
Functionalized 10-Membered Aza- and Oxaenediynes through the Nicholas Reaction
 , , , and
, , , and
Abstract
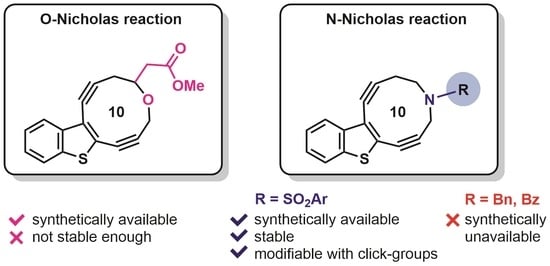
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Synthesis of Oxaenediyne I
2.2. Synthesis of Azaenediynes II–V
2.3. Biological Activity of Cyclic Enediynes
3. Discussion
4. Materials and Methods
4.1. General Information and Methods
4.2. Experimental Details: General Procedures
4.2.1. General Procedure (A) for the Synthesis of Acyclic Enediynes 11a,b; 16a,b
4.2.2. General Procedure (B) for the Synthesis of Acyclic Enediyne Co2(CO)6 Complexes 4; 12a,b; 17a,b
4.2.3. General Procedure (C) for the Synthesis of Cyclic Co2(CO)6-Complexes 5; 13a,b; 18 by the Nicholas Reaction and the Synthesis of Pyrroline Derivative 19
4.2.4. General Procedure (D) for the Synthesis of 10-Mebered Enediynes I–III, 7, 14 by the Deprotection of Acyclic Co2(CO)6-complexes from Cobalt
4.3. Experimental Details: Specific Procedures
4.4. Cell Culture
4.5. Antiproliferative Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nicolaou, K.C.; Dai, W.-M. Chemistry and Biology of the Enediyne Anticancer Antibiotics. Angew. Chem. Int. Ed. 1991, 30, 1387–1416. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Enediynes, enyne-allenes, their reactions, and beyond. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 285–324. [Google Scholar] [CrossRef]
- Basak, A.; Roy, S.; Roy, B.; Basak, A. Synthesis of Highly Strained Enediynes and Dienediynes. Curr. Top. Med. Chem. 2008, 8, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Jean, M.; Tomasi, S.; van de Weghe, P. When the nine-membered enediynes play hide and seek. Org. Biomol. Chem. 2012, 10, 7453–7456. [Google Scholar] [CrossRef]
- Hamann, P.R.; Upeslacis, J.; Borders, D.B. Anticancer Agents from Natural Products; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; CRC Press: Boca Raton, FL, USA, 2011; ISBN 9780429130854. [Google Scholar]
- Shen, B.; Liu, W.; Nonaka, K. Enediyne Natural Products: Biosynthesis and Prospect Towards Engineering Novel Antitumor Agents. Curr. Med. Chem. 2003, 10, 2317–2325. [Google Scholar] [CrossRef]
- Yan, X. Anthraquinone-fused enediynes: Discovery, biosynthesis and development. Nat. Prod. Rep. 2022, 39, 703–728. [Google Scholar] [CrossRef]
- Konishi, M.; Ohkuma, H.; Saitoh, K.-I.; Kawaguchi, H.; Golik, J.; Dubay, G.; Groenewold, G.; Krishman, B.; Doyle, T.W. Esperamicins, a novel class of potent antitumor antibiotics. I. Physico-chemical data and partial structure. J. Antibiot. 1985, 38, 1605–1609. [Google Scholar] [CrossRef]
- Lee, M.D.; Dunne, T.S.; Siegel, M.M.; Chang, C.C.; Morton, G.O.; Borders, D.B. Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin γlI. J. Am. Chem. Soc. 1987, 109, 3464–3466. [Google Scholar] [CrossRef]
- Lee, M.D.; Dunne, T.S.; Chang, C.C.; Ellestad, G.A.; Siegel, M.M.; Morton, G.O.; McGahren, W.J.; Borders, D.B. Calichemicins, a novel family of antitumor antibiotics. 2. Chemistry and structure of calichemicin γlI. J. Am. Chem. Soc. 1987, 109, 3466–3468. [Google Scholar] [CrossRef]
- Igarashi, M.; Sawa, R.; Umekita, M.; Hatano, M.; Arisaka, R.; Hayashi, C.; Ishizaki, Y.; Suzuki, M.; Kato, C. Sealutomicins, new enediyne antibiotics from the deep-sea actinomycete Nonomuraea sp. MM565M-173N2. J. Antibiot. 2021, 74, 291–299. [Google Scholar] [CrossRef]
