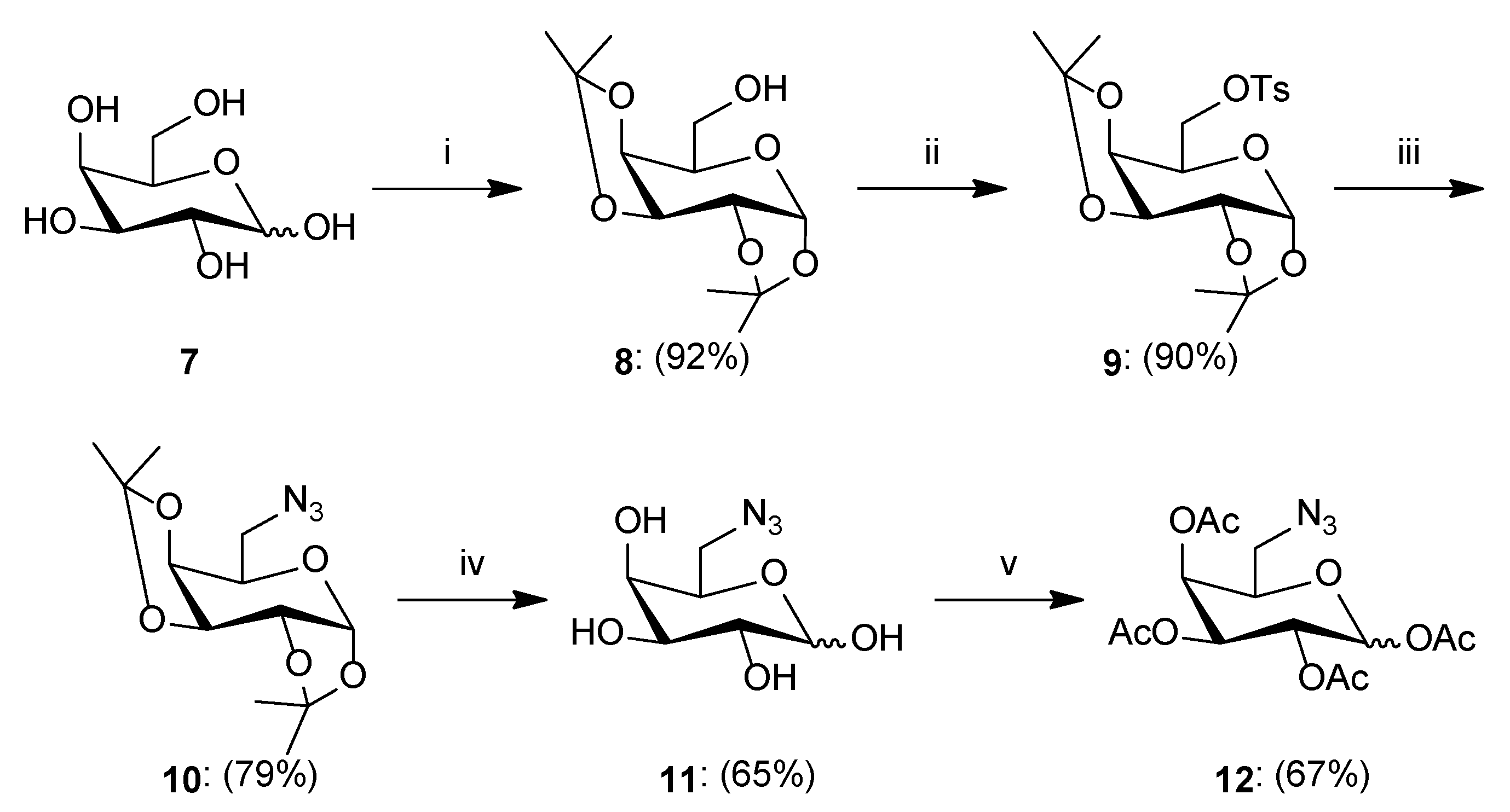
3.2.1. General Procedure for the Synthesis of Sugar Derivatives
- Synthesis of 1,2,3,4-Tetra-O-acetyl-6-O-triphenylmethyl-d-glucopyranose2: d-Glucose 1 (1.507 g, 8.365 mmol) was dissolved in pyridine (8 mL), and then trityl chloride (2.350 g, 8.430 mmol) and DMAP (0.180 g, 1.473 mmol) were added. The reaction mixture was stirred for 24 h at room temperature. After this time, acetyl chloride (4 mL) was added in portions to the reaction mixture and stirred for 1 h at room temperature. Then, the reaction diluted with dichloromethane (100 mL) and washed with brine (3 × 30 mL). The organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 100:1 to 10:1) to give product 2 as a white solid (4.155 g, 84%); ratio of anomers (α:β = 1:1.3). 1H NMR (400 MHz, CDCl3): δ 1.73, 1.74, 2.00, 2.01, 2.03, 2.04, 2.15, 2.15 (8s, 24H, 4 × CH3-α, 4 × CH3-β), 3.02 (dd, 1H, J = 3.9 Hz, J = 10.7 Hz, H6a-α), 3.07 (dd, 1H, J = 4.2 Hz, J = 10.6 Hz, H6a-β), 3.32 (dd, 1H, J = 2.2 Hz, J = 10.7 Hz, H6b-α), 3.34 (dd, 1H, J = 2.5 Hz, J = 10.6 Hz, H6a-β), 3.69 (ddd, 1H, J = 2.5 Hz, J = 4.2 Hz, J = 9.8 Hz, H5-β), 4.02 (ddd, 1H, J = 2.2 Hz, J = 3.9 Hz, J = 10.2 Hz, H5-α), 5.14–5.29 (m, 4H, H2-α, H4-α, H2-β, H4-β), 5.31 (dd~t, 1H, J = 9.6 Hz, J = 10.1 Hz, H3-β), 5.41 (dd~t, 1H, J = 9.7 Hz, J = 10.0 Hz, H3-α), 5.72 (d, 1H, J = 8.0 Hz, H1-β), 6.44 (d, 1H, J = 3.7 Hz, H1-α), 7.12–7.46 (m, 30H, 3 × Ph-α, 3 × Ph-β); 13C NMR (100 MHz, CDCl3): δ 20.42, 20.45, 20.49, 20.60, 20.64, 20.74, 20.87, 20.93 (4 × CH3-α, 4 × CH3-β), 61.24, 61.69 (C6-α, C6-β), 68.29, 68.34, 69.50, 70.36, 70.56, 71.26, 73.19, 74.07 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 86.54, 86.66 (C-Ph3-α, C-Ph3-β), 89.36, 91.94 (C1-α, C1-β), 127.02, 127.02, 127.79, 127.81, 128.70, 128.72 (C2Ph-α, C3Ph-α, C4Ph-α, C2Ph-β, C3Ph-β, C4Ph-β), 143.46, 143.48 (C1Ph-α, C1Ph-β), 168.88, 168.89, 168.93, 168.97, 169.33, 169.76, 170.24, 170.43 (4 × CO-α, 4 × CO-β); HRMS (ESI-TOF): calcd for C33H34O10Na ([M + Na]+): m/z 613.2050; found: m/z 613.2050.
- Synthesis of 1,2,3,4-Tetra-O-acetyl-d-glucopyranose3: Compound 2 (1.251 g, 2.116 mmol) was suspended in glacial acetic acid (10 mL). The solution was cooled to 0 °C and 33% solution of hydrogen bromide in acetic acid (0.370 mL, 6.494 mmol) was added. Stirring was continued at room temperature for 1 h. After completion, the reaction was diluted with chloroform (50 mL) and water (50 mL). The aqueous phase was washed with chloroform (2 × 30 mL), and the combined organic phases were washed with NaHCO3 (2 × 50 mL) and brine (1 × 50 mL). Next, the organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 20:1 to 1:1) to give product 3 as a yellow oil (0.597 g, 81%); ratio of anomers (α:β = 1:3.3). 1H NMR (400 MHz, CDCl3): δ 2.02, 2.03, 2.03, 2.04, 2.07, 2.08, 2.11, 2.18 (8s, 24H, 4 × CH3-α, 4 × CH3-β), 3.54–3.62 (m, 2H, H6a-α, H6a-β), 3.65 (ddd, 1H, J = 2.2 Hz, J = 4.2 Hz, J = 9.9 Hz, H5-β), 3.69–3.81 (m, 2H, H6b-α, H6b-β), 3.93 (ddd, 1H, J = 2.3 Hz, J = 4.0 Hz, J = 10.2 Hz, H5-α), 5.04–5.15 (m, 4H, H2-α, H4-α, H2-β, H4-β), 5.31 (dd~t, 1H, J = 9.5 Hz, J = 9.5 Hz, H3-β), 5.53 (dd~t, 1H, J = 9.9 Hz, J = 10.0 Hz, H3-α), 5.73 (d, 1H, J = 8.3 Hz, H1-β), 6.35 (d, 1H, J = 3.7 Hz, H1-α); 13C NMR (100 MHz, CDCl3): δ 20.46, 20.56, 20.60, 20.63, 20.69, 20.81, 20.89, 21.33 (4 × CH3-α, 4 × CH3-β), 60.85, 62.44 (C6-α, C6-β), 68.20, 68.31, 69.38, 69.60, 70.43, 72.05, 72.64, 74.92 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 89.12, 91.74 (C1-α, C1-β), 168.89, 168.98, 169.06, 169.26, 169.66, 170.09, 170.19, 170.26 (4 × CO-α, 4 × CO-β); HRMS (ESI-TOF): calcd for C14H20O10Na ([M + Na]+): m/z 371.0954; found: m/z 371.0951.
