

In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations

Abstract
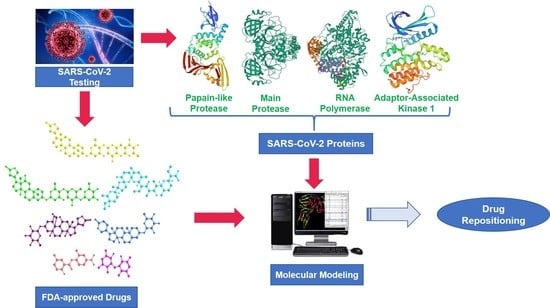
:
1. Introduction
2. Results and Discussion
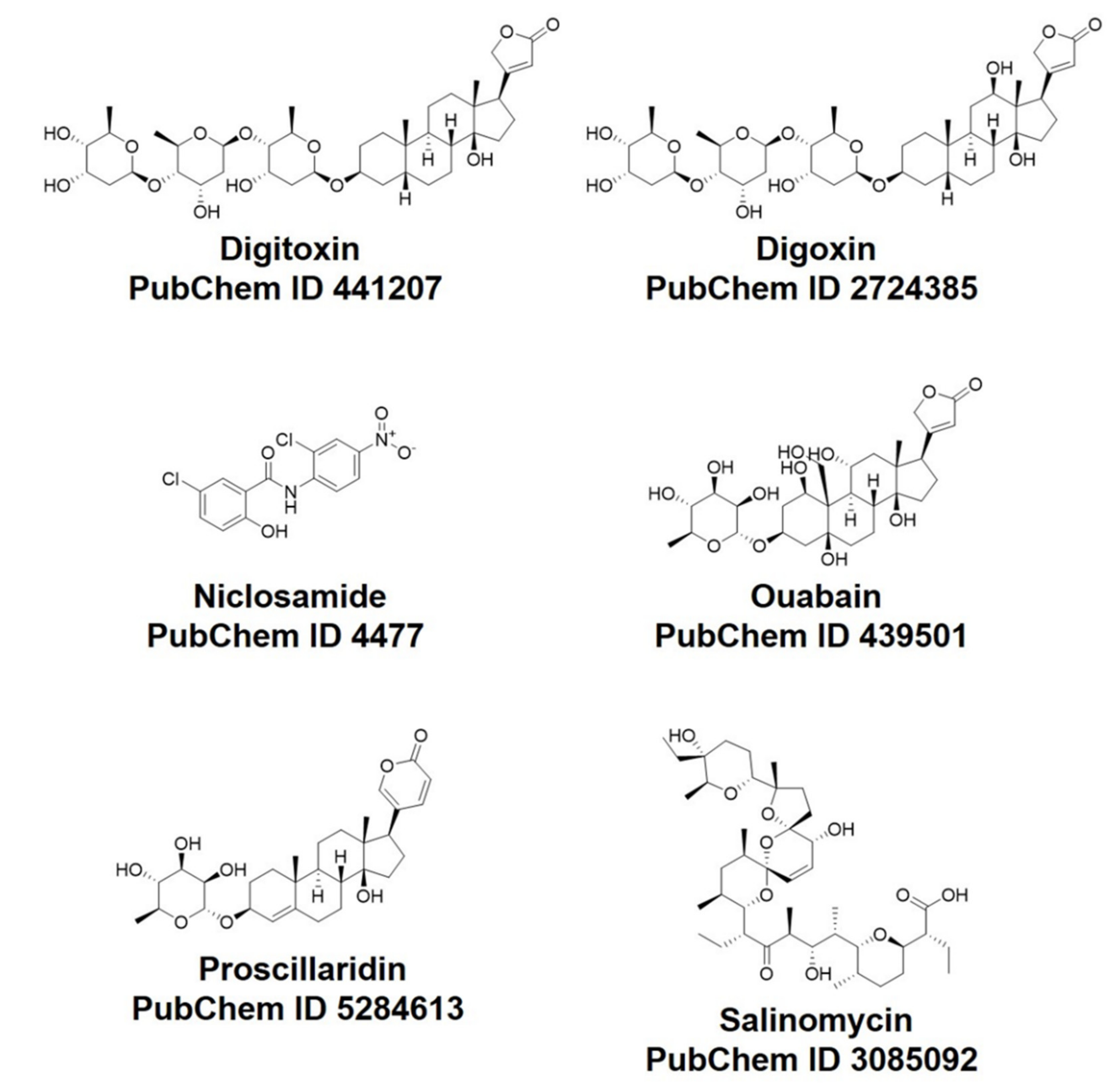
2.1. Rationale of Drug and Proteins Selection
2.2. Molecular Docking