- Low, Z.J.; Ma, G.-L.; Tran, H.T.; Zou, Y.; Xiong, J.; Pang, L.; Nuryyeva, S.; Ye, H.; Hu, J.-F.; Houk, K.N.; et al. Sungeidines from a Non-canonical Enediyne Biosynthetic Pathway. J. Am. Chem. Soc. 2020, 142, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.-G. Pharmacology and Therapeutic Applications of Enediyne Antitumor Antibiotics. Curr. Mol. Pharmacol. 2008, 1, 50–60. [Google Scholar] [CrossRef]
- Jones, R.R.; Bergman, R.G. p-Benzyne. Generation as an Intermediate in a Thermal Isomerization Reaction and Trapping Evidence for the 1,4-Benzenediyl Structure. J. Am. Chem. Soc. 1972, 94, 660–661. [Google Scholar] [CrossRef]
- Li, J.J. Bergman Cyclization. In Name Reactions; Springer International Publishing: Cham, Switzerland, 2021; pp. 35–37. ISBN 9783030508647. [Google Scholar]
- Cosgrove, J.P.; Dedon, P.C. Binding and Reaction of Calicheamicin and Other Enediyne Antibiotics with DNA. In Small Molecule DNA and RNA Binders; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; Volume 2, pp. 609–642. ISBN 3527305955. [Google Scholar]
- Nicolaou, K.C.; Rigol, S.; Pitsinos, E.N.; Das, D.; Lu, Y.; Rout, S.; Schammel, A.W.; Holte, D.; Lin, B.; Gu, C.; et al. Uncialamycin-based antibody–drug conjugates: Unique enediyne ADCs exhibiting bystander killing effect. Proc. Natl. Acad. Sci. USA 2021, 118, e2107042118. [Google Scholar] [CrossRef]
- Adhikari, A.; Shen, B.; Rader, C. Challenges and opportunities to develop enediyne natural products as payloads for antibody-drug conjugates. Antib. Ther. 2021, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Teijaro, C.N.; Yan, X.; Chang, C.-Y.; Gui, C.; Liu, Y.-C.; Crnovcic, I.; Yang, D.; Annaval, T.; Rader, C.; et al. Characterization of TnmH as an O-Methyltransferase Revealing Insights into Tiancimycin Biosynthesis and Enabling a Biocatalytic Strategy to Prepare Antibody–Tiancimycin Conjugates. J. Med. Chem. 2020, 63, 8432–8441. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Basak, A.; Mandal, S.; Bag, S.S. Chelation-Controlled Bergman Cyclization: Synthesis and Reactivity of Enediynyl Ligands. Chem. Rev. 2003, 103, 4077–4094. [Google Scholar] [CrossRef]
- Joshi, M.C.; Rawat, D.S. Recent developments in enediyne chemistry. Chem. Biodivers. 2012, 9, 459–498. [Google Scholar] [CrossRef]
- Kar, M.; Basak, A. Design, Synthesis, and Biological Activity of Unnatural Enediynes and Related Analogues Equipped with pH-Dependent or Phototriggering Devices. Chem. Rev. 2007, 107, 2861–2890. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Giofre, S.V.; Chiacchio, M.A. Synthesis and Biological Activity of Unnatural Enediynes. Curr. Med. Chem. 2017, 24, 3433–3484. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Ge, H.; Huang, T.; Hindra; Yang, D.; Teng, Q.; Crnovčić, I.; Li, X.; Rudolf, J.D.; Lohman, J.R.; et al. Strain Prioritization and Genome Mining for Enediyne Natural Products. MBio 2016, 7, e02104-16. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Basak, A.; Campbell, A.; Alabugin, I.V. Photochemical Activation of Enediyne Warheads: A Potential Tool for Targeted Antitumor Therapy. Mol. Pharm. 2018, 15, 768–797. [Google Scholar] [CrossRef]
- Lessi, M.; Panattoni, A.; Guglielmero, L.; Minei, P.; Bellina, F. Imidazole-Fused Enediynes by Selective C5–C4 Alkynylations of 4,5-Dibromoimidazoles. Synthesis. 2019, 51, 933–943. [Google Scholar]