- Synthesis of 1,2,3,4-Tetra-O-acetyl-6-O-p-toluenesulfonyl-d-glucopyranose4: To a solution of 3 (0.338 g, 0.970 mmol) in pyridine (5 mL), p-toluenosulfonyl chloride (0.461 g, 2.418 mmol) and DMAP (23.8 mg, 0.195 mmol) were added. The reaction mixture was stirred at room temperature for 24 h. After completion, the reaction was diluted with water (50 mL) and washed with chloroform (3 × 50 mL). The organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 30:1 to 2:1) to give product 4 as a white solid (0.352 g, 72%); ratio of anomers (α:β = 1:4). 1H NMR (400 MHz, CDCl3): δ 1.98, 1.99, 2.00, 2.01, 2.02, 2.02, 2.09, 2.16 (8s, 24H, 4 × CH3-α, 4 × CH3-β), 2.46 (s, 6H, Ar-CH3-α, Ar-CH3-β), 3.84 (m, 1H, H5-β), 4.04–5.25 (m, 5H, H5-α, H6a-α, H6b-α, H6a-β, H6b-β), 4.93–5.09 (m, 4H, H2-α, H4-α, H2-β, H4-β), 5.20 (dd~t, 1H, J = 9.4 Hz, J = 9.4 Hz, H3-β), 5.42 (dd~t, 1H, J = 9.6 Hz, J = 10.2 Hz, H3-α), 5.65 (d, 1H, J = 8.2 Hz, H1-β), 6.21 (d, 1H, J = 3.7 Hz, H1-α), 7.32–7.38 (m, 4H, Ar-H-α, Ar-H-β), 7.75–7.80 (m, 4H, Ar-H-α, Ar-H-β); 13C NMR (100 MHz, CDCl3): δ 20.40, 20.49, 20.52, 20.55, 20.58, 20.64, 20.74, 20.84 (4 × CH3-α, 4 × CH3-β), 21.68 (Ar-CH3-α, Ar-CH3-β), 66.74, 67.16, 67.95, 68.11, 69.00, 69.61, 70.05, 70.29, 72.17, 72.58 (C2-α, C3-α, C4-α, C5-α, C6-α, C2-β, C3-β, C4-β, C5-β, C6-β), 88.70, 91.53 (C1-α, C1-β), 128.16, 129.85, 132.43, 145.13 (Ar-α, Ar-β), 168.62, 168.74, 169.11, 169.26, 169.29, 169.55, 170.08, 170.20 (4 × CO-α, 4 × CO-β); HRMS (ESI-TOF): calcd for C21H26O12SNa ([M + Na]+): m/z 525.1043; found: m/z 525.1043.
- Synthesis of 1,2,3,4-Tetra-O-acetyl-6-azido-6-deoxy-d-glucopyranose5: To a solution of 4 (0.319 g, 0.635 mmol) in dry DMF (5 mL), sodium azide (0.208 g, 3.200 mmol) was added. The reaction mixture was stirred at 80 °C for 2 h. After this time, the solution was diluted with chloroform (50 mL) and water (50 mL). The aqueous phase was washed with chloroform (2 × 30 mL), and the combined organic phases were washed with brine (2 × 30 mL). Next, the organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated at reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 30:1 to 10:1) to give product 5 as a white solid (0.216 g, 91%); ratio of anomers (α:β = 1.7:1). 1H NMR (400 MHz, CDCl3): δ 2.02, 2.02, 2.03, 2.04, 2.04, 2.05, 2.12, 2.19 (8s, 24H, 4 × CH3-α, 4 × CH3-β), 3.27–3.45 (m, 4H, H6a-α, H6b-α, H6a-β, H6b-β), 3.81 (ddd, 1H, J = 3.4 Hz, J = 5.2 Hz, J = 9.9 Hz, H5-β), 4.08 (ddd, 1H, J = 2.8 Hz, J = 5.5 Hz, J = 10.1 Hz, H5-α), 5.04–5.17 (m, 4H, H2-α, H4-α, H2-β, H4-β), 5.25 (dd~t, 1H, J = 9.4 Hz, J = 9.4 Hz, H3-β), 5.47 (dd~t, 1H, J = 9.7 Hz, J = 10.0 Hz, H3-α), 5.73 (d, 1H, J = 8.3 Hz, H1-β), 6.36 (d, 1H, J = 3.7 Hz, H1-α); 13C NMR (100 MHz, CDCl3): δ 20.44, 20.55, 20.57, 20.59, 20.66, 20.75, 20.82, 20.85 (4 × CH3-α, 4 × CH3-β), 50.65, 50.73 (C6-α, C6-β), 69.01, 69.07, 69.17, 69.68, 70.15, 70.92, 72.69, 73.84 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 88.88, 91.54 (C1-α, C1-β), 168.69, 168.93, 169.20, 169.42, 169.61, 169.93, 170.11, 170.24 (4 × CO-α, 4 × CO-β); HRMS (ESI-TOF): calcd for C14H19N3O9Na ([M + Na]+): m/z 396.1019; found: m/z 396.1022.
- Synthesis of 6-Azido-6-deoxy-d-glucopyranose6: Compound 5 (0.479 g, 1.283 mmol) was suspended in MeOH (10 mL), and then 1 M solution of MeONa in MeOH (1.3 mL, 1.3 mmol) was added. The reaction mixture was stirred for 0.5 h at room temperature. The reaction progress was monitored on TLC. After the reaction was complete, the mixture was neutralized with Amberlyst-15 ion exchange resin, filtered off, and the filtrate was concentrated under reduced pressure to give product 6 as a white solid (0.218 g, 83%); ratio of anomers (α:β = 1:1). 1H NMR (400 MHz, DMSO): δ 2.93 (dd, 1H, J = 7.7 Hz, J = 8.9 Hz, H2-β), 2.99–3.05 (m, 2H, H4-α, H4-β), 3.10–3.17 (m, 2H, H2-α, H3-β), 3.23–3.31 (m, 2H, H3-α, H5-β), 3.32–3.42 (m, 2H, H6a-α, H6a-β), 3.43–3.52 (m, 2H, H6b-α, H6b-β), 3.74 (ddd, 1H, J = 2.4 Hz, J = 6.4 Hz, J = 9.3 Hz, H5-α), 4.31 (d, 1H, J = 7.7 Hz, H1-β), 4.93 (d, 1H, J = 3.6 Hz, H1-α); 13C NMR (100 MHz, DMSO): δ51.60, 51.69 (C6-α, C6-β), 70.45, 70.93, 71.34, 72.22, 72.71, 74.60, 74.64, 76.34 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 92.43, 97.06 (C1-α, C1-β); HRMS (ESI-TOF): calcd for C6H11N3O5Na ([M + Na]+): m/z 228.0596; found: m/z 228.0604.
- Synthesis of 1,2:3,4-Di-O-isopropylidene-α-d-galactopyranose8: To a solution of d-galactose 7 (3.122 g, 17.329 mmol) in dry acetone (150 mL), iodine (0.923 g, 3.637 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. After the completion reaction, 10% Na2S2O3 aqueous solution (18 mL) was added to the reaction mixture and then the solvent was evaporated under reduced pressure. The residue was diluted with dichloromethane (150 mL) and washed with brine (2 × 50 mL). The organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 44:1 to 2:1) to give product 8 as a yellow oil (4.150 g, 92%). [α]26D = −50.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.34, 1.34, 1.46, 1.54 (4s, 12H, 4 × CH3), 3.75 (m, 1H, H5), 3.83–3.91 (m, 2H, H6a, H6b), 4.28 (dd, 1H, J = 1.7 Hz, J = 7.9 Hz, H4), 4.34 (dd, 1H, J = 2.4 Hz, J = 5.0 Hz, H2), 4.62 (dd, 1H, J = 2.4 Hz, J = 7.9 Hz, H3), 5.57 (d, 1H, J = 5.0 Hz, H1); 13C NMR (100 MHz, CDCl3): δ 24.45, 25.08, 26.07, 26.18 (4xCH3), 62.49 (C6), 68.23, 70.73, 70.91, 71.76 (C2, C3, C4, C5), 96.44 (C1), 108.82, 109.62 (2 × C(CH3)2); HRMS (ESI-TOF): calcd for C12H20O6Na ([M + Na]+): m/z 283.1158; found: m/z 283.1162.
- Synthesis of 1,2:3,4-Di-O-isopropylidene-6-O-p-toluenesulfonyl-α-d-galactopyranose9: To a solution of compound 8 (1.324 g, 5.087 mmol) in pyridine (10 mL), p-toluenosulfonyl chloride (2.378 g, 12.473 mmol) and DMAP (0.120 g, 0.982 mmol) were added. The reaction mixture was stirred at room temperature for 24 h. After completion, the reaction was diluted with water (80 mL) and washed with chloroform (3 × 80 mL). The organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 100:1 to 5:1) to give product 9 as a yellow oil (1.886 g, 90%). [α]26D = −24.4 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.28, 1.32, 1.35, 1.50 (4s, 12H, 4 × CH3), 2.44 (s, 3H, Ar-CH3), 4.02–4.13 (m, 2H, H6a, H6b), 4.17–4.23 (m, 2H, H4, H5), 4.29 (dd, 1H, J = 2.5 Hz, J = 5.0 Hz, H2), 4.59 (dd, 1H, J = 2.5 Hz, J = 7.9 Hz, H3), 5.46 (d, 1H, J = 5.0 Hz, H1), 7.33 (d, 2H, J = 8.2 Hz, Ar-H), 7.81 (d, 2H, J = 8.2 Hz, Ar-H); 13C NMR (100 MHz, CDCl3): δ 21.63 (Ar-CH3), 24.36, 24.92, 25.82, 25.99 (4 × CH3), 65.88 (C6), 68.20, 70.38, 70.42, 70.53 (C2, C3, C4, C5), 96.13 (C1), 108.95, 109.59 (2 × C(CH3)2), 128.12, 129.74, 132.84, 144.75 (Ar); HRMS (ESI-TOF): calcd for C19H26O8SNa ([M + Na]+): m/z 437.1246; found: m/z 473.1248.
- Synthesis of 1,2:3,4-Di-O-isopropylidene-6-azido-6-deoxy-α-d-galactopyranose10: To a solution of compound 9 (0.798 g, 1.925 mmol) in dry DMF (7 mL), sodium azide (0.507 g, 7.799 mmol) was added. The reaction mixture was stirred at 120 °C for 24 h. After this time, the reaction was diluted with chloroform (30 mL) and water (30 mL). The aqueous phase was washed with chloroform (2 × 30 mL), and the combined organic phases were washed with water (1 × 30 mL) and brine (1 × 30 mL). Next, the organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 30:1 to 10:1) to give product 10 as a colorless oil (0.432 g, 79%). [α]24D = −104.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.34, 1.35, 1.46, 1.55 (4s, 12H, 4 × CH3), 3.37 (dd, 1H, J = 5.4 Hz, J = 12.7 Hz, H6a), 3.52 (dd, 1H, J = 7.8 Hz, J = 12.7 Hz, H6b), 3.92 (ddd, 1H, J = 2.0 Hz, J = 5.4 Hz, J = 7.8 Hz, H5), 4.20 (dd, 1H, J = 2.0 Hz, J = 7.9 Hz, H4), 4.34 (dd, 1H, J = 2.5 Hz, J = 5.0 Hz, H2), 4.63 (dd, 1H, J = 2.5 Hz, J = 7.9 Hz, H3), 5.55 (d, 1H, J = 5.0 Hz, H1); 13C NMR (100 MHz, CDCl3): δ 24.44, 24.89, 25.95, 26.03 (4 × CH3), 50.68 (C6), 67.00, 70.40, 70.81, 71.17 (C2, C3, C4, C5), 96.35 (C1), 108.81, 109.63 (2 × C(CH3)2); HRMS (ESI-TOF): calcd for C12H19N3O5Na ([M + Na]+): m/z 308.1222; found: m/z 308.1224.