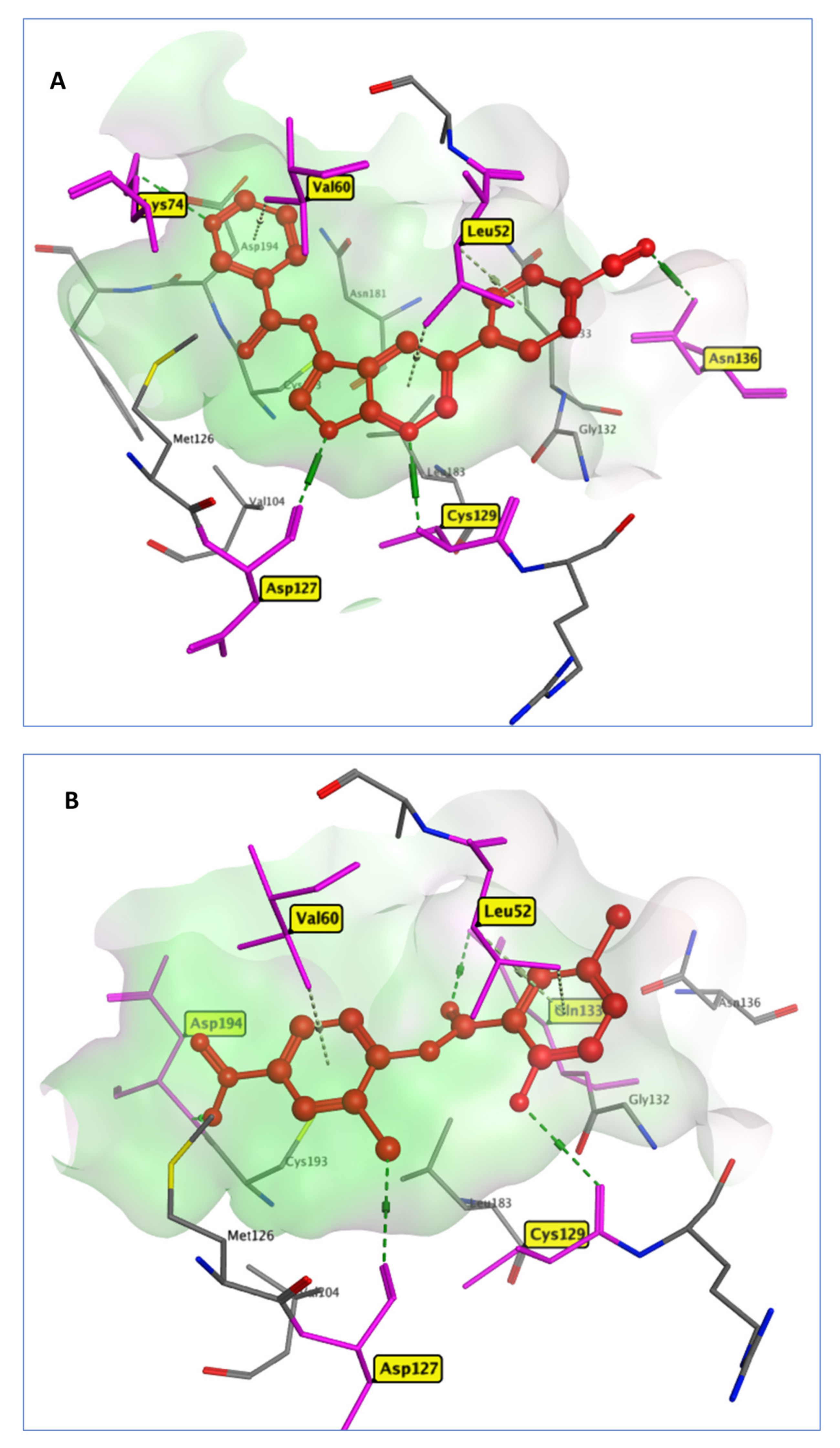
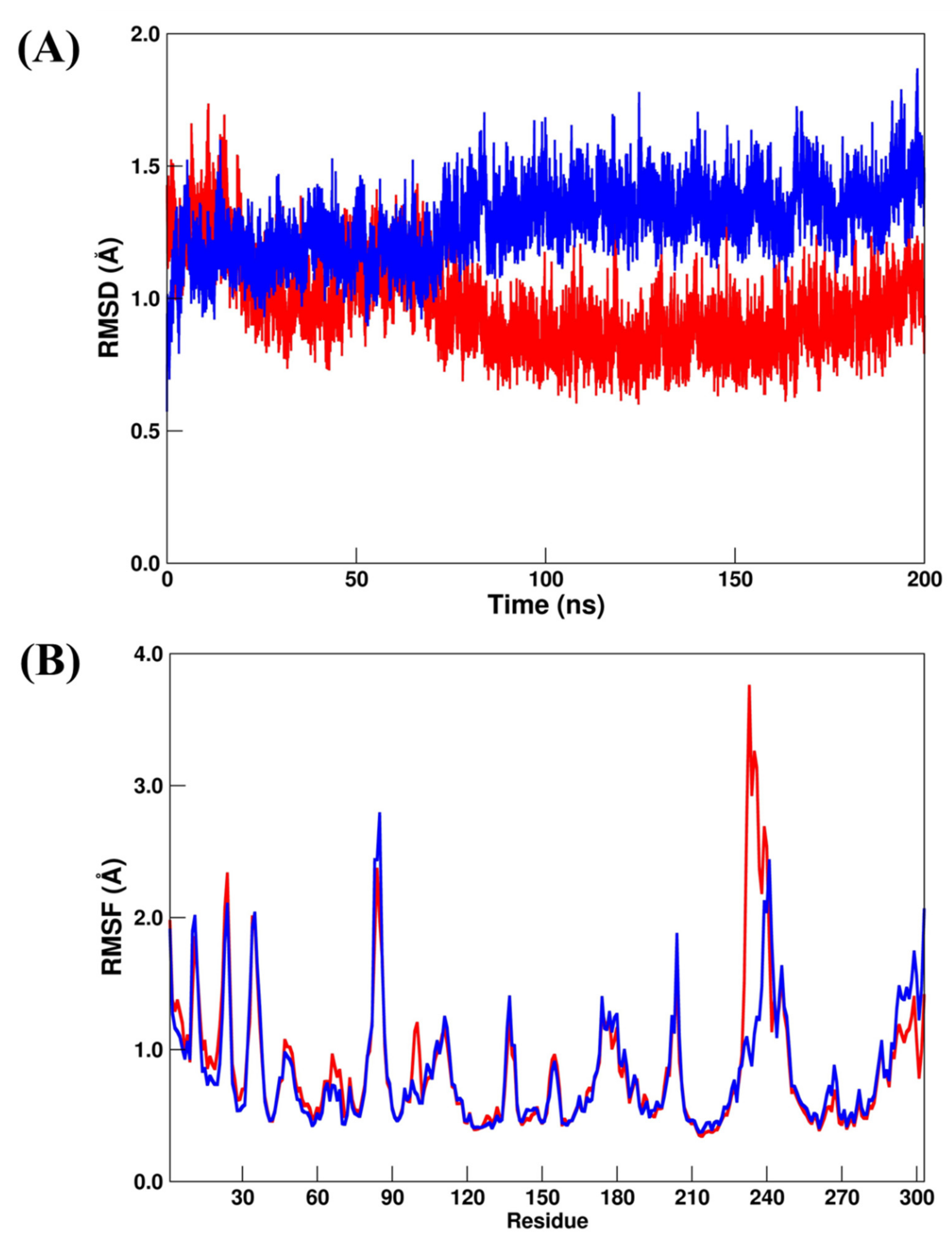
2.3. Analysis of the Intermolecular Binding Pattern of Repurposed Systems by Docking and MD Simulations
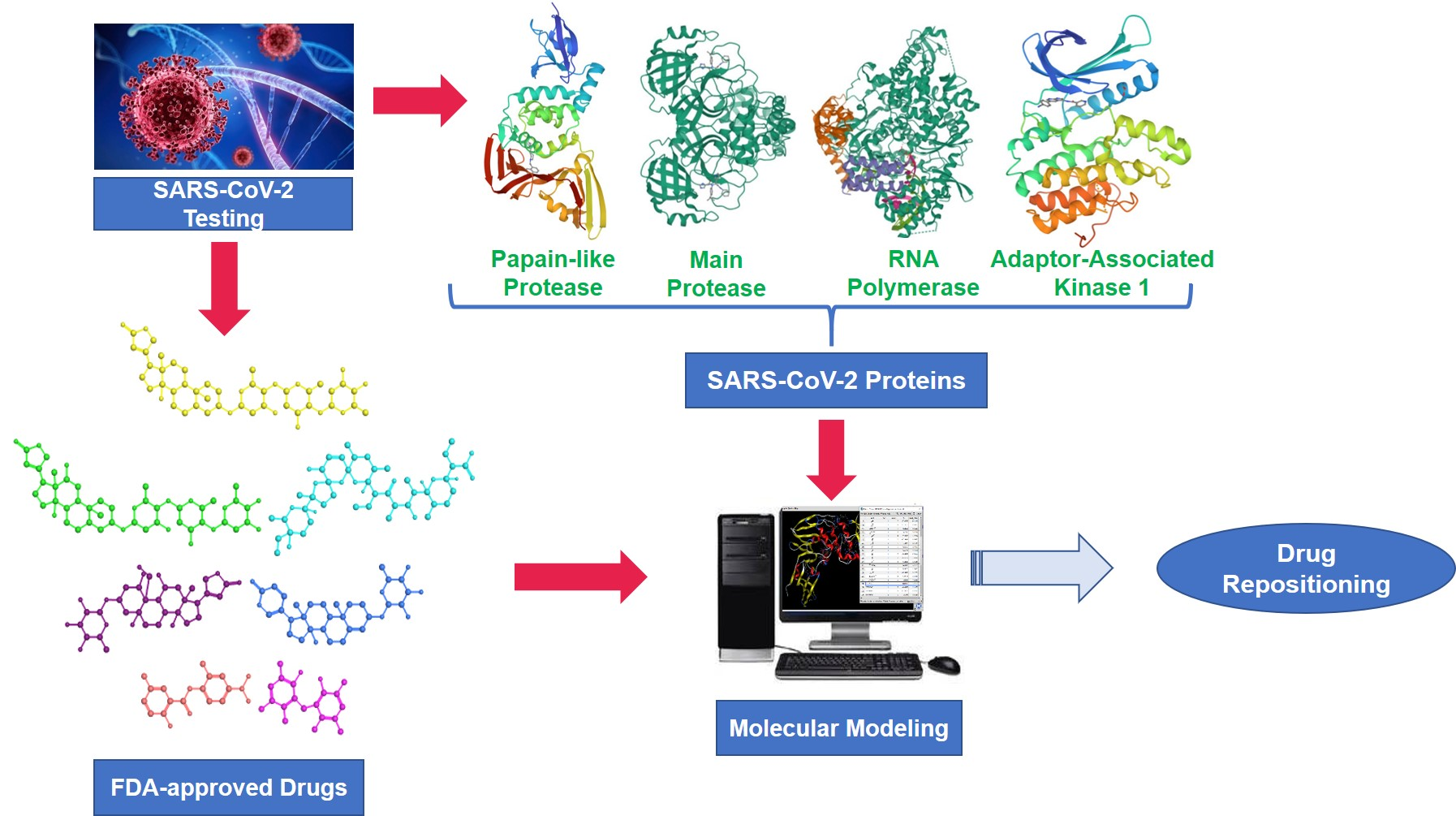
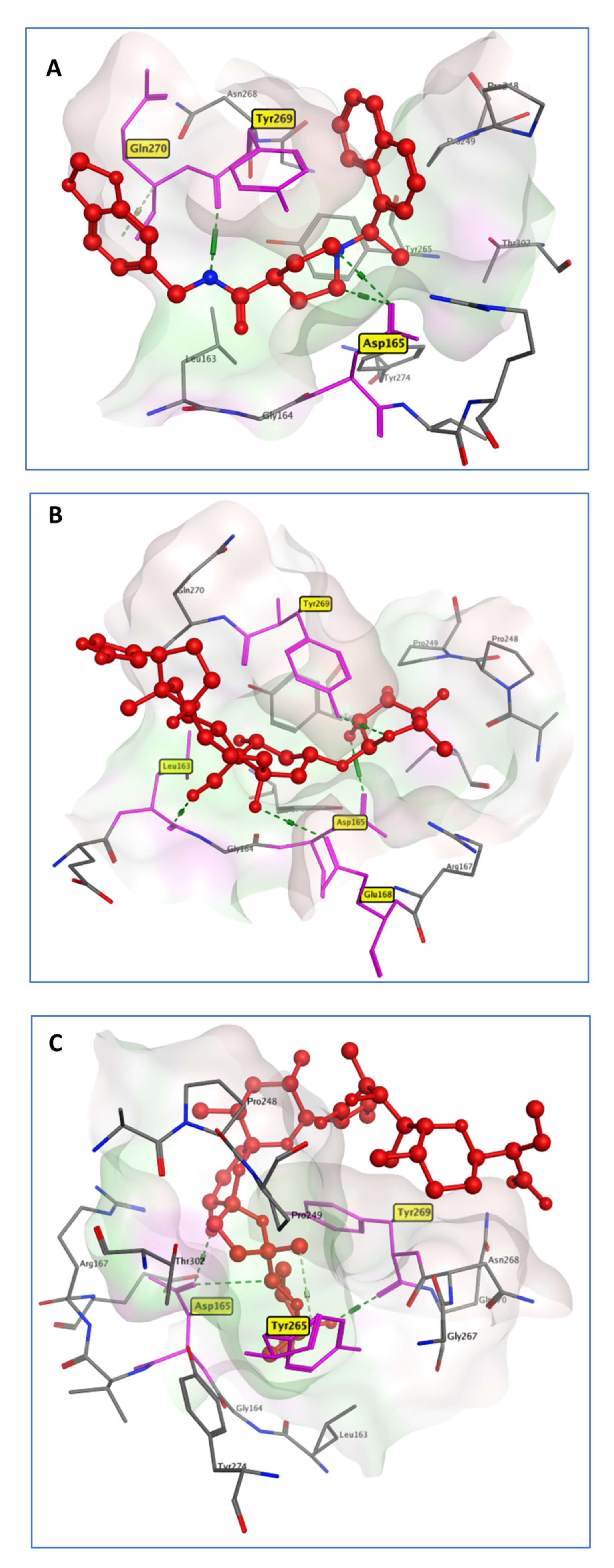
