5.1. Chemistry
5.1.1. General
Starting materials were obtained from commercial suppliers and used without further purification. IR spectra were recorded on a Perkin-Elmer Spectrum 65 or Smart Orbit, Nicolet 5700 thermo electron FT-IR spectrometer ( in cm−1). NMR spectra, performed on a Bruker AVANCE 400 III HD (1H: 400 MHz, 13C: 101 MHz) or a Bruker AVANCE III HD 500 (1H: 500 MHz, 13C: 126 MHz), are reported in ppm using the solvent residual peak as an internal standard; the following abbreviations are used: singlet (s), doublet (d), triplet (t), quadruplet (q), quintuplet (quint), hexuplet (hex), heptuplet (hept), multiplet (m) and broad signal (br s). Coupling constants are expressed in Hertz. High resolution mass spectra were determined on a high-resolution Waters Micro Q-Tof or Thermo Scientific Q Exactive Q-Orbitrap apparatus (UCA START, Université Clermont Auvergne, Clermont-Ferrand, France). Chromatographic purifications were performed by column chromatography using 40–63 μm silica gel or by preparative TLC using silica gel-coated glass plates 60 F254 from Macherey Nagel. Reactions were monitored by TLC using fluorescent silica gel plates (60 F254 from Macherey Nagel). Melting points were measured on a Stuart SMP3 apparatus and were uncorrected.
The purity of all tested compounds was established by HPLC analysis using either a VWR Hitachi chromatograph (for 1a–1c, 1a’, 2a–2c, 3e, 4) or an Agilent 1100 series G1315A (for 3a–3d) with DAD detector. A Macherey Nagel Nucleodur gravity column (4.6 mm × 250 mm, 5 µM) was used for all compounds. The flow rate was 0.5 mL/min, and the analysis was performed at 25 °C at 240 or 270 nm as the detection wavelength for each compound. Solvents were (A) water/0.1% formic acid, (B) Acetonitrile. Two methods were designed: method A was a gradient of 5:95 A/B for 5 min to 95:5 A/B in 25 min, whereas method B was an isocratic mode using 60/40 water/acetonitrile.