- Synthesis of 6-Azido-6-deoxy-d-galactopyranose11: Compound 10 (0.410 g, 1.437 mmol) was suspended in water (0.9 mL). The solution was cooled to 0 °C and trifluoroacetic acid (3.5 mL) was added in portions. Stirring was continued at room temperature for 2 h. After completion, the reaction mixture was concentrated under reduced pressure with the addition of i-propanol. Next, the residue was purified by column chromatography (dry loading: chloroform/methanol; gradient 50:1 to 5:1) to give product 11 as a white solid (0.193 g, 65%); ratio of anomers (α:β = 6.3:1). 1H NMR (400 MHz, DMSO): δ 3.17 (m, 1H, H6a-β), 3.28 (dd, 1H, J = 4.3 Hz, J = 12.7 Hz, H6a-α), 3.37–3.47 (m, 2H, H6b-α, H6b-β), 3.48–3.59 (m, 4H, H2-α, H2-β, H3-α, H3-β), 3.61 (m, 1H, H5-α), 3.68 (m, 1H, H5-β), 3.95 (dd, 1H, J = 4.2 Hz, J = 8.6 Hz, H4-α), 4.09 (dd, 1H, J = 5.2 Hz, J = 10.5 Hz, H4-β), 4.26 (t, 1H, J = 7.1 Hz, OH-β), 4.33 (d, 1H, J = 6.6 Hz, OH-α), 4.53 (d, 1H, J = 5.2 Hz, OH-α), 4.54 (d, 1H, J = 6.7 Hz, OH-α), 4.58 (d, 1H, J = 4.5 Hz, OH-β), 4.71 (d, 1H, J = 5.2 Hz, OH-β), 4.75 (d, 1H, J = 4.3 Hz, OH-β), 4.95 (t, 1H, J = 4.1 Hz, OH-α), 6.29 (d, 1H, J = 4.8 Hz, H1-α), 6.62 (d, 1H, J = 6.9 Hz, H1-β); 13C NMR (100 MHz, DMSO): δ 51.36, 51.46 (C6-α, C6-β), 68.40, 68.83, 68.97, 69.60 (C2-α, C3-α, C4-α, C5-α), 71.72, 71.74, 72.97, 73.08 (C2-β, C3-β, C4-β, C5-β), 92.71 (C1-α), 97.42 (C1-β); HRMS (ESI-TOF): calcd for C6H11N3O5Na ([M + Na]+): m/z 228.0596; found: m/z 228.0595.
- Synthesis of 1,2,3,4-Tetra-O-acetyl-6-azido-6-deoxy-d-galactopyranose12: Unprotected compound 11 (0.308 g, 1.501 mmol) was suspended in acetic anhydride (5 mL) and sodium acetate (0.286 g, 3.487 mmol) was added. The reaction mixture was heated to reflux for 1 h. After completion, the reaction was diluted with water (30 mL) and chloroform (30 mL). The aqueous phase was washed with chloroform (3 × 30 mL), and the combined organic phases were washed with NaHCO3 (1 × 30 mL) and brine (1 × 30 mL). Next, the organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 30:1 to 10:1) to give product 12 as a colorless oil (0.377 g, 67%); ratio of anomers (α:β = 1:2.6). 1H NMR (400 MHz, CDCl3): δ 2.00, 2.05, 2.12, 2.19 (4s, 12H, 4xCH3-β), 2.01, 2.02, 2.17, 2.18 (4s, 12H, 4 × CH3-α), 3.22 (dd, 1H, J = 5.4 Hz, J = 12.8 Hz, H6a-β), 3.23 (dd, 1H, J = 5.5 Hz, J = 12.8 Hz, H6a-α), 3.45 (dd, 1H, J = 7.4 Hz, J = 12.8 Hz, H6b-α), 3.53 (dd, 1H, J = 7.4 Hz, J = 12.8 Hz, H6b-β), 3.95 (m, 1H, H5-β), 4.23 (m, 1H, H5-α), 5.08 (dd, 1H, J = 3.4 Hz, J = 10.4 Hz, H3-β), 5.30–5.37 (m, 3H, H2-β, H2-α, H3-α), 5.41 (dd, 1H, J = 1.1 Hz, J = 3.4 Hz, H4-β), 5.48 (m, 1H, H4-α), 5.72 (d, 1H, J = 8.3 Hz, H1-β), 6.40 (bs, 1H, H1-α); 13C NMR (100 MHz, CDCl3): δ 20.52, 20.53, 20.61, 20.63, 20.65, 20.68, 20.76, 20.88 (4 × CH3-α, 4 × CH3-β), 50.08, 50.32 (C6-α, C6-β), 66.36, 67.37, 67.52, 67.69, 68.14, 70.13, 70.83, 73.20 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 89.63, 92.13 (C1-α, C1-β), 168.86, 168.88, 169.36, 169.39, 169.87, 169.88, 170.05, 170.08 (4 × CO-α, 4 × CO-β); HRMS (ESI-TOF): calcd for C14H19N3O9Na ([M + Na]+): m/z 396.1019; found: m/z 396.1020.
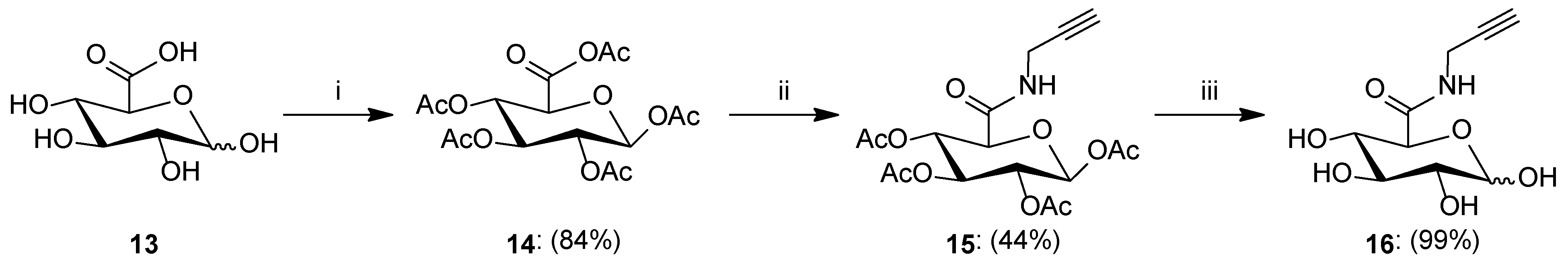
- Synthesis of 1,2,3,4-Tetra-O-acetyl-β-d-glucopyranuronic acetic anhydride14: d-Glucuronic acid 13 (2.003 g, 10.317 mmol) was suspended in acetic anhydride (30 mL). The solution was cooled to 0 °C and iodine (150.0 mg, 0.591 mmol) was added. Stirring was continued at 0 °C for 1 h and further at room temperature for 3 h. After the completion reaction, acetic anhydride was removed under reduced pressure, and the residue was diluted with dichloromethane (40 mL) and washed with 1M Na2S2O3 (2 × 40 mL). The organic layer was dried over anhydrous MgSO4, filtered off, and the solvent was concentrated under reduced pressure and recrystallized from dichloromethane/hexane to give product 14 as a white solid (3.511 g, 84%). 1H NMR (400 MHz, CDCl3): δ 2.04, 2.06, 2.07, 2.13, 2.28 (5s, 15H, 5 × CH3), 4.34 (d, 1H, J = 9.2 Hz, H5), 5.13 (dd, 1H, J = 6.9 Hz, J = 8.6 Hz, H2), 5.30 (dd~t, 1H, J = 8.6 Hz, J = 9.1 Hz, H3), 5.38 (dd~t, 1H, J = 9.1 Hz, J = 9.2 Hz, H4), 5.82 (d, 1H, J = 6.9 Hz, H1); 13C NMR (100 MHz, CDCl3): δ 20.58, 20.64, 20.64, 20.81, 22.19 (5 × CH3), 68.20, 70.19, 71.44, 73.13 (C2, C3, C4, C5), 91.49 (C1), 162.69 (C6), 164.87, 168.80, 169.33, 169.48, 169.97 (5 × COCH3); HRMS (ESI-TOF): calcd for C16H20O12Na ([M + Na]+): m/z 427.0852; found: m/z 427.0851.
- Synthesis of 1,2,3,4-Tetra-O-acetyl-N-(prop-2-yn-1-yl)-β-d-glucopyranuronic acid amide15: To a solution of anhydride 14 (1.7 g, 4.205 mmol) in dry dichloromethane (20 mL), propargyl amine (400 µL, 6.245 mmol) was added. The reaction mixture was stirred overnight at room temperature. Afterward, the solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (toluene/ethyl acetate; gradient 50:1 to 5:1) to give product 15 as a white solid (0.745 g, 44%). 1H NMR (400 MHz, CDCl3): δ 2.03, 2.05, 2.08, 2.15 (4s, 12H, 4 × CH3), 2.26 (t, 1H, J = 2.6 Hz, CCH), 4.03 (m, 2H, CH2), 4.12 (d, 1H, J = 9.6 Hz, H5), 5.11 (dd, 1H, J = 7.9 Hz, J = 8.9 Hz, H2), 5.22 (dd~t, 1H, J = 9.2 Hz, J = 9.6 Hz, H4), 5.31 (dd~t, 1H, J = 8.9 Hz, J = 9.2 Hz, H3), 5.77 (d, 1H, J = 7.9 Hz, H1), 6.50 (t, 1H, J = 5.1 Hz, NH); 13C NMR (100 MHz, CDCl3): δ 20.54, 20.54, 20.68, 20.78 (4 × CH3), 28.92 (CH2), 68.75, 70.23, 71.94, 72.12 (C2, C3, C4, C5), 72.88 (CCH), 78.62 (CCH), 91.40 (C1), 165.71 (CONH), 168.78, 169.23, 169.61, 169.81 (4 × COCH3); HRMS (ESI-TOF): calcd for C17H21NO10Na ([M + Na]+): m/z 422.1063; found: m/z 422.1064.