2.3.1. PLpro Systems
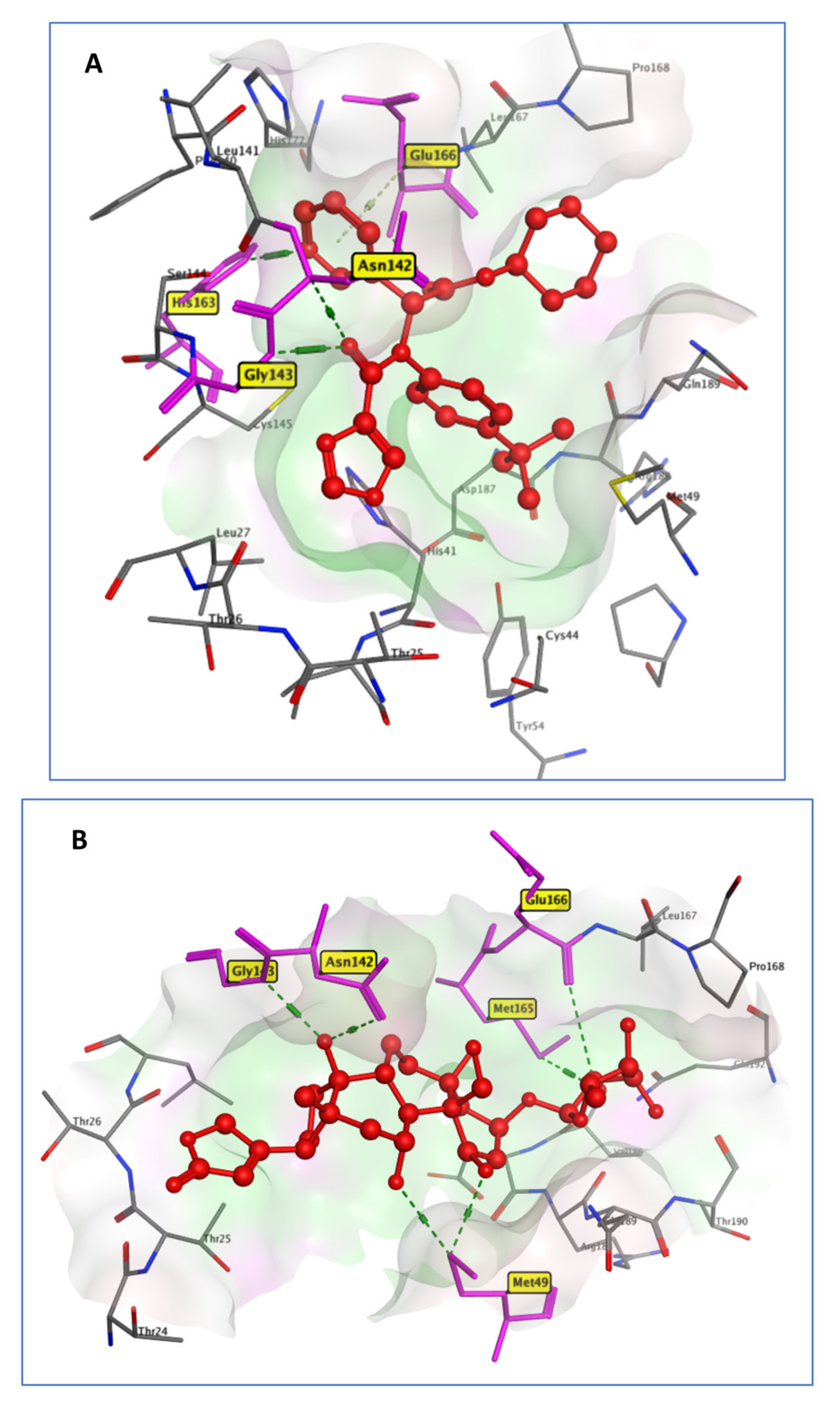
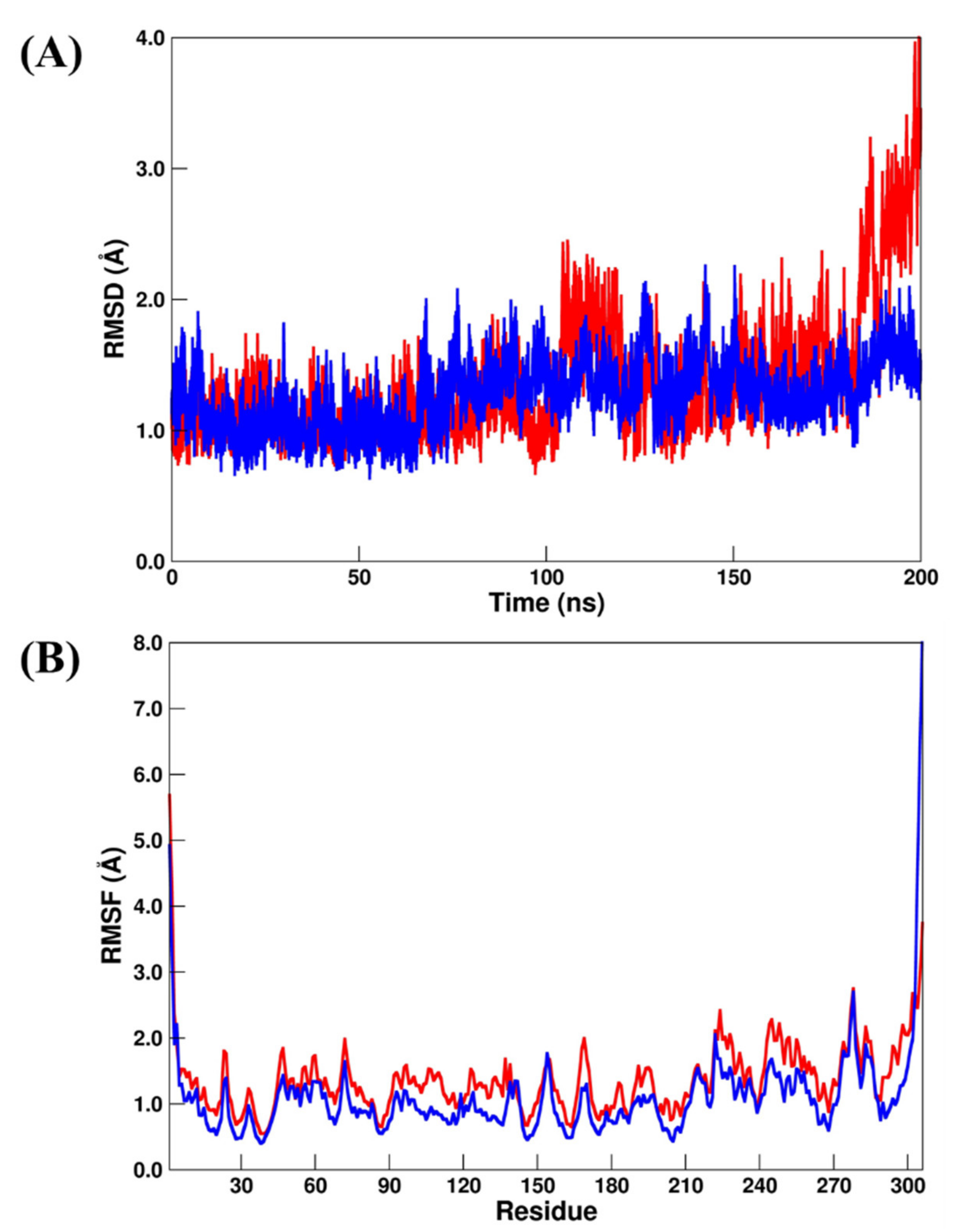
2.3.2. Mpro Systems
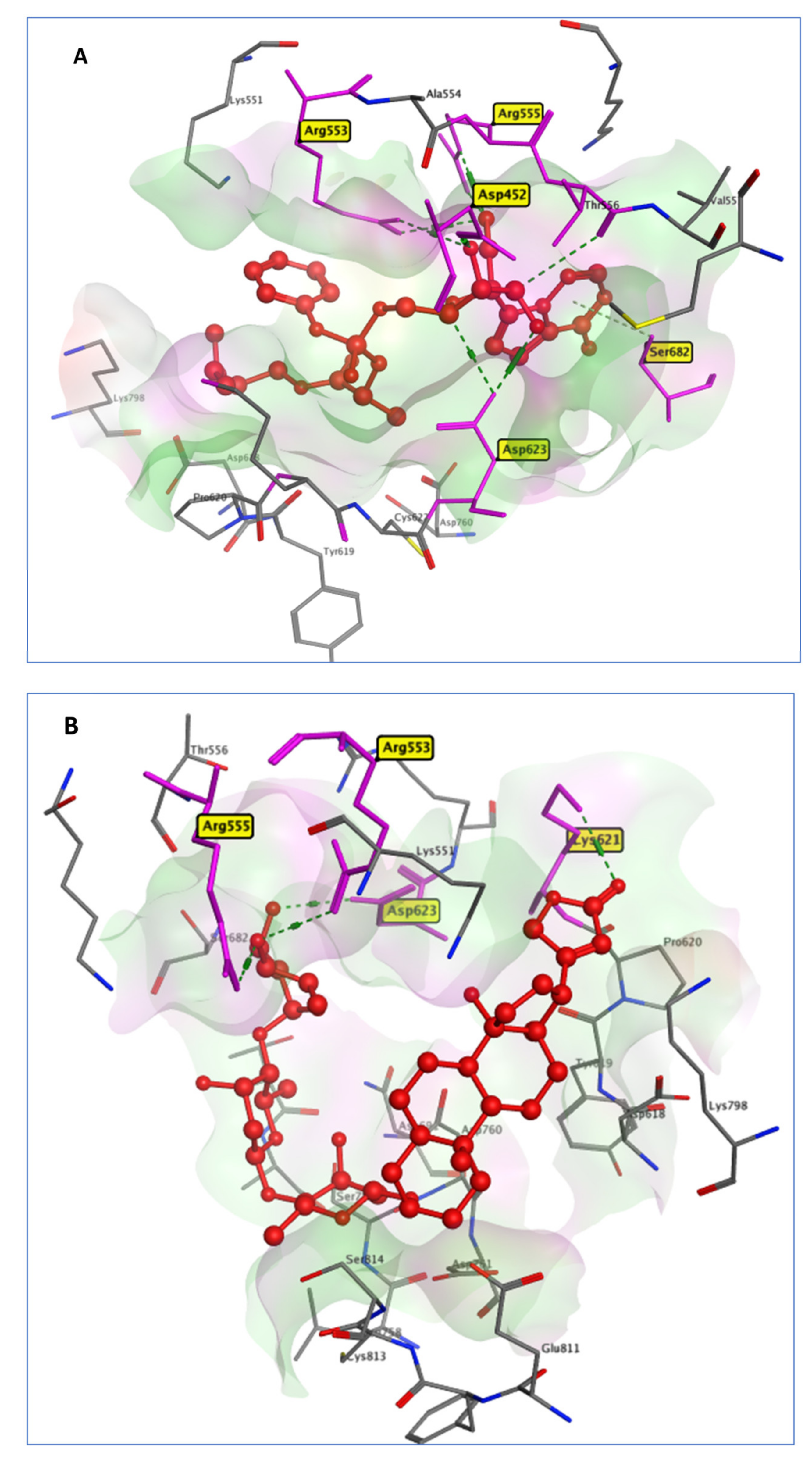
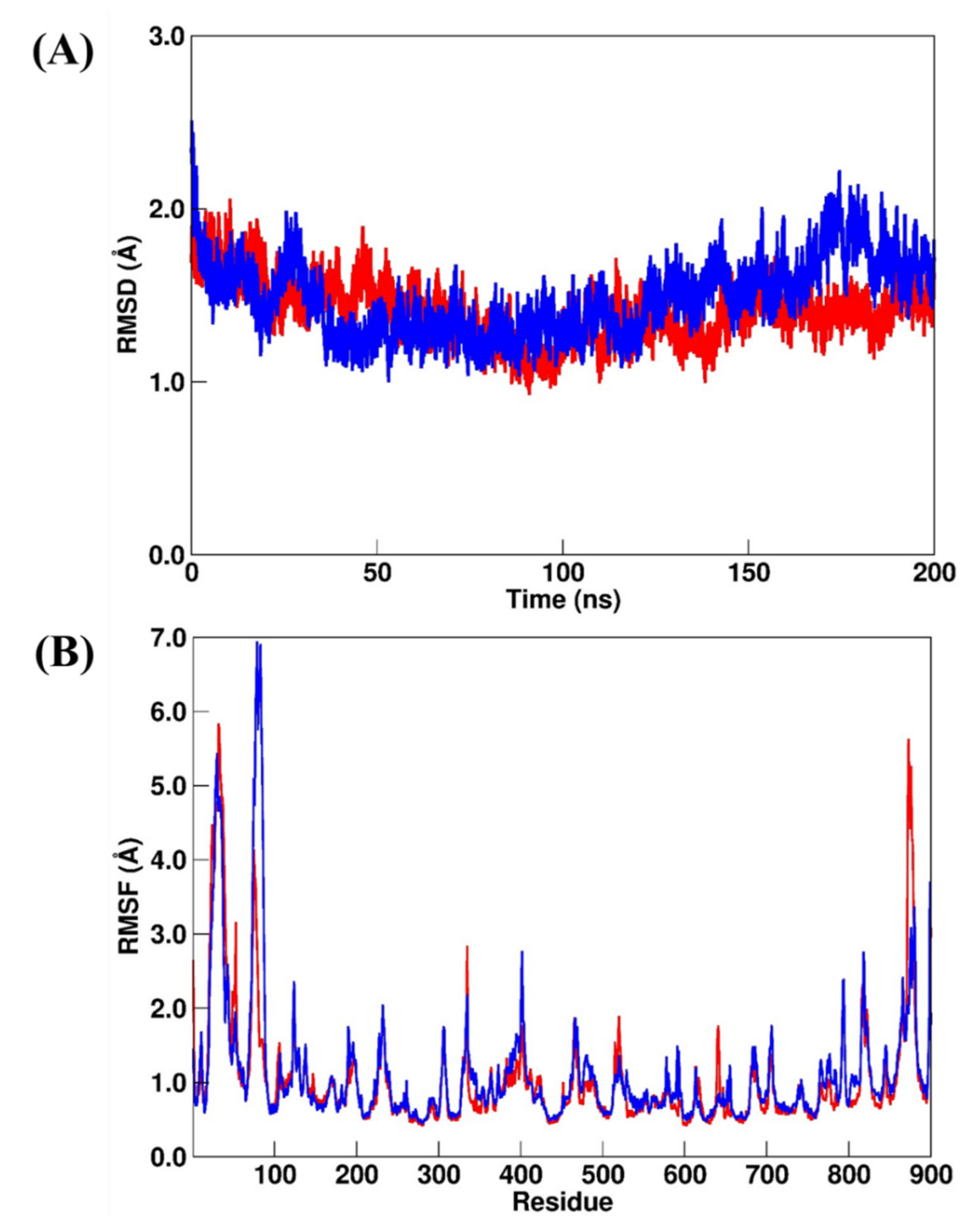
2.3.3. RdRp Systems
2.3.4. AAK1 Systems