- Li, B.; Zhang, M.; Lu, H.; Ma, H.; Wang, Y.; Chen, H.; Ding, Y.; Hu, A. Coordination-Accelerated Radical Formation from Acyclic Enediynes for Tumor Cell Suppression. Chem. Asian J. 2019, 14, 4352–4357. [Google Scholar] [CrossRef]
- Li, B.; Wu, Y.; Wang, Y.; Zhang, M.; Chen, H.; Li, J.; Liu, R.; Ding, Y.; Hu, A. Light-Cross-linked Enediyne Small-Molecule Micelle-Based Drug-Delivery System. ACS Appl. Mater. Interfaces 2019, 11, 8896–8903. [Google Scholar] [CrossRef]
- Lu, H.; Ma, H.; Li, B.; Zhang, M.; Chen, H.; Wang, Y.; Li, X.; Ding, Y.; Hu, A. Facilitating Myers–Saito cyclization through acid-triggered tautomerization for the development of maleimide-based antitumor agents. J. Mater. Chem. B 2020, 8, 1971–1979. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Das, D.; Lu, Y.; Rout, S.; Pitsinos, E.N.; Lyssikatos, J.; Schammel, A.; Sandoval, J.; Hammond, M.; Aujay, M.; et al. Total Synthesis and Biological Evaluation of Tiancimycins A and B, Yangpumicin A, and Related Anthraquinone-Fused Enediyne Antitumor Antibiotics. J. Am. Chem. Soc. 2020, 142, 2549–2561. [Google Scholar] [CrossRef]
- Zhang, M.; Li, B.; Chen, H.; Lu, H.; Ma, H.; Cheng, X.; Wang, W.; Wang, Y.; Ding, Y.; Hu, A. Triggering the Antitumor Activity of Acyclic Enediyne through Maleimide-Assisted Rearrangement and Cycloaromatization. J. Org. Chem. 2020, 85, 9808–9819. [Google Scholar] [CrossRef]
- Singha, M.; Bhattacharya, P.; Ray, D.; Basak, A. Sterically hindering the trajectory of nucleophilic attack towards p-benzynes by a properly oriented hydrogen atom: An approach to achieve regioselectivity. Org. Biomol. Chem. 2021, 19, 5148–5154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, B.; Chen, H.; Lu, H.; Ma, H.; Cheng, X.; Wang, W.; Wang, Y.; Ding, Y.; Hu, A. Maleimides Allow for Cycloaromatization of Acyclic Enediynes. Synfacts 2020, 16, 1232. [Google Scholar]
- Wang, W.; Lu, H.; Zhang, M.; Ma, H.; Cheng, X.; Ding, Y.; Hu, A. Synthesis of maleimide-based enediynes with cyclopropane moieties for enhanced cytotoxicity under normoxic and hypoxic conditions. J. Mater. Chem. B 2021, 9, 4502–4509. [Google Scholar] [CrossRef]
- Das, E.; Basak, S.; Anoop, A.; Chand, S.; Basak, A. How To Achieve High Regioselectivity in Barrier-less Nucleophilic Addition to p -Benzynes Generated via Bergman Cyclization of Unsymmetrical Cyclic Azaenediyne? J. Org. Chem. 2019, 84, 2911–2921. [Google Scholar] [CrossRef]
- Das, E.; Basak, A. Regioselective Synthesis of Benzo-Fused Tetrahydroisoquinoline-Based Biaryls through a Tandem One-Pot Halogenation of p -Benzynes from Enediynes and Suzuki-Miyaura Coupling. J. Org. Chem. 2020, 85, 2697–2703. [Google Scholar] [CrossRef] [PubMed]
- Shrinidhi, A.; Perrin, C.L. Cyclohexeno[3,4]cyclodec-1,5-diyne-3-ene: A Convenient Enediyne. Org. Lett. 2021, 23, 6911–6915. [Google Scholar] [CrossRef]
- Zhang, M.; Ma, H.; Li, B.; Sun, K.; Lu, H.; Wang, W.; Cheng, X.; Li, X.; Ding, Y.; Hu, A. Nucleophilic Addition to Diradicals Derived From Cycloaromatization of Maleimide-Based Enediynes. Asian J. Org. Chem. 2021, 10, 1454–1462. [Google Scholar] [CrossRef]
- Lyapunova, A.G.; Danilkina, N.A.; Rumyantsev, A.M.; Khlebnikov, A.F.; Chislov, M.V.; Starova, G.L.; Sambuk, E.V.; Govdi, A.I.; Bräse, S.; Balova, I.A. Relative Reactivity of Benzothiophene-Fused Enediynes in the Bergman Cyclization. J. Org. Chem. 2018, 83, 2788–2801. [Google Scholar] [CrossRef]