- Synthesis of N-(prop-2-yn-1-yl)-d-glucopyranuronic acid amide16: Acetylated amide 15 (0.526 g, 1.317 mmol) was suspended in MeOH (20 mL), and then 1 M solution of MeONa in MeOH (1.3 mL, 1.3 mmol) was added. The reaction mixture was stirred for 0.5 h at room temperature. The reaction progress was monitored on TLC. After the reaction was complete, the mixture was neutralized with Amberlyst-15 ion exchange resin, filtered off, and the filtrate was concentrated under reduced pressure to give product 16 as a white solid (0.301 g, 99%); ratio of anomers (α:β = 1.3:1). 1H NMR (400 MHz, CD3OD): δ 2.55–2.60 (m, 2H, CCH-α, CCH-β), 3.18 (dd, 1H, J = 7.8 Hz, J = 9.2 Hz, H2-β), 3.36–3.52 (m, 4H, H2-α, H3-β, H4-α, H4-β), 3.69 (d, 1H, J = 9.7 Hz, H5-β), 3.72 (dd~t, 1H, J = 9.2 Hz, J = 9.6 Hz, H3-α), 3.98–4.02 (m, 4H, CH2-α, CH2-β), 4.18 (d, 1H, J = 9.9 Hz, H5-α), 4.53 (d, 1H, J = 7.8 Hz, H1-β), 5.18 (d, 1H, J = 3.7 Hz, H1-α), 8.52 (s, 2H, NH-α, NH-β); 13C NMR (100 MHz, CD3OD): δ 29.17, 29.24 (CH2-α, CH2-β), 71.91, 72.19, 73.30, 73.50, 73.98, 74.41, 75.80, 76.29 (C2-α, C3-α, C4-α, C5-α, C2-β, C3-β, C4-β, C5-β), 72.13, 72.33 (CCH-α, CCH-β), 80.32, 80.43 (CCH-α, CCH-β), 94.27, 98.52 (C1-α, C1-β), 169.97, 171.69 (CONH-α, CONH-β); HRMS (ESI-TOF): calcd for C9H13NO6Na ([M + Na]+): m/z 254.0641; found: m/z 254.0641.
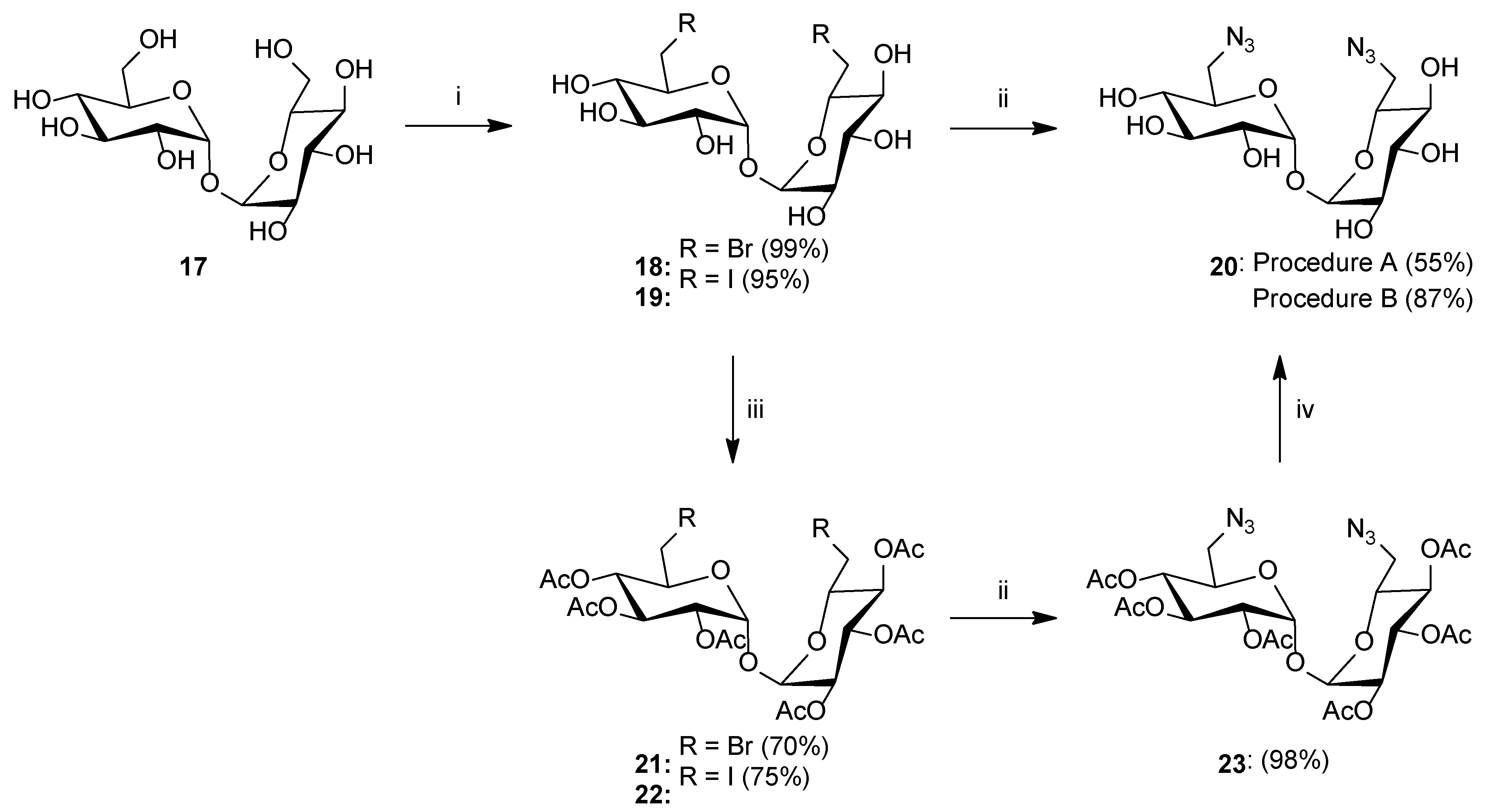
- Synthesis of 6,6′-Dibromo-6,6′-dideoxy-d-trehalose18: To a solution of d-trehalose 17 (1.008 g, 2.945 mmol) in dry DMF (10 mL), triphenylphosphine (3.325 g, 12.677 mmol) and NBS (2.268 g, 12.742 mmol) were added. The reaction mixture was stirred at room temperature for 72 h. Afterward, the solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (chloroform/methanol; gradient 50:1 to 1:1) to give product 18 as a white solid (1.368 g, 99%). m.p.: 131–132 °C; [α]24D = 109.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 3.11 (dd~t, 2H, J = 9.1 Hz, J = 9.2 Hz, 2 × H4), 3.27 (dd, 2H, J = 3.6 Hz, J = 9.5 Hz, 2 × H2), 3.52–3.63 (m, 4H, 2 × H3, 2 × H6a), 3.69 (dd, 2H, J = 2.3 Hz, J = 10.8 Hz, 2 × H6b), 3.87 (ddd, 2H, J = 2.3 Hz, J = 5.5 Hz, J = 9.3 Hz, 2 × H5), 4.92 (d, 2H, J = 3.6 Hz, 2 × H1); 13C NMR (100 MHz, DMSO): δ 35.57 (2 × C6), 70.35, 71.35, 72.02, 72.41 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 93.40 (2 × C1); HRMS (ESI-TOF): calcd for C12H20Br2O9Na ([M + Na]+): m/z 488.9372; found: m/z 488.9367.
- Synthesis of 6,6′-Diiodo-6,6′-dideoxy-d-trehalose19: To a solution of d-trehalose 17 (1.018 g, 2.974 mmol) in dry DMF (10 mL), triphenylphosphine (3.281 g, 12.509 mmol) and iodine (3.224 g, 12.702 mmol) were added. The reaction mixture was stirred at room temperature for 72 h. Afterward, the solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (chloroform/methanol; gradient 50:1 to 1:1) to give product 19 as a white solid (1.588 g, 95%). m.p.: 62–63 °C; [α]24D = 95.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 3.00 (m, 2H, 2 × H4), 3.26–3.35 (m, 4H, 2 × H2, 2 × H6a), 3.45–3.52 (m, 4H, 2 × H3, 2 × H6b), 3.59 (m, 2H, 2 × H5), 4.81 (d, 2H, J = 6.2 Hz, 2 × OH), 4.91 (m, 2H, 2 × OH), 4.96 (d, 2H, J = 3.7 Hz, 2 × H1), 5.16 (d, 2H, J = 5.4 Hz, 2 × OH); 13C NMR (100 MHz, DMSO): δ 10.59 (2 × C6), 69.92, 71.39, 72.12, 73.96 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 93.19 (2 × C1); HRMS (ESI-TOF): calcd for C12H20I2O9Na ([M + Na]+): m/z 584.9094; found: m/z 584.9090.
- Synthesis of 6,6′-Diazido-6,6′-dideoxy-d-trehalose20(Procedure A): To a solution of 18 (1.234 g, 2.637 mmol) in dry DMF (20 mL), sodium azide (1.030 g, 15.844 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h. After completion reaction, the residual salt was filtered off and the solvent was concentrated under reduced pressure. The residue was purified by column chromatography (chloroform/methanol; gradient 20:1 to 1:1) to give product 20 as a white solid (0.568 g, 55%).
- Synthesis of 6,6′-Diazido-6,6′-dideoxy-d-trehalose20(Procedure B): Compound 23 (0.202 g, 0.313 mmol) was suspended in MeOH (10 mL), and then 1 M solution of MeONa in MeOH (0.5 mL, 0.5 mmol) was added. The reaction mixture was stirred for 1 h at room temperature. The reaction progress was monitored on TLC. After the reaction was complete, the mixture was neutralized with Amberlyst-15 ion exchange resin, filtered off, and the filtrate was concentrated under reduced pressure to give product 20b as a white solid (0.107 g, 87%). m.p.: 144 °C; [α]24D = 47.0 (c = 1.0, DMSO); 1H NMR (400 MHz, DMSO): δ 3.10 (m, 2H, 2 × H4), 3.29 (m, 2H, 2 × H2), 3.39 (m, 4H, 2 × H6a, 2 × H6b), 3.54 (m, 2H, 2 × H3), 3.95 (m, 2H, 2 × H5), 4.86–4.93 (m, 6H, 2 × H1, 4 × OH), 5.12 (d, 2H, J = 5.3 Hz, 2 × OH); 13C NMR (100 MHz, DMSO): δ 51.26 (2 × C6), 70.95, 71.30, 71.37, 72.46 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 93.91 (2 × C1); HRMS (ESI-TOF): calcd for C12H20N6O9Na ([M + Na]+): m/z 415.1189; found: m/z 415.1193.