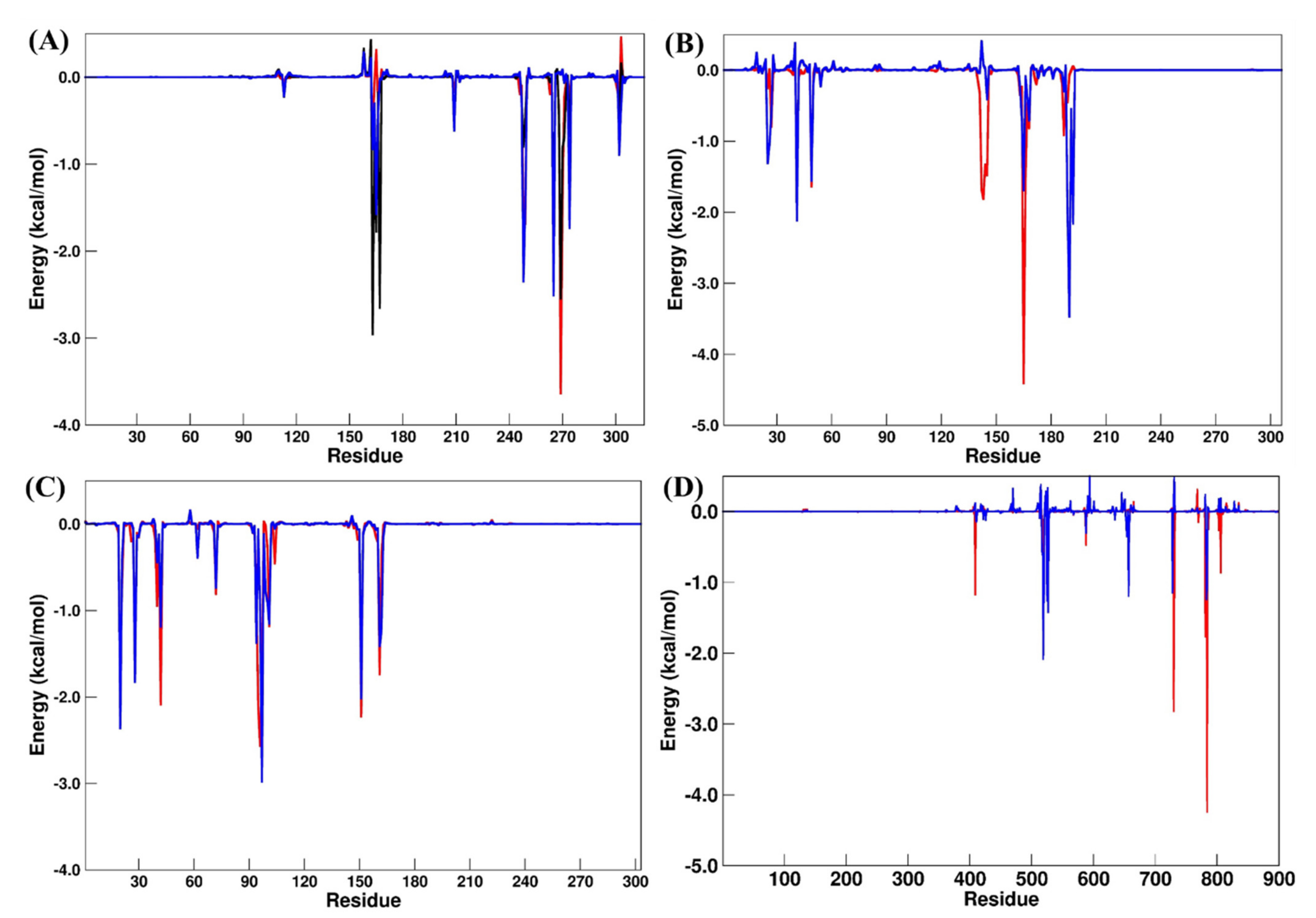
2.4. Binding Free Energy Calculations
3. Computational Methods
3.1. FDA-Approved Small Molecule Selection Rationale
3.2. Target Selection from Identified SARS-CoV-2 Protein Crystal Structures
3.3. In Silico Docking Protocol Validation