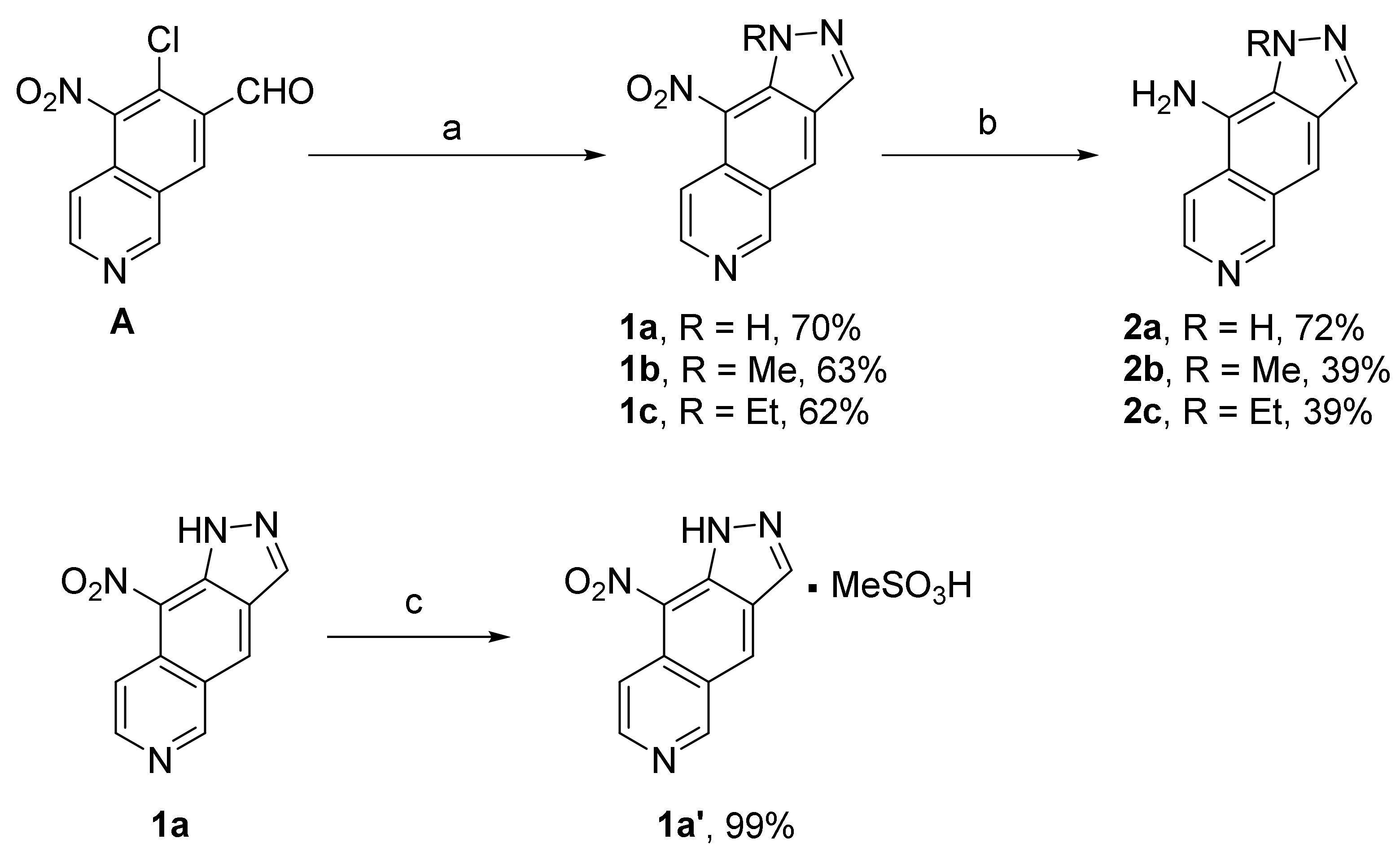
5.1.2. 9-Nitro-1H-pyrazolo[3,4-g]isoquinoline (1a)
To a solution of 6-chloro-5-nitroisoquinoline-7-carbaldehyde A (100 mg, 0.423 mmol) in EtOH (2.4 mL) was added a solution of hydrazine hydrate 98% (25.3 µL, 0.519 mmol, 1.2 eq.). The reaction mixture was stirred 1 h at room temperature before refluxing for 2 h. After filtration, washing with EtOH and a mixture CH2Cl2/Acetone (9:1, 50 mL), compound 1a was obtained as a yellow solid (64 mg, 0.299 mmol, 70%). Rf = 0.1 (CH2Cl2/Acetone 9:1). Mp > 250 °C. IR (ATR): 3225-1988, 1624, 1602, 1524, 1455, 1368 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (1H, d, J = 6.4 Hz), 8.87 (1H, s), 8.97 (1H, d, J = 6.4 Hz), 9.36 (1H, s), 9.68 (1H, s), 14.18 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6) δ 115.2, 133.9, 138.6, 147.5, 155.9 (CHarom), 99.6, 127.6, 128.1, 135.6, 160.9 (Carom). HRMS (ESI+) calcd for C10H7N4O2 (M + H)+ 215.0563, found 215.0556. HPLC: purity > 95%, λ = 240 nm, tR = 18.6 min (Method B).
5.1.3. 9-Nitro-1H-pyrazolo[3,4-g]isoquinoline Methanesulfonate (1a’)
To a solution of 1a (34.2 mg, 0.159 mmol), in anhydrous diethyl ether (10 mL), was slowly added methanesulfonic acid (10 µL, 0.154 mmol, 1.1 eq.). After 10 min of stirring, the brown precipitate formed was filtered off. After trituration with pentane and washing with diethyl ether, 1a’ was obtained as a brown solid (49.3 mg, 0.159 mmol, 99%). Mp > 250 °C. IR (ATR): 3300-2944, 1627, 1529, 1338, 1275 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 2.32 (3H, s), 8.81 (1H, d, J = 5.2 Hz), 8.96 (1H, s), 9.12 (1H, d, J = 5.2 Hz), 9.48 (1H, s), 9.89 (1H, s), 14.37 (1H, br s, NH). 13C NMR (100 MHz, DMSO-d6) δ 117.4, 135.4, 139.2, 140.7, 153.3 (CHarom), 122.5, 128.9, 129.0, 136.4, 147.8 (Carom). CH3 signal under solvent. HRMS (ESI+) calcd for C10H7N4O2 (M + H)+ 215.0563, found 215.0561. HPLC: purity > 97%, λ = 240 nm, tR = 18.6 min (Method A).