- Synthesis of 2,3,4,2′,3′,4′-Hexa-O-acetyl-6,6′-dibromo-6,6′-dideoxy-d-trehalose21: Compound 18 (1 g, 2.136 mmol) was dissolved in pyridine (3 mL), the solution was cooled to 0 °C and Ac2O (3 mL) was added. Stirring was continued overnight at room temperature. After completion, the reaction solution was poured into ice water. The precipitated crystals are filtered off under reduced pressure and recrystallized from methanol to give product 21 as a white solid (1.072 g, 70%). m.p.: 161 °C; [α]24D = 114.0 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.03, 2.08, 2.13 (3s, 18H, 6 × CH3), 3.32 (dd, 2H, J = 7.8 Hz, J = 11.2 Hz, 2 × H6a), 3.39 (dd, 2H, J = 2.6 Hz, J = 11.2 Hz, 2 × H6b), 4.12 (ddd, 2H, J = 2.6 Hz, J = 7.8 Hz, J = 10.1 Hz, 2 × H5), 4.97 (dd, 2H, J = 9.2 Hz, J = 10.1 Hz, 2 × H4), 5.16 (dd, 2H, J = 3.9 Hz, J = 10.2 Hz, 2 × H2), 5.38 (d, 2H, J = 3.9 Hz, 2 × H1), 5.49 (dd, 2H, J = 9.2 Hz, J = 10.2 Hz, 2 × H3); 13C NMR (100 MHz, CDCl3): δ 20.66, 20.68, 20.94 (6 × CH3), 30.40 (2 × C6), 69.36, 69.80, 69.96, 71.23 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 91.91 (2 × C1), 169.47, 169.57, 169.92 (6 × CO); HRMS (ESI-TOF): calcd for C24H32Br2O15Na ([M + Na]+): m/z 741.0006; found: m/z 741.0008.
- Synthesis of 2,3,4,2′,3′,4′-Hexa-O-acetyl-6,6′-diiodo-6,6′-dideoxy-d-trehalose22: Compound 19 (1.646 g, 2.928 mmol) was dissolved in pyridine (5 mL), the solution was cooled to 0 °C and Ac2O (5 mL) was added. Stirring was continued overnight at room temperature. After completion, the reaction solution was poured into ice water. The precipitated crystals are filtered off under reduced pressure and recrystallized from methanol to give product 22 as a white solid (1.788 g, 75%). m.p.: 190 °C; [α]23D = 84.2 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.02, 2.08, 2.15 (3s, 18H, 6 × CH3), 3.07 (dd, 2H, J = 9.0 Hz, J = 10.9 Hz, 2 × H6a), 3.23 (dd, 2H, J = 2.5 Hz, J = 10.9 Hz, 2 × H6b), 3.96 (ddd, 2H, J = 2.5 Hz, J = 9.0 Hz, J = 9.8 Hz, 2 × H5), 4.90 (dd, 2H, J = 9.2 Hz, J = 9.8 Hz, 2 × H4), 5.20 (dd, 2H, J = 3.9 Hz, J = 10.2 Hz, 2 × H2), 5.42 (d, 2H, J = 3.9 Hz, 2 × H1), 5.48 (dd, 2H, J = 9.2 Hz, J = 10.2 Hz, 2 × H3); 13C NMR (100 MHz, CDCl3): δ 2.49 (2 × C6), 20.64, 20.70, 21.20 (6 × CH3), 69.30, 69.75, 69.96, 72.34 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 91.78 (2 × C1), 169.45, 169.58, 169.91 (6 × CO); HRMS (ESI-TOF): calcd for C24H32I2O15Na ([M + Na]+): m/z 836.9728; found: m/z 836.9733.
- Synthesis of 2,3,4,2′,3′,4′-Hexa-O-acetyl-6,6′-diazido-6,6′-dideoxy-d-trehalose23: To a solution of 21 or 22 (0.484 g, 0.672 mmol) in dry DMF (10 mL), sodium azide (0.273 g, 4.199 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h. After completion, the reaction was diluted with ethyl acetate (30 mL) and water (30 mL). The aqueous phase was washed with ethyl acetate (2 × 30 mL), and the combined organic phases were washed with water (1 × 30 mL) and brine (1 × 30 mL). Next, the organic layer was dried over anhydrous MgSO4, filtered, and the solvent was concentrated under reduced pressure. The crude product was purified by column chromatography (toluene/ethyl acetate; gradient 20:1 to 1:1) to give product 23 as a white solid (0.425 g, 98%). m.p.: 150–151 °C; [α]24D = 39.5 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.03, 2.07, 2.13 (3s, 18H, 6 × CH3), 3.17 (dd, 2H, J = 2.5 Hz, J = 13.3 Hz, 2 × H6a), 3.37 (dd, 2H, J = 7.3 Hz, J = 13.3 Hz, 2 × H6b), 4.09 (ddd, 2H, J = 2.5 Hz, J = 7.3 Hz, J = 10.2 Hz, 2 × H5), 5.00 (dd, 2H, J = 9.3 Hz, J = 10.2 Hz, 2 × H4), 5.09 (dd, 2H, J = 3.9 Hz, J = 10.3 Hz, 2 × H2), 5.34 (d, 2H, J = 3.9 Hz, 2 × H1), 5.48 (dd, 2H, J = 9.3 Hz, J = 10.3 Hz, 2 × H3); 13C NMR (100 MHz, CDCl3): δ 20.64, 20.66, 20.67 (6 × CH3), 50.99 (2 × C6), 69.67, 69.72, 69.83, 69.92 (2 × C2, 2 × C3, 2 × C4, 2 × C5), 93.01 (2 × C1), 169.62, 169.65, 169.96 (6 × CO); HRMS (ESI-TOF): calcd for C24H32N6O15Na ([M + Na]+): m/z 667.1823; found: m/z 667.1819.
3.2.2. General Procedure for the Synthesis of Glycoconjugates
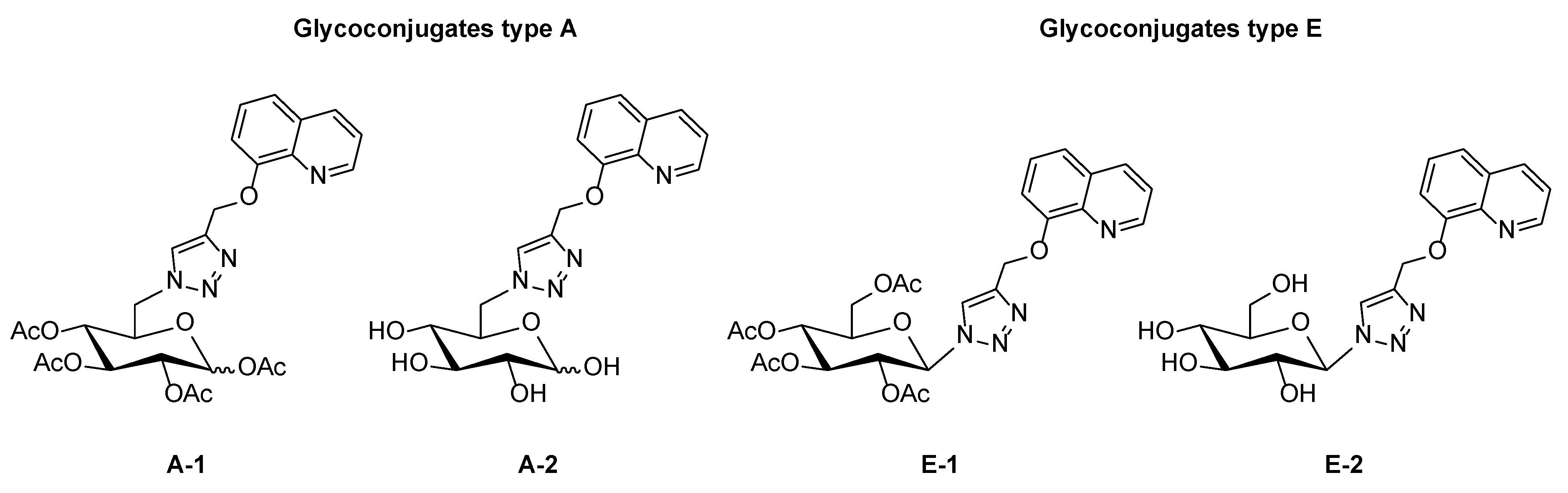
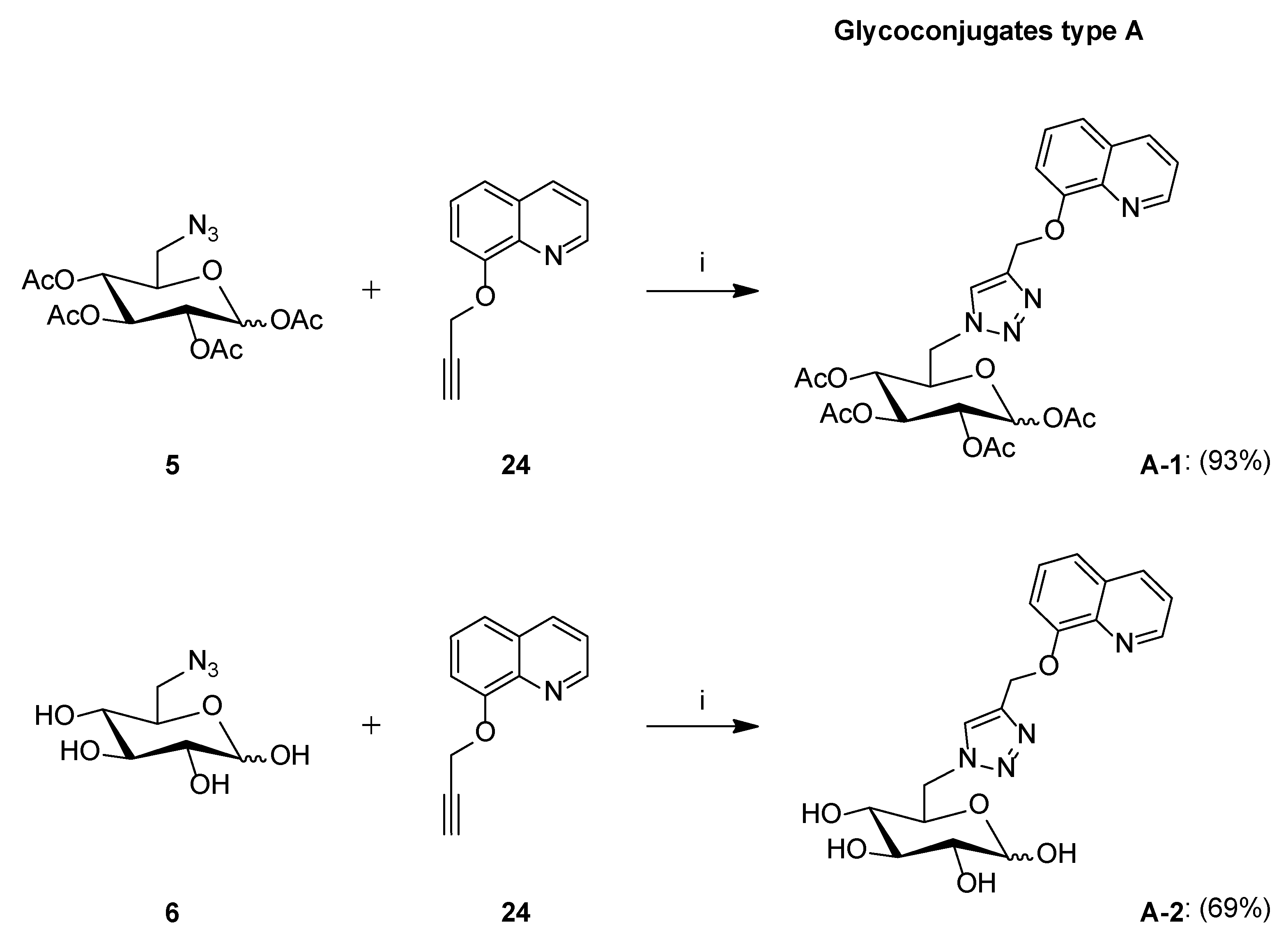
Synthesis of Glycoconjugates Type A
- Glycoconjugate A-1
- Glycoconjugate A-2
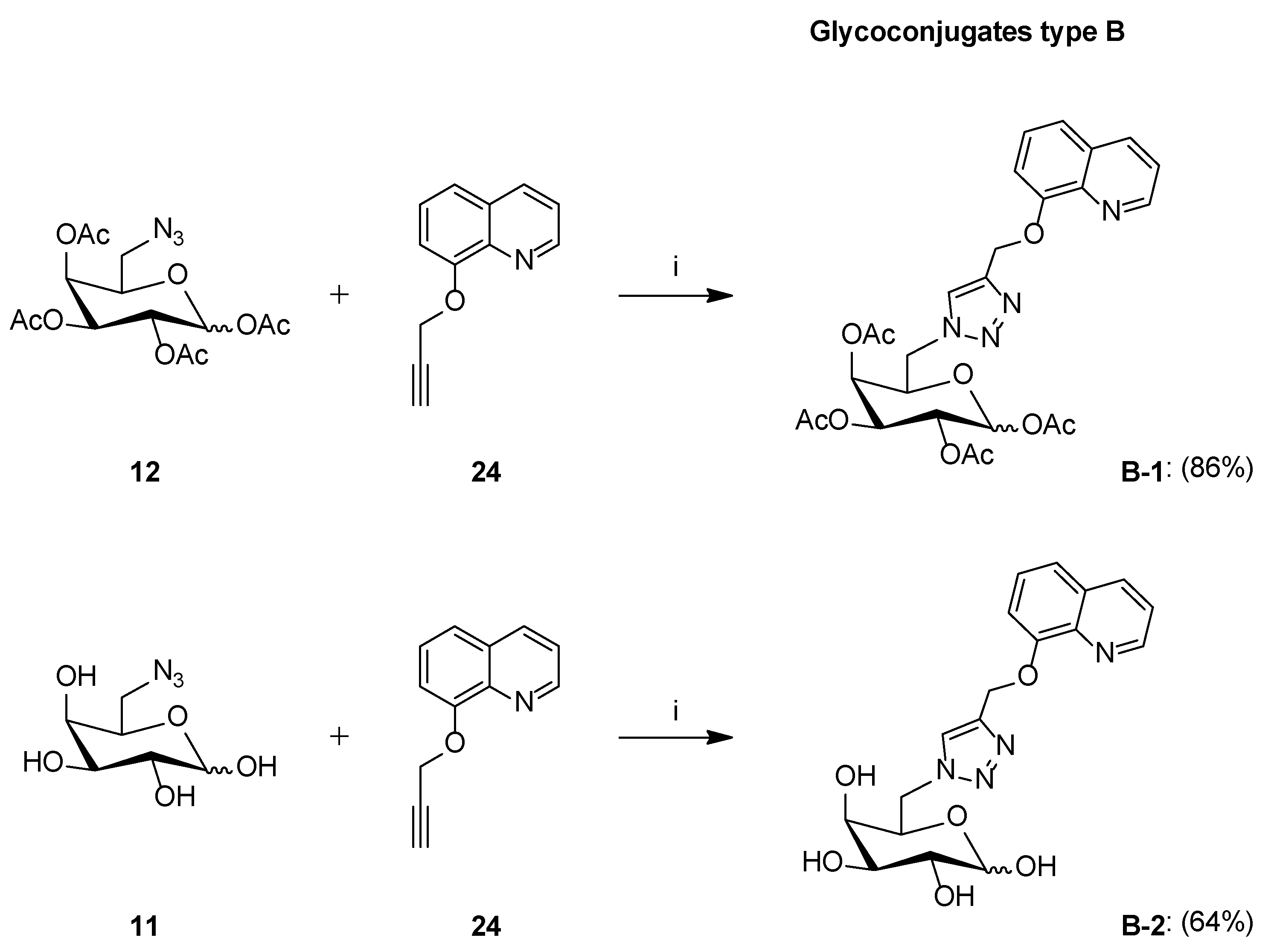
Synthesis of Glycoconjugates Type B
- Glycoconjugate B-1
- Glycoconjugate B-2
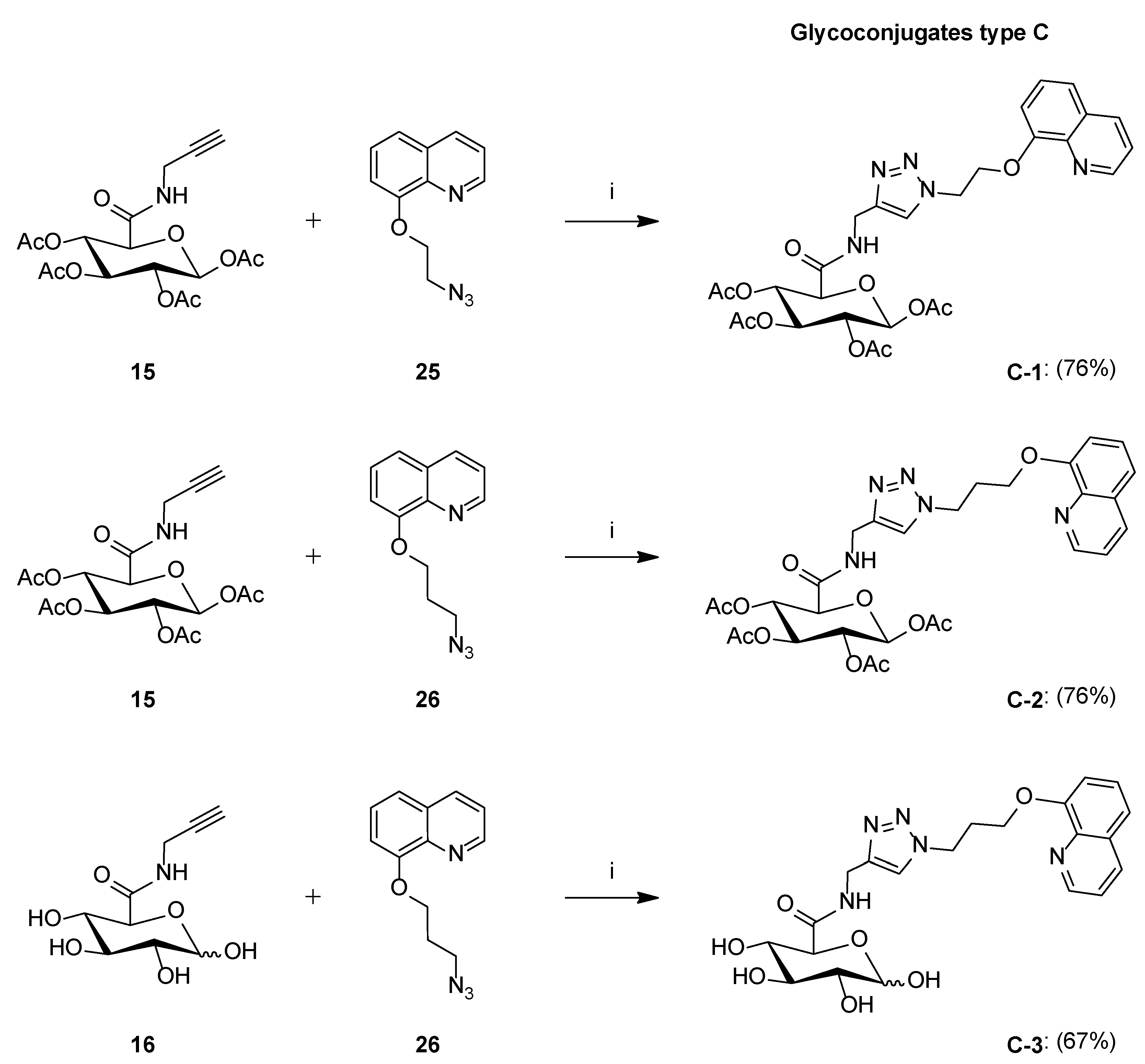
Synthesis of Glycoconjugates Type C
- Glycoconjugate C-1
- Glycoconjugate C-2
- Glycoconjugate C-3
Synthesis of Glycoconjugates Type D
- Glycoconjugate D-1
- Glycoconjugate D-2
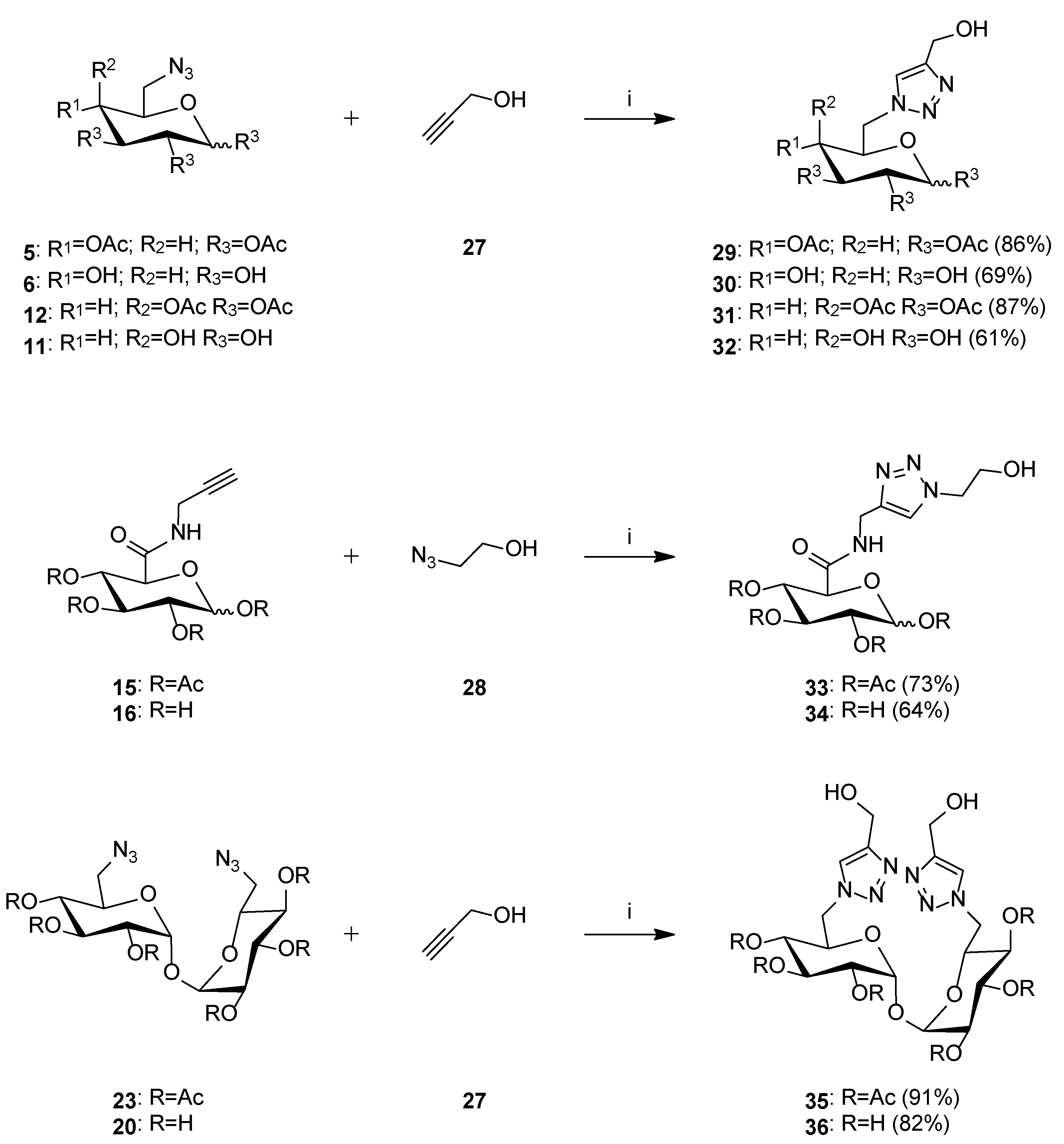
3.2.3. General Procedure for the Synthesis of Metabolites of Glycoconjugates
- Metabolite 29
- Metabolite 30
- Metabolite 31
- Metabolite 32
- Metabolite 33
- Metabolite 34
- Metabolite 35
- Metabolite 36
3.3. Biological Evaluation
3.3.1. Cell Cultures
3.3.2. MTT Cytotoxicity Assay
3.3.3. Clonogenic Assay
3.3.4. Wound-Healing Assay