3.4. Molecular Docking
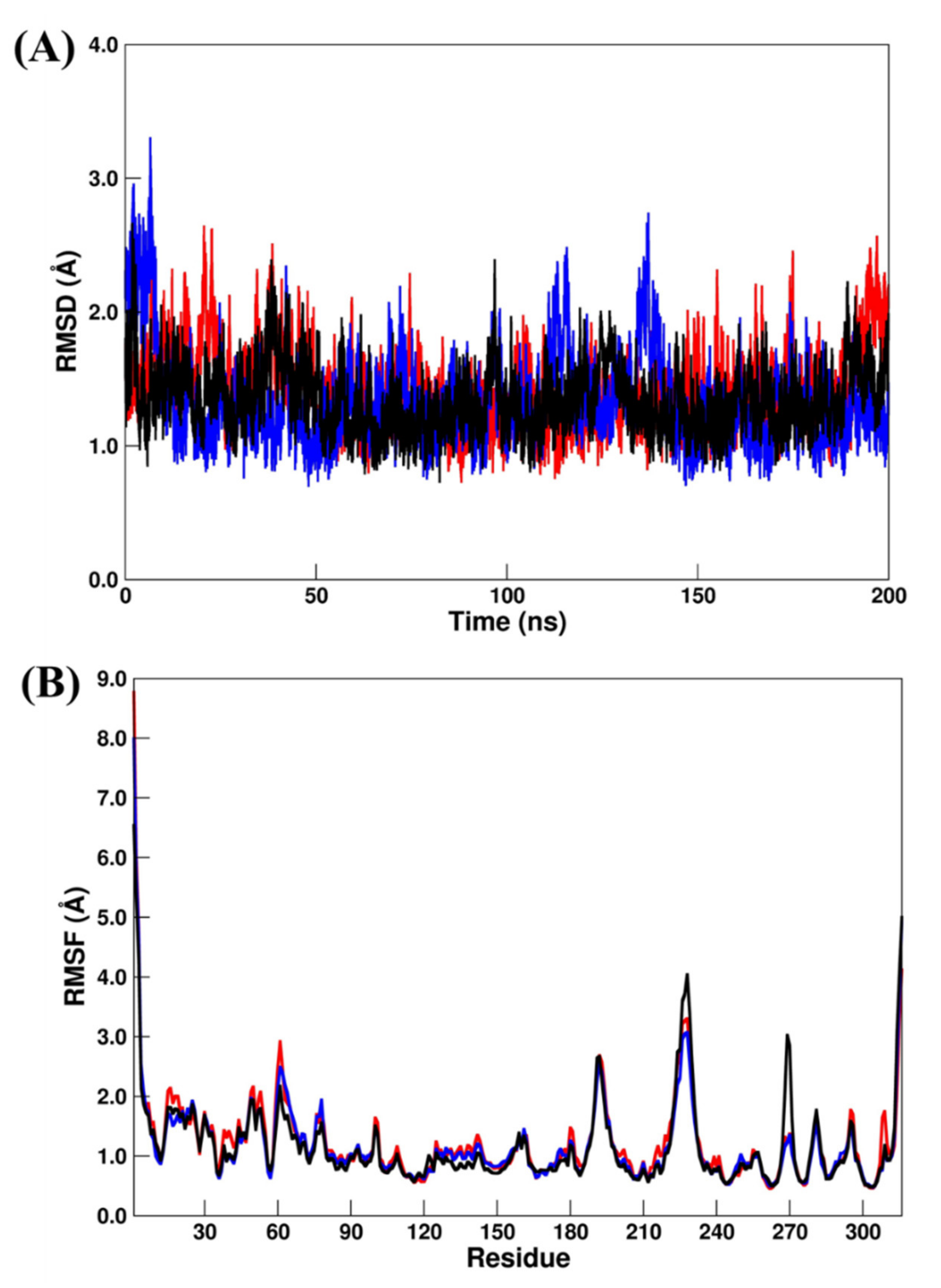
3.5. Molecular Dynamic (MD) Simulations and Binding Free Energy
4. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Arshad Ali, S.; Baloch, M.; Ahmed, N.; Arshad Ali, A.; Iqbal, A. The outbreak of Coronavirus Disease 2019 (COVID-19)—An emerging global health threat. J. Infect. Public Health 2020, 13, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef]
- WHO Coronavirus Disease (COVID-19). Available online: https://covid19.who.int/ (accessed on 4 January 2022).