- Danilkina, N.; Rumyantsev, A.; Lyapunova, A.; D’yachenko, A.; Khlebnikov, A.; Balova, I. 10-Membered Azaenediyne Fused to a Benzothiophene through the Nicholas Macrocyclization: Synthesis and DNA Cleavage Ability. Synlett 2019, 30, 161–166. [Google Scholar]
- Danilkina, N.A.; D’yachenko, A.S.; Govdi, A.I.; Khlebnikov, A.F.; Kornyakov, I.V.; Bräse, S.; Balova, I.A. Intramolecular Nicholas Reactions in the Synthesis of Heteroenediynes Fused to Indole, Triazole, and Isocoumarin. J. Org. Chem. 2020, 85, 9001–9014. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ogawa, Y.; Zuccarello, G.; Schweiger, E.J.; Kumazawa, T. Cyclic conjugated enediynes related to calicheamicins and esperamicins: Calculations, synthesis, and properties. J. Am. Chem. Soc. 1988, 110, 4866–4868. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Chakraborty, S.; Balaji, A.; Basak, A. Angle distortion model for predicting enediyne activation towards Bergman cyclization: An alternate to the distance theory. RSC Adv. 2022, 12, 23552–23565. [Google Scholar] [CrossRef] [PubMed]
- Lyapunova, A.G.; Danilkina, N.A.; Khlebnikov, A.F.; Köberle, B.; Bräse, S.; Balova, I.A. Oxaenediynes through the Nicholas-Type Macrocyclization Approach. Eur. J. Org. Chem. 2016, 2016, 4842–4851. [Google Scholar] [CrossRef]
- Danilkina, N.A.; Lyapunova, A.G.; Khlebnikov, A.F.; Starova, G.L.; Bräse, S.; Balova, I.A. Ring-Closing Metathesis of Co2(CO)6 –Alkyne Complexes for the Synthesis of 11-Membered Dienediynes: Overcoming Thermodynamic Barriers. J. Org. Chem. 2015, 80, 5546–5555. [Google Scholar] [CrossRef] [PubMed]
- Kulyashova, A.E.; Ponomarev, A.V.; Selivanov, S.I.; Khlebnikov, A.F.; Popik, V.V.; Balova, I.A. Cr(II)-promoted internal cyclization of acyclic enediynes fused to benzo[b]thiophene core: Macrocycles versus 2-methylenecycloalkan-1-ols formation. Arab. J. Chem. 2019, 12, 151–167. [Google Scholar] [CrossRef]
- Nicholas, K.M.; Pettit, R. On the stability of α-(alkynyl)dicobalt hexacarbonyl carbonium ions. J. Organomet. Chem. 1972, 44, C21–C24. [Google Scholar] [CrossRef]
- Magnus, P. A general strategy using eta2Co2(CO)6 acetylene complexes for the synthesis of the enediyne antitumor agents esperamicin, calicheamicin, dynemicin and neocarzinostatin. Tetrahedron 1994, 50, 1397–1418. [Google Scholar] [CrossRef]
- Teobald, B.J. The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis. Tetrahedron 2002, 58, 4133–4170. [Google Scholar] [CrossRef]
- Kann, N. Applications of the Nicholas Reaction in the Synthesis of Natural Products. Curr. Org. Chem. 2012, 16, 322–334. [Google Scholar] [CrossRef]
- Röse, P.; Hilt, G. Cobalt-Catalysed Bond Formation Reactions; Part 2. Synthesis. 2015, 48, 463–492. [Google Scholar]
- Green, J.R.; Nicholas, K.M. Propargylic Coupling Reactions via Bimetallic Alkyne Complexes: The Nicholas Reaction. In Organic Reactions; Johnson, J.B., Ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 931–1326. [Google Scholar]
- Danilkina, N.A.; Kulyashova, A.E.; Khlebnikov, A.F.; Bräse, S.; Balova, I.A. Electrophilic Cyclization of Aryldiacetylenes in the Synthesis of Functionalized Enediynes Fused to a Heterocyclic Core. J. Org. Chem. 2014, 79, 9018–9045. [Google Scholar] [CrossRef] [PubMed]