3.3.5. Apoptosis and Cell Cycle Analyses by Flow Cytometry
3.3.6. Intercalation Study
3.3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Srinivasarao, M.; Low, P.S. Ligand-Targeted Drug Delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef] [PubMed]
- Srinivasarao, M.; Galliford, C.V.; Low, P.S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discov. 2015, 14, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Muro, S. Challenges in design and characterization of ligand-targeted drug delivery systems. J. Control. Release 2012, 164, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Nomura, N.; Pastorino, S.; Jiang, P.; Lambert, G.; Crawford, J.R.; Gymnopoulos, M.; Piccioni, D.; Juarez, T.; Pingle, S.C.; Makale, M.; et al. Prostate specific membrane antigen (PSMA) expression in primary gliomas and breast cancer brain metastases. Cancer Cell Int. 2014, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Teske, S.; Vessella, R.L.; True, L.D.; Zakrajsek, B.A. Expression of prostate specific membrane antigen and three alternatively spliced variants of PSMA in prostate cancer patients. Int. J. Cancer 2003, 107, 323–329. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Sig. Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Kratz, F.; Müller, I.A.; Ryppa, C.; Warnecke, A. Prodrug Strategies in Anticancer Chemotherapy. Chem. Med. Chem. 2008, 3, 20–53. [Google Scholar] [CrossRef]
- Domiński, A.; Konieczny, T.; Duale, K.; Krawczyk, M.; Pastuch-Gawołek, G.; Kurcok, P. Stimuli-Responsive Aliphatic Polycarbonate Nanocarriers for Tumor-Targeted Drug Delivery. Polymers 2020, 12, 2890. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Tanasova, M.; Fedie, J.R. Molecular Tools for Facilitative Carbohydrate Transporters (Gluts). ChemBioChem 2017, 18, 1774–1788. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef] [Green Version]
- La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Cardona, F.; Merlo, S.; Sironi, E.; D’Orazio, G.; Nicotra, F. Natural glycoconjugates with antitumor activity. Nat. Prod. Rep. 2011, 28, 630–648. [Google Scholar] [CrossRef]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef]
- Pohl, J.; Bertram, B.; Hilgard, P.; Nowrousian, M.R.; Stüben, J.; Wieβler, M. D-19575—A sugar-linked isophosphoramide mustard derivative exploiting transmembrane glucose transport. Cancer Chemother. Pharmacol. 1995, 35, 364–370. [Google Scholar] [CrossRef]
- Shimizu, T.; Okamoto, I.; Tamura, K.; Satoh, T.; Miyazaki, M.; Akashi, Y.; Ozaki, T.; Fukuoka, M.; Nakagawa, K. Phase I clinical and pharmacokinetic study of the glucose-conjugated cytotoxic agent D-19575 (glufosfamide) in patients with solid tumors. Cancer Chemother. Pharmacol. 2010, 65, 243–250. [Google Scholar] [CrossRef]
- Lacombe, D. Glufosfamide: Can We Improve the Process of Anticancer Agent Development? Expert Opin. Investig. Drugs 2012, 21, 749–754. [Google Scholar] [CrossRef]
- Wu, M.; Li, H.; Liu, R.; Gao, X.; Zhang, M.; Liu, P.; Fu, Z.; Yang, J.; Zhang-Negrerie, D.; Gao, Q. Galactose conjugated platinum(II) complex targeting the Warburg effect for treatment of non-small cell lung cancer and colon cancer. Eur. J. Med. Chem. 2016, 110, 32–42. [Google Scholar] [CrossRef]
- Halmos, T.; Santarromana, M.; Antonakis, K.; Scherman, D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur. J. Pharmacol. 1996, 318, 477–484. [Google Scholar] [CrossRef]
- Lin, Y.S.; Tungpradit, R.; Sinchaikul, S.; An, F.M.; Liu, D.Z.; Phutrakul, S.; Chen, S.T. Targeting the Delivery of Glycan-Based Paclitaxel Prodrugs to Cancer Cells via Glucose Transporters. J. Med. Chem. 2008, 51, 7428–7441. [Google Scholar] [CrossRef]
- Cao, J.; Cui, S.; Li, S.; Du, C.; Tian, J.; Wan, S.; Qian, Z.; Gu, Y.; Chen, W.R.; Wang, G. Targeted Cancer Therapy with a 2-Deoxyglucose–Based Adriamycin Complex. Cancer Res. 2013, 73, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Shustov, G.; Liang, H.; Khlebnikov, V.; Zheng, W.; Yang, X.H.; Cheeseman, C.; Wiebe, L.I. Design, Synthesis, and Preliminary Biological Evaluation of 6-O-Glucose–Azomycin Adducts for Diagnosis and Therapy of Hypoxic Tumors. J. Med. Chem. 2012, 55, 6033–6046. [Google Scholar] [CrossRef]
- Woźniak, M.; Pastuch-Gawołek, G.; Makuch, S.; Wiśniewski, J.; Krenács, T.; Hamar, P.; Gamian, A.; Szeja, W.; Szkudlarek, D.; Krawczyk, M.; et al. In Vitro and In Vivo Efficacy of a Novel Glucose–Methotrexate Conjugate in Targeted Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1748. [Google Scholar] [CrossRef]
- Akam, E.A.; Tomat, E. Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators. Bioconjugate Chem. 2016, 27, 1807–1812. [Google Scholar] [CrossRef]
- Zhao, F.Q.; Keating, A.F. Functional Properties and Genomics of Glucose Transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.; Nieto, O.; Rivas, E.; Montenegro, G.; Fontenla, J.A.; Fernández-Mayoralas, A. Synthesis and biological studies of glycosyl dopamine derivatives as potential antiparkinsonian agents. Carbohydr. Res. 2000, 327, 353–365. [Google Scholar] [CrossRef]