- Chakraborty, I.; Maity, P. COVID-19 outbreak: Migration, effects on society, global environment and prevention. Sci. Total Environ. 2020, 728, 138882. [Google Scholar] [CrossRef]
- Vellingiri, B.; Jayaramayya, K.; Iyer, M.; Narayanasamy, A.; Govindasamy, V.; Giridharan, B.; Ganesan, S.; Venugopal, A.; Venkatesan, D.; Ganesan, H.; et al. COVID-19: A promising cure for the global panic. Sci. Total Environ. 2020, 725, 138277. [Google Scholar] [CrossRef]
- Malik, J.A.; Ahmed, S.; Mir, A.; Shinde, M.; Bender, O.; Alshammari, F.; Ansari, M.; Anwar, S. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J. Infect. Public Health 2022, 15, 228–240. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Kiani, A.K.; Dhuli, K.; Anpilogov, K.; Bressan, S.; Dautaj, A.; Dundar, M.; Beccari, T.; Ergoren, M.C.; Bertelli, M. Natural compounds as inhibitors of SARS-CoV-2 endocytosis: A promising approach against COVID-19. Acta Biomed. 2020, 91, e2020008. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S.; et al. Drug for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56. [Google Scholar] [CrossRef]
- Subissi, L.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-encoded nonstructural proteins 12–16: Replicative enzymes as antiviral targets. Antivir. Res. 2014, 101, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Laufer, S. Kinases as potential therapeutic targets for anti-coronaviral therapy. J. Med. Chem. 2022, 65, 955–982. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.R.; Shakya, A.K.; Aladwan, S.M.; El-Tanani, M. Kinase inhibitors as potential therapeutic agents in the treatment of COVID-19. Front. Pharmacol. 2022, 13, 806568. [Google Scholar] [CrossRef]
- Talevi, A. Drug repurposing. In Comprehensive Pharmacology; Kenakin, T.B.T.-C.P., Ed.; Elsevier: Oxford, UK, 2022; pp. 813–824. ISBN 978-0-12-820876-2. [Google Scholar]
- Sayed, A.M.; Khalaf, A.M.; Abdelrahim, M.E.A.; Elgendy, M.O. Repurposing of some anti-infective drugs for COVID-19 treatment: A surveillance study supported by an in silico investigation. Int. J. Clin. Pract. 2021, 75, e13877. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Panwar, U.; Dinesh, D.C.; Boura, E.; Singh, P.; Dubey, V.K.; Singh, S.K. Microsecond MD simulation and multiple-conformation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Front. Chem. 2021, 8, 595273. [Google Scholar] [CrossRef] [PubMed]
- Arantes, P.R.; Saha, A.; Palermo, G. Fighting COVID-19 using molecular dynamics simulations. ACS Cent. Sci. 2020, 6, 1654–1656. [Google Scholar] [CrossRef]
- Kandeel, M.; Abdelrahman, A.H.M.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A.A. Repurposing of FDA-approved antivirals, antibiotics, anthelmintics, antioxidants, and cell protectives against SARS-CoV-2 papain-like protease. J. Biomol. Struct. Dyn. 2021, 39, 5129–5136. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Hegazy, M.-E.F. Natural-like products as potential SARS-CoV-2 M pro inhibitors: In-silico drug discovery. J. Biomol. Struct. Dyn. 2021, 39, 5722–5734. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Mohamed, T.A.; Atia, M.A.M.; Al-Hammady, M.A.M.; Abdeljawaad, K.A.A.; Elkady, E.M.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; et al. In silico mining of terpenes from red-sea invertebrates for SARS-CoV-2 main protease (Mpro) inhibitors. Molecules 2021, 26, 2082. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.-E.F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Abhinand, C.S.; Nair, A.S.; Krishnamurthy, A.; Oommen, O.V.; Sudhakaran, P.R. Potential protease inhibitors and their combinations to block SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 903–917. [Google Scholar] [CrossRef]
- Daoud, S.; Alabed, S.J.; Dahabiyeh, L.A. Identification of potential COVID-19 main protease inhibitors using structure-based pharmacophore approach, molecular docking and repurposing studies. Acta Pharm. 2021, 71, 163–174. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Ismail, M.I.; Bauer, M.R.; Bekhit, A.A.; Boeckler, F.M. Supporting SARS-CoV-2 papain-like protease drug discovery: In silico methods and benchmarking. Front. Chem. 2020, 8, 592289. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- García-Cárceles, J.; Caballero, E.; Gil, C.; Martínez, A. Kinase inhibitors as underexplored antiviral agents. J. Med. Chem. 2022, 65, 935–954. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Takayama, J.; Rao, K.V.; Ratia, K.; Chaudhuri, R.; Mulhearn, D.C.; Lee, H.; Nichols, D.B.; Baliji, S.; Baker, S.C.; et al. Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: Design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 2010, 53, 4968–4979. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Verdonck, S.; Pu, S.-Y.; Sorrell, F.J.; Elkins, J.M.; Froeyen, M.; Gao, L.-J.; Prugar, L.I.; Dorosky, D.E.; Brannan, J.M.; Barouch-Bentov, R.; et al. Synthesis and structure-activity relationships of 3,5-disubstituted-pyrrolo[2,3-b]pyridines as inhibitors of adaptor-associated kinase 1 with antiviral activity. J. Med. Chem. 2019, 62, 5810–5831. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Lesyng, B. Generalized born model: Analysis, refinement, and applications to proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E. 01; Gaussian: Wallingford, CT, USA, 2009; ISBN 9781935522027. [Google Scholar]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural survey of zinc-containing proteins and development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate—DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Srivastava, M.; Mittal, L.; Kumari, A.; Asthana, S. Molecular dynamics simulations reveal the interaction fingerprint of remdesivir triphosphate pivotal in allosteric regulation of SARS-CoV-2 RdRp. Front. Mol. Biosci. 2021, 8, 639614. [Google Scholar] [CrossRef]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of SARS-CoV-2 peptide binders with better predicted binding affinities than human ACE2 receptor. Sci. Rep. 2021, 11, 15650. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Yu, W.; Wu, X.; Zhao, Y.; Chen, C.; Yang, Z.; Zhang, X.; Ren, J.; Wang, Y.; Wu, C.; Li, C.; et al. Computational simulation of HIV protease inhibitors to the main protease (Mpro) of SARS-CoV-2: Implications for COVID-19 drugs design. Molecules 2021, 26, 7385. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.A.; Urban, J.; Araújo, E.; Lameira, J.; Moliner, V.; Alves, C.N. Exploring the catalytic mechanism of the RNA cap modification by nsp16-nsp10 complex of SARS-CoV-2 through a QM/MM approach. Int. J. Mol. Sci. 2022, 23, 300. [Google Scholar] [CrossRef]