5.1.4. 1-Methyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (1b)
As described for 1a, using intermediate A (70 mg, 0.296 mmol), EtOH (2 mL), methylhydrazine sulfate (51.3 mg, 0.356 mmol, 1.2 eq.), compound 1b was obtained as a brown solid (42.8 mg, 0.187 mmol, 63%). Rf = 0.4 (EtOAc). Mp: 202–203 °C. IR (ATR): 1619, 1534, 1458, 1345, 1137 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 4.09 (3H, s), 8.21 (1H, d, J = 6.8 Hz), 8.67 (1H, d, J = 6.8 Hz), 8.97 (1H, s), 9.38 (1H, s), 9.92 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 38.6 (CH3), 115.6, 131.5, 137.3, 139.0, 153.2 (CHarom), 121.5, 126.4, 126.6, 129.5, 132.2 (Carom). HRMS (ESI+) calcd for C11H9N4O2 (M + H)+ 229.0720, found 229.0717. HPLC: purity > 95%, λ = 240 nm, tR = 20.8 min (Method A).
5.1.5. 1-Ethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (1c)
As described for 1a, using intermediate A (500 mg, 2.113 mmol), EtOH (34 mL), ethylhydrazine oxalate (381 mg, 2.536 mmol, 1.2 eq.), the reaction mixture was refluxed for 6 h. The solvent was removed under reduced pressure, and the residue was partitioned between water and ethyl acetate. The organic phase was dried over MgSO4 and evaporated. Purification by flash chromatography using CH2Cl2/Acetone 9:1 as eluent afforded compound 1c as a brown solid (320.7 mg, 1.325 mmol, 62%). Rf = 0.55 (CH2Cl2 /Acetone 9:1). Mp: 123–124 °C. IR (ATR): 1625, 1510, 1341, 1291, 1213 cm−1. 1H NMR (400 MHz, CD3OD) δ 1.43 (3H, t, J = 7.2 Hz), 4.48 (2H, q, J = 7.2 Hz), 7.94 (1H, d, J = 6.4 Hz), 8.54 (1H, d, J = 6.4 Hz), 8.66 (1H, s), 9.05 (1H, s), 9.52 (1H, s). 13C NMR (100 MHz, CD3OD) δ 15.3 (CH3), 47.5 (CH2), 115.1, 129.3, 137.6, 144.9, 156.0 (CHarom), 119.5, 124.3, 127.6, 130.4, 131.7(Carom). HRMS (ESI+) calcd for C12H11N4O2 (M + H)+ 243.0876, found 243.0875. HPLC: purity > 95%, λ = 240 nm, tR = 22.1 min (Method A).
5.1.6. 1-H-pyrazolo[3,4-g]isoquinolin-9-amine (2a)
To a solution of 1a (50 mg, 0.233 mmol) in a mixture of CH2Cl2/MeOH (1:1) (20 mL) was added Pd/C (10 wt%, 9 mg, 0.084 mmol). The reaction mixture was then stirred under H2 atmosphere (1 bar) at room temperature for 2 h 30. After filtration over Celite, washing with EtOAc and solvent evaporation, 2a was obtained as a yellow powder (31 mg, 0.168 mmol, 72%). Rf = 0.4 (EtOAc/MeOH 9:1). Mp > 250 °C. IR (ATR): 3407-2386, 1645, 1599, 1489, 1366 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 6.21 (2H, br s, NH2), 7.80 (1H, s), 7.97 (1H, d, J = 6.0 Hz), 8.13 (1H, d, J = 6.4 Hz), 8.33 (1H, s), 9.23 (1H, s), 12.82 (1H, br s, NH); 13C NMR (100 MHz, DMSO-d6) δ 105.8, 115.0, 134.8, 137.3, 154.7 (CHarom), 117.3, 124.7, 125.5, 126.2, 128.9 (Carom). HRMS (ESI+) calcd for C10H9N4 (M + H)+ 185.0827, found 185.0824. HPLC: purity > 95%, λ = 270 nm, tR = 18.5 min (Method B).