- Patra, M.; Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. A Potent Glucose–Platinum Conjugate Exploits Glucose Transporters and Preferentially Accumulates in Cancer Cells. Angew. Chem. Int. Ed. 2016, 55, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, M.; Pastuch-Gawolek, G.; Mrozek-Wilczkiewicz, A.; Kuczak, M.; Skonieczna, M.; Musiol, R. Synthesis of 8-hydroxyquinoline glycoconjugates and preliminary assay of their β1,4-GalT inhibitory and anti-cancer properties. Bioorg. Chem. 2019, 84, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Pastuch-Gawołek, G.; Pluta, A.; Erfurt, K.; Domiński, A.; Kurcok, P. 8-Hydroxyquinoline Glycoconjugates: Modifications in the Linker Structure and Their Effect on the Cytotoxicity of the Obtained Compounds. Molecules 2019, 24, 4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, M.; Pastuch-Gawołek, G.; Hadasik, A.; Erfurt, K. 8-Hydroxyquinoline Glycoconjugates Containing Sulfur at the Sugar Anomeric Position—Synthesis and Preliminary Evaluation of Their Cytotoxicity. Molecules 2020, 25, 4174. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Dev. Ther. 2013, 7, 1157–1178. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A privileged structure with a broad-ranging pharmacological potential. Med. Chem. Commun. 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Oliveri, V.; Vecchio, G. 8-Hydroxyquinolines in medicinal chemistry: A structural perspective. Eur. J. Med. Chem. 2016, 120, 252–274. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Gaur, K.; Vázquez-Salgado, A.M.; Duran-Camacho, G.; Dominguez-Martinez, I.; Benjamín-Rivera, J.A.; Fernández-Vega, L.; Carmona Sarabia, L.; Cruz García, A.; Pérez-Deliz, F.; Méndez Román, J.A.; et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6, 126. [Google Scholar] [CrossRef] [Green Version]
- Nagelkerke, A.; Bussink, J.; Rowan, A.E.; Span, P.N. The mechanical microenvironment in cancer: How physics affects tumours. Semin. Cancer Biol. 2015, 35, 62–70. [Google Scholar] [CrossRef]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. Innovative Linker Strategies for Tumor-Targeted Drug Conjugates. Chem. Eur. J. 2019, 25, 14740–14757. [Google Scholar] [CrossRef]
- Dheer, D.; Sing, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Lei, Z.; Wang, J.; Mao, G.; Wen, Y.; Tian, Y.; Wu, H.; Li, Y.; Xu, H. Glucose Positions Affect the Phloem Mobility of Glucose–Fipronil Conjugates. J. Agric. Food Chem. 2014, 62, 6065–6071. [Google Scholar] [CrossRef]
- Arai, M.A.; Yamaguchi, Y.; Ishibashi, M. Total synthesis of agalloside, isolated from Aquilaria agallocha, by the 5-O-glycosylation of flavan. Org. Biomol. Chem. 2017, 15, 5025–5032. [Google Scholar] [CrossRef]
- Işılar, Ö.; Bulut, A.; Yaglioglu, A.S.; Demirtaş, İ.; Arat, E.; Türk, M. Synthesis and biological evaluation of novel urea, thiourea and squaramide diastereomers possessing sugar backbone. Carbohydr. Res. 2020, 492, 107991. [Google Scholar] [CrossRef]
- Zemplén, G.; Pacsu, E. Über die Verseifung acetylierter Zucker und verwandter Substanzen. Ber. Dtsch. Chem. Ges. (A B Ser.) 1929, 62, 1613–1614. [Google Scholar] [CrossRef]
- Campo, V.L.; Carvalho, I.; Da Silva, C.H.T.P.; Schenkman, S.; Hill, L.; Nepogodiev, S.A.; Field, R.A. Cyclooligomerisation of azido-alkyne-functionalised sugars: Synthesis of 1,6-linked cyclic pseudo-galactooligosaccharides and assessment of their sialylation by Trypanosoma cruzi trans-sialidase. Chem. Sci. 2010, 1, 507–514. [Google Scholar] [CrossRef]
- Laurent, P.; Razafindralambo, H.; Wathelet, B.; Blecker, C.; Wathelet, J.P.; Paquot, M. Synthesis and Surface-Active Properties of Uronic Amide Derivatives, Surfactants from Renewable Organic Raw Materials. J. Surfactants Deterg. 2011, 14, 51–63. [Google Scholar] [CrossRef]
- Menger, F.M.; Mbadugha, B.N.A. Gemini Surfactants with a Disaccharide Spacer. J. Am. Chem. Soc. 2001, 123, 875–885. [Google Scholar] [CrossRef]
- Wang, M.; Xu, Z.; Tu, P.; Yu, X.; Xiao, S.; Yang, M. α,α-Trehalose derivatives bearing guanidino groups as inhibitors to HIV-1 Tat–TAR RNA interaction in human cells. Bioorg. Med. Chem. Lett. 2004, 14, 2585–2588. [Google Scholar] [CrossRef]