- Silva, J.R.A.; Kruger, H.G.; Molfetta, F.A. Drug repurposing and computational modeling for discovery of inhibitors of the main protease (Mpro) of SARS-CoV-2. RSC Adv. 2021, 11, 23450–23458. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Jeon, S.; Ryu, W.-S.; Kim, S. Comparative analysis of antiviral efficacy of FDA-approved drugs against SARS-CoV-2 in human lung cells. J. Med. Virol. 2021, 93, 1403–1408. [Google Scholar] [CrossRef]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hegazy, M.-E.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 5756–5767. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Majerová, T.; Novotný, P. Precursors of viral proteases as distinct drug targets. Viruses 2021, 13, 1981. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Alqahtani, S.M.; Froeyen, M.; Chen, L.-L. Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. J. Pharm. Anal. 2020, 10, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Pindiprolu, S.K.S.; Kumar, C.S.P.; Golla, V.S.K.; Likitha, P.; Chandra, S.; SK, E.B.; Ramachandra, R.K. Pulmonary delivery of nanostructured lipid carriers for effective repurposing of salinomycin as an antiviral agent. Med. Hypotheses 2020, 143, 109858. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wei, P.; Huang, C.; Tan, L.; Liu, Y.; Lai, L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006, 281, 13894–13898. [Google Scholar] [CrossRef] [PubMed]
- Tam, N.M.; Nam, P.C.; Quang, D.T.; Tung, N.T.; Vu, V.V.; Ngo, S.T. Binding of inhibitors to the monomeric and dimeric SARS-CoV-2 Mpro. RSC Adv. 2021, 11, 2926–2934. [Google Scholar] [CrossRef]
- Tam, N.M.; Pham, M.Q.; Ha, N.X.; Nam, P.C.; Phung, H.T.T. Computational estimation of potential inhibitors from known drugs against the main protease of SARS-CoV-2. RSC Adv. 2021, 11, 17478–17486. [Google Scholar] [CrossRef]
- Dömling, A.; Gao, L. Chemistry and biology of SARS-CoV-2. Chemistry 2020, 6, 1283–1295. [Google Scholar] [CrossRef]
- Parvez, M.S.A.; Karim, M.A.; Hasan, M.; Jaman, J.; Karim, Z.; Tahsin, T.; Hasan, M.N.; Hosen, M.J. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int. J. Biol. Macromol. 2020, 163, 1787–1797. [Google Scholar] [CrossRef]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a drug with many (re)purposes. ChemMedChem 2018, 13, 1088–1091. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef]
- Karim, M.; Saul, S.; Ghita, L.; Sahoo, M.K.; Ye, C.; Bhalla, N.; Lo, C.-W.; Jin, J.; Park, J.-G.; Martinez-Gualda, B.; et al. Numb-associated kinases are required for SARS-CoV-2 infection and are cellular targets for antiviral strategies. Antivir. Res. 2022, 204, 105367. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Tsai, T.F. Oral disease-modifying antirheumatic drugs and immunosuppressants with antiviral potential, including SARS-CoV-2 infection: A review. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X2094729. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Generic Name | Clinical Use | (IC50, μM) in Vero Cell | (IC50, μM) in Calu Cell |
|---|---|---|---|
| Ouabain (OUB) | Cardiovascular agents | <0.097 | 0.1 |
| Digoxin (DGX) | Cardiovascular agents | 0.19 | 0.72 |
| Digitoxin (DIG) | Cardiovascular agents | 0.23 | 0.16 |
| Salinomycin (SLM) | Antibacterial agents | 0.24 | 0.5 |
| Niclosamide (NIS) | Antiparasitic agents | 0.28 | 0.84 |
| Proscillaridin (PRO) | Cardiovascular agents | 2.04 | 5.95 |
| Drug Name | PLpro | Mpro | RdRp | AAK1 |
|---|---|---|---|---|
| Ouabain (OUB) | −9.52 | −8.21 | −6.35 | −8.17 |
| Digoxin (DGX) | −7.88 | −7.64 | −7.63 | −9.08 |
| Digitoxin (DIG) | −8.01 | −6.56 | −7.69 | −9.9 |
| Salinomycin (SLM) | −9.02 | −4.97 | −5.19 | −9.05 |
| Niclosamide (NIS) | −6.43 | −5.87 | −5.66 | −11.84 |
| Proscillaridin (PRO) | −6.22 | −7.28 | −7.11 | −8.25 |
| Cocrystallized ligand GRM | −8.46 | |||
| Cocrystallized ligand X77 | −8.05 | |||
| Cocrystallized ligand RDV | −7.8 | |||
| Cocrystallized ligand LKB | −11.72 |
| System | |||||
|---|---|---|---|---|---|
| GRM–PLpro | −39.7 (0.1) | 0.7 (0.1) | 13.9 (0.1) | −4.6 (0.1) | −29.7 (0.1) |
| OUB–PLpro | −38.2 (0.2) | −55.4 (0.3) | 61.9 (0.2) | −5.7 (0.1) | −37.4 (0.2) |
| PLpro–SLM | −36.7 (0.1) | 52.9 (0.2) | −41.4 (0.2) | −4.6 (0.1) | −29.8 (0.1) |
| Mpro–X77 | −52.9 (0.1) | −22.8 (0.2) | 39.1 (0.1) | −6.2 (0.1) | −42.8 (0.1) |
| Mpro–OUB | −41.9 (0.1) | −29.3 (0.2) | 47.7 (0.1) | −5.2 (0.1) | −28.7 (0.1) |
| RdRp–RDV | −36.9 (0.1) | −219.8 (0.8) | 250.2 (0.7) | −6.5 (0.1) | −13.0 (0.1) |
| RdRp–DIG | −50.6 (0.1) | −34.6 (0.3) | 76.8 (0.3) | −6.3 (0.1) | −14.7 (0.1) |
| AAK1–LKB | −44.7 (0.1) | −31.3 (0.1) | 40.3 (0.1) | −5.7 (0.1) | −41.4 (0.1) |
| AAK1–NIS | −41.0 (0.1) | −22.0 (0.1) | 29.1 (0.1) | −5.3 (0.1) | −39.2 (0.1) |
| PLpro systems | |||
| GRM | OUB | SLM | |
| Leu163 | −0.68 | −2.96 | −0.83 |
| Asp165 | 0.31 | −1.78 | −1.58 |
| Arg167 | −0.12 | −2.65 | −0.02 |
| Pro248 | −1.60 | −0.80 | −2.35 |
| Tyr265 | −2.15 | −1.75 | −2.51 |
| Tyr269 | −3.64 | −2.55 | 0.03 |
| Gln270 | −1.47 | −0.81 | 0.09 |
| Mpro systems | |||
| X77 | OUB | ||
| His41 | −1.55 | −2.12 | |
| Met49 | −1.64 | −1.56 | |
| Asn142 | −1.68 | 0.41 | |
| Gly143 | −1.81 | 0.07 | |
| Ser144 | −1.33 | 0.02 | |
| Cys145 | −1.48 | −0.41 | |
| Met165 | −4.41 | −1.69 | |
| Glu166 | −1.56 | −0.09 | |
| Gln189 | −0.45 | −1.98 | |
| Thr190 | −0.07 | −3.47 | |
| Gln192 | 0.05 | −2.16 | |
| RdRp systems | |||
| RDV | DIG | ||
| Ser550 | −0.25 | −2.08 | |
| Val558 | 0.00 | −1.42 | |
| Thr688 | 0.03 | −1.20 | |
| Glu730 | −2.82 | 0.30 | |
| Cys731 | −1.45 | 0.50 | |
| Glu812 | −1.77 | 0.24 | |
| Ser815 | −4.24 | −1.02 | |
| AAK1 systems | |||
| LKB | NIS | ||
| Leu20 | −1.99 | −2.36 | |
| Val28 | −1.65 | −1.83 | |
| Lys42 | −2.09 | −1.19 | |
| Asp95 | −2.11 | −0.06 | |
| Phe96 | −2.56 | −0.95 | |
| Cys97 | −1.69 | −2.98 | |
| Leu151 | −2.23 | −2.02 | |
| Cys161 | −1.74 | −1.41 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qayed, W.S.; Ferreira, R.S.; Silva, J.R.A. In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations. Molecules 2022, 27, 5988. https://doi.org/10.3390/molecules27185988
Qayed WS, Ferreira RS, Silva JRA. In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations. Molecules. 2022; 27(18):5988. https://doi.org/10.3390/molecules27185988
Chicago/Turabian StyleQayed, Wesam S., Rafaela S. Ferreira, and José Rogério A. Silva. 2022. "In Silico Study towards Repositioning of FDA-Approved Drug Candidates for Anticoronaviral Therapy: Molecular Docking, Molecular Dynamics and Binding Free Energy Calculations" Molecules 27, no. 18: 5988. https://doi.org/10.3390/molecules27185988