5.1.7. 1-Methyl-1H-pyrazolo[3,4-g]isoquinolin-9-amine (2b)
As described for 2a, using 1b (70 mg, 0.306 mmol), CH2Cl2/MeOH (1:1) (20 mL), Pd/C (10 wt%, 14 mg, 0.131 mmol), the reaction mixture, protected from light, was stirred under H2 atmosphere (1 bar) at room temperature for 1 h 30. After filtration over Celite and solvent evaporation, the crude mixture was purified by flash chromatography using CH2Cl2/NH3 (7N methanolic solution) 98:2 to give 2b as a brown solid (24 mg, 0.121 mmol, 39%). Rf = 0.7 (EtOAc/MeOH 9:1). Mp: 174–175 °C. IR (ATR): 3354-2854, 1654, 1584, 1453, 1387, 1182 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 4.42 (3H, s), 6.00 (2H, br s, NH2), 7.83 (1H, s), 8.10 (1H, d, J = 6.4 Hz), 8.15 (1H, d, J = 6.4 Hz), 8.24 (1H, s), 9.21 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 39.1 (CH3), 107.5, 114.9, 133.2, 137.9, 154.6 (CHarom), 119.2, 125.3, 126.7, 127.2, 129.2 (Carom). HRMS (ESI+) calcd for C11H11N4 (M + H)+ 199.0978, found 199.0976. HPLC: purity > 95%, λ = 270 nm, tR = 19.1 min (Method A).
5.1.8. 1-Ethyl-1H-pyrazolo[3,4-g]isoquinolin-9-amine (2c)
As described for 2b, using 1c (51 mg, 0.211 mmol), CH2Cl2/MeOH (1:1) (12 mL), Pd/C (8 mg, 0.075 mmol). Flash chromatography using EtOAc to give 2c as a yellow gum (17.7 mg, 0.083 mmol, 39%). Rf = 0,4 (AcOEt). IR (ATR): 3411-2851, 1647, 1571, 1484, 1380 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.32 (3H, t, J = 7.2 Hz), 4.80 (2H, q, J = 7.2 Hz), 5.97 (2H, br s, NH2), 7.85 (1H, s), 8.13 (1H, d, J = 6.4 Hz), 8.16 (1H, d, J = 6.4 Hz), 8.29 (1H, s), 9.22 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 16.7 (CH3), 46.2 (CH2), 107.6, 115.0, 134.0, 138.0, 154.5 (CHarom), 119.5, 125.3, 126.8, 127.2, 128.4 (Carom). HRMS (ESI+) calcd for C12H13N4 (M + H)+ 213.1135, found 213.1135. HPLC: purity > 95%, λ = 240 nm, tR = 19.8 min (Method A).
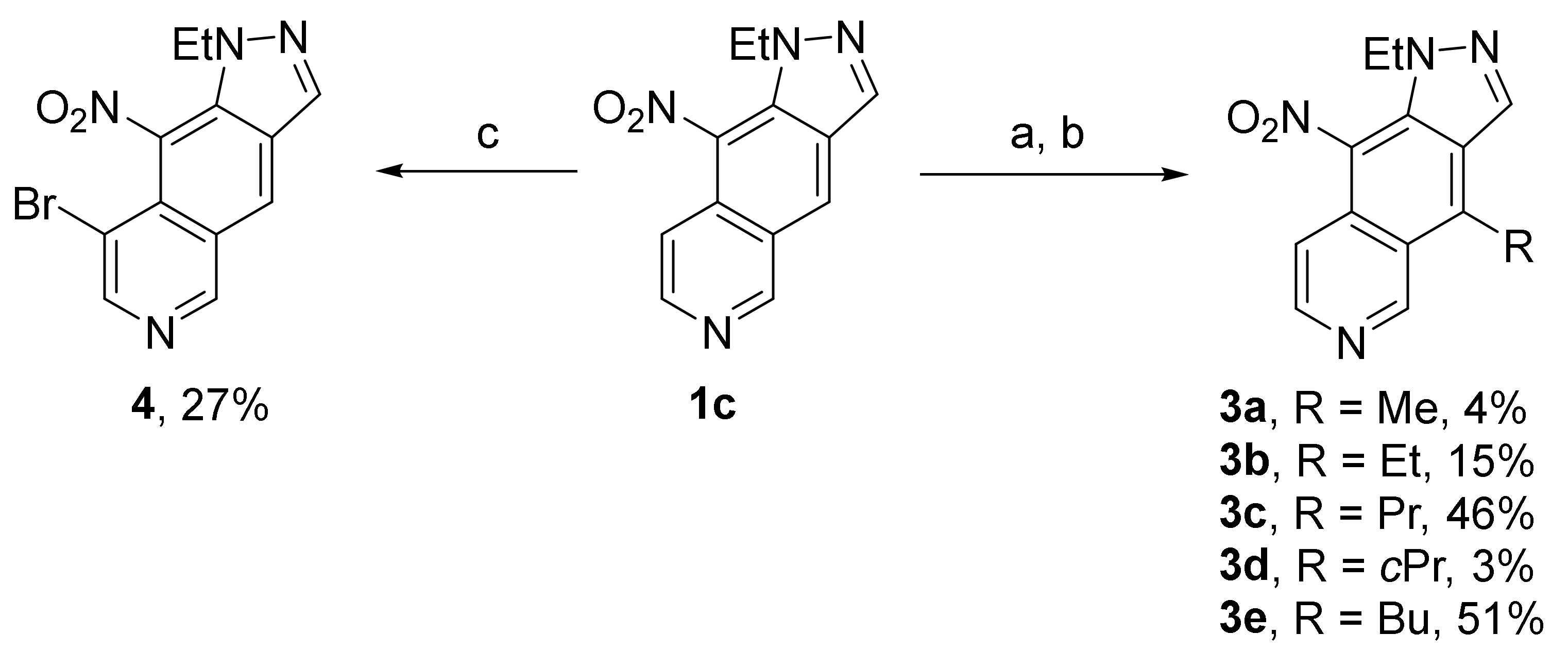
5.1.9. General Procedure for the Alkylation of 1-Ethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline 1c
Preparation of Grignard reagents:
Except for methylmagnesium bromide (1.92 M in diethyl ether) and ethylmagnesium bromide (0.33 M in THF) solutions that were purchased from Sigma-Aldrich, all other organomagnesium reagents were prepared according to the following procedure.