- Srinivasachari, S.; Liu, Y.; Zhang, G.; Prevette, L.; Reineke, T.M. Trehalose Click Polymers Inhibit Nanoparticle Aggregation and Promote pDNA Delivery in Serum. J. Am. Chem. Soc. 2006, 128, 8176–8184. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Domińska, M.; Pastuch-Gawołek, G.; Domiński, A.; Kurcok, P.; Erfurt, K. Synthesis and Preliminary Evaluation of the Cytotoxicity of Potential Metabolites of Quinoline Glycoconjugates. Molecules 2022, 27, 1040. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.; Rodermond, H.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Cory, G. Scratch-wound assay. Methods Mol. Biol. 2011, 769, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hulkower, K.I.; Herber, R.L. Cell Migration and Invasion Assays as Tools for Drug Discovery. Pharmaceutics 2011, 3, 107–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauria, A.; La Monica, G.; Bono, A.; Martorana, A. Quinoline anticancer agents active on DNA and DNA-interacting proteins: From classical to emerging therapeutic targets. Eur. J. Med. Chem. 2021, 220, 113555. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Nhili, R.; Cindric, M.; Bertosa, B.; Vusak, D.; Martin-Kleiner, I.; Laine, W.; Karminski-Zamola, G.; Kralj, M.; David-Cordonnier, M.H.; et al. Amino substituted benzimidazo [1,2-a]quinolines: Antiproliferative potency, 3D QSAR study and DNA binding properties. Eur. J. Med. Chem. 2016, 122, 530–545. [Google Scholar] [CrossRef] [Green Version]
- Loganathan, R.; Ramakrishnan, S.; Suresh, E.; Riyasdeen, A.; Akbarsha, M.A.; Palaniandavar, M. Mixed Ligand Copper(II) Complexes of N,N-Bis(benzimidazol-2-ylmethyl)amine (BBA) with Diimine Co-Ligands: Efficient Chemical Nuclease and Protease Activities and Cytotoxicity. Inorg. Chem. 2012, 51, 5512–5532. [Google Scholar] [CrossRef]
- Ma, T.; Xu, J.; Wang, Y.; Yu, H.; Yang, Y.; Liu, Y.; Ding, W.; Zhu, W.; Chen, R.; Ge, Z.; et al. Ternary copper(II) complexes with amino acid chains and heterocyclic bases: DNA binding, cytotoxic and cell apoptosis induction properties. J. Inorg. Biochem. 2015, 144, 38–46. [Google Scholar] [CrossRef]
- Semenov, S.N.; Belding, L.; Cafferty, B.J.; Mousavi, M.P.S.; Finogenova, A.M.; Cruz, R.S.; Skorb, E.V.; Whitesides, G.M. Autocatalytic Cycles in a Copper-Catalyzed Azide–Alkyne Cycloaddition Reaction. J. Am. Chem. Soc. 2018, 140, 10221–10232. [Google Scholar] [CrossRef]
- Da Silva, C.M.; Silva, M.M.; Reis, F.S.; Ruiz, A.L.T.G.; de Carvalho, J.E.; Santos, J.C.C.; Figueiredo, I.M.; Alves, R.B.; Modolo, L.V.; de Fatima, A. Studies on free radical scavenging, cancer cell antiproliferation, and calf thymus DNA interaction of Schiff bases. J. Photochem. Photobiol. B Biol. 2017, 172, 129–138. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activity IC50 [µM] a | |
|---|---|---|
| HCT-116 b | MCF-7 c | |
| 5 | >800 | 201.12 ± 2.09 |
| 6 | >800 | >800 |
| 11 | >800 | >800 |
| 12 | >800 | 641.24 ± 1.79 |
| 15 | 584.30 ± 9.39 | 713.59 ± 15.69 |
| 16 | >800 | >800 |
| 20 | >800 | >800 |
| 23 | >800 | >800 |
| 24 | >800 | 95.95 ± 4.29 |
| 25 | 83.02 ± 1.83 | 27.27 ± 0.06 |
| 26 | 461.39 ± 1.34 | 244.44 ± 1.34 |
| 29 | >800 | >800 |
| 30 | >800 | >800 |
| 31 | >800 | >800 |
| 32 | >800 | >800 |
| 33 | >800 | >800 |
| 34 | >800 | >800 |
| 35 | >800 | >800 |
| 36 | >800 | >800 |
| Compound | Activity IC50 [µM] a | |||
|---|---|---|---|---|
| HCT-116 b | HCT-116 c | MCF-7 c | NHDF-Neo c | |
| A-1 | 83.51 ± 1.91 | 11.98 ± 0.90 | 52.84 ± 0.75 | 129.73 ± 1.49 |
| A-2 | 191.15 ± 2.46 | 22.32 ± 0.70 | 54.92 ± 8.71 | 317.22 ± 9.46 |
| B-1 | 75.08 ± 1.30 | - | 36.67 ± 0.03 | 81.42 ± 0.52 |
| B-2 | 293.19 ± 8.00 | - | 155.73 ± 7.40 | 271.60 ± 0.35 |
| C-1 | 173.40 ± 6.68 | 81.49 ± 3.36 | 223.67 ± 7.34 | 215.24 ± 3.10 |
| C-2 | 94.24 ± 1.48 | 62.35 ± 0.32 | 157.15 ± 7.86 | 318.53 ± 3.09 |
| C-3 | >800 | 767.00 ± 2.39 | 706.60 ± 2.87 | >800 |
| D-1 | 46.26 ± 3.12 | 26.33 ± 0.72 | 30.71 ± 0.99 | 34.48 ± 0.39 |
| D-2 | 53.44 ± 5.14 | 21.28 ± 0.94 | 34.04 ± 0.95 | 70.66 ± 0.75 |
| E-1 | 69.00 ± 2.53 | 45.73 ± 1.81 | 57.69 ± 3.32 | 55.28 ± 5.08 |
| E-2 | 212.00 ± 7.71 | 162.47 ± 5.97 | 185.34 ± 2.21 | 232.75 ± 9.44 |
| Compound | Selectivity Index (SI) a | |
|---|---|---|
| HCT-116 | MCF-7 | |
| A-1 | 10.83 | 2.46 |
| A-2 | 14.21 | 5.78 |
| B-1 | 1.08 b | 2.22 |
| B-2 | 0.93 b | 1.74 |
| C-1 | 2.64 | 0.96 |
| C-2 | 5.11 | 2.03 |
| C-3 | - | - |
| D-1 | 1.31 | 1.12 |
| D-2 | 3.32 | 2.08 |
| E-1 | 1.21 | 0.96 |
| E-2 | 1.43 | 1.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domińska, M.; Pastuch-Gawołek, G.; Skonieczna, M.; Szeja, W.; Domiński, A.; Kurcok, P. Glycoconjugation of Quinoline Derivatives Using the C-6 Position in Sugars as a Strategy for Improving the Selectivity and Cytotoxicity of Functionalized Compounds. Molecules 2022, 27, 6918. https://doi.org/10.3390/molecules27206918
Domińska M, Pastuch-Gawołek G, Skonieczna M, Szeja W, Domiński A, Kurcok P. Glycoconjugation of Quinoline Derivatives Using the C-6 Position in Sugars as a Strategy for Improving the Selectivity and Cytotoxicity of Functionalized Compounds. Molecules. 2022; 27(20):6918. https://doi.org/10.3390/molecules27206918
Chicago/Turabian StyleDomińska, Monika, Gabriela Pastuch-Gawołek, Magdalena Skonieczna, Wiesław Szeja, Adrian Domiński, and Piotr Kurcok. 2022. "Glycoconjugation of Quinoline Derivatives Using the C-6 Position in Sugars as a Strategy for Improving the Selectivity and Cytotoxicity of Functionalized Compounds" Molecules 27, no. 20: 6918. https://doi.org/10.3390/molecules27206918