- Lyapunova, A.G.; D’yachenko, A.S.; Danilkina, N.A. Potassium fluoride for one-pot desilylation and the Sonogashira coupling of ethynylsilanes and buta-1,3-diynylsilanes. Russ. J. Org. Chem. 2017, 53, 800–804. [Google Scholar] [CrossRef]
- Ni, R.; Mitsuda, N.; Kashiwagi, T.; Igawa, K.; Tomooka, K. Heteroatom-embedded Medium-Sized Cycloalkynes: Concise Synthesis, Structural Analysis, and Reactions. Angew. Chem. Int. Ed. 2015, 54, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.D.; Mueller, U. Nicholas reactions of amines. Tetrahedron Lett. 1993, 34, 2919–2922. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kazantsev, A.; Bakulina, O.; Dar’in, D.; Kantin, G.; Bunev, A.; Krasavin, M. Unexpected Ring Contraction of Homophthalic Anhydrides under Diazo Transfer Conditions. Org. Lett. 2022, 24, 4762–4765. [Google Scholar] [CrossRef]
- Mulay, S.V.; Fernandes, R.A. Synthetic Studies on Actinorhodin and γ-Actinorhodin: Synthesis of Deoxyactinorhodin and Deoxy-γ-actinorhodin/Crisamicin A Isomer. Chem. A Eur. J. 2015, 21, 4842–4852. [Google Scholar] [CrossRef]
- Van Rooden, E.J.; Kreekel, R.; Hansen, T.; Janssen, A.P.A.; Van Esbroeck, A.C.M.; Den Dulk, H.; Van Den Berg, R.J.B.H.N.; Codée, J.D.C.; Van Der Stelt, M. Two-step activity-based protein profiling of diacylglycerol lipase. Org. Biomol. Chem. 2018, 16, 5250–5253. [Google Scholar] [CrossRef]
- Hess, W.; Burton, J.W. Palladium-Catalysed Cyclisation of N-Alkynyl Aminomalonates. Chem. Eur. J. 2010, 16, 12303–12306. [Google Scholar] [CrossRef]
- Shen, S.; Hadley, M.; Ustinova, K.; Pavlicek, J.; Knox, T.; Noonepalle, S.; Tavares, M.T.; Zimprich, C.A.; Zhang, G.; Robers, M.B.; et al. Discovery of a New Isoxazole-3-hydroxamate-Based Histone Deacetylase 6 Inhibitor SS-208 with Antitumor Activity in Syngeneic Melanoma Mouse Models. J. Med. Chem. 2019, 62, 8557–8577. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Organic Chemicals. In Purification of Laboratory Chemicals; Elsevier: Amsterdam, The Netherlands, 2017; p. 135. ISBN 9780128054574. [Google Scholar]
- Kim, S.; Chung, Y.K. Rhodium-Catalyzed Carbonylative [3 + 2 + 1] Cycloaddition of Alkyne-Tethered Alkylidenecyclopropanes to Phenols in the Presence of Carbon Monoxide. Org. Lett. 2014, 16, 4352–4355. [Google Scholar] [CrossRef] [PubMed]
- Kalbarczyk, K.P.; Diver, S.T. Enyne Metathesis/Brønsted Acid-Promoted Heterocyclization. J. Org. Chem. 2009, 74, 2193–2196. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilkina, N.A.; Khmelevskaya, E.A.; Lyapunova, A.G.; D’yachenko, A.S.; Bunev, A.S.; Gasanov, R.E.; Gureev, M.A.; Balova, I.A. Functionalized 10-Membered Aza- and Oxaenediynes through the Nicholas Reaction. Molecules 2022, 27, 6071. https://doi.org/10.3390/molecules27186071
Danilkina NA, Khmelevskaya EA, Lyapunova AG, D’yachenko AS, Bunev AS, Gasanov RE, Gureev MA, Balova IA. Functionalized 10-Membered Aza- and Oxaenediynes through the Nicholas Reaction. Molecules. 2022; 27(18):6071. https://doi.org/10.3390/molecules27186071
Chicago/Turabian StyleDanilkina, Natalia A., Ekaterina A. Khmelevskaya, Anna G. Lyapunova, Alexander S. D’yachenko, Alexander S. Bunev, Rovshan E. Gasanov, Maxim A. Gureev, and Irina A. Balova. 2022. "Functionalized 10-Membered Aza- and Oxaenediynes through the Nicholas Reaction" Molecules 27, no. 18: 6071. https://doi.org/10.3390/molecules27186071