Magnesium turnings (100 mg, 4.1 mmol, 1 eq) in 2 mL of anhydrous THF were activated by adding a small amount of iodine. Then the suitable bromide (4.1 mmol, 1 eq) was added dropwise at 0 °C. Once the addition finished, the mixture was heated to reflux for 45 min. The concentration of the obtained organomagnesium solution was determined as follows: to a solution of iodine (100 mg) in anhydrous THF was added dropwise the organomagnesium solution until the complete disappearance of iodine color.
The reaction of organomagnesium/organolithium reagents with compound 1c:
To a solution of compound 1c in anhydrous THF (0.05 mmol/mL) was added dropwise a solution of the corresponding organomagnesium derivative. The mixture was stirred at a suitable temperature under an argon atmosphere until all the starting material was consumed (TLC control, from 3 to 18 h). The mixture was then treated with a saturated aqueous NH4Cl solution, made alkaline with a saturated aqueous NaHCO3 solution and extracted with EtOAc. Combined organic layers were dried over MgSO4 and concentrated. The crude material was used in the next step without purification as follows: the residue was solubilized in a mixture of DCM/MeOH (3/2, 0.01 mmol/mL), then DDQ (2,3-dichloro-5,6-dicyano-p-benzoquinone) (1 eq) was added. The solution was stirred at room temperature until the TLC control showed that the additional product was totally oxidized (1 h). The reaction mixture was then concentrated under reduced pressure. The residue was purified by flash chromatography or/and preparative TLC to give the corresponding 5-substituted derivative.
5.1.10. 1-Ethyl-4-methyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (3a)
Compound 1c (50 mg, 0.206 mmol), methylmagnesium bromide (1.92 M/Et2O, 0.43 mL, 0.826 mmol), reflux, 12 h. Then DDQ (187 mg, 0.826 mmol) in DCM/MeOH 3:2 mixture (60 mL). Purification by preparative TLC using DCM/Acetone 10% (two runs) to give compound 3a (2.1 mg, 0.008 mmol, 4%) as a yellow powder. Rf = 0.45 (DCM/MeOH 9:1). Mp. 92–93 °C. IR (ATR): 2931, 1675, 1506, 1456, 1377, 1340 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.32 (3H, t, J = 7.2 Hz), 3.21 (3H, s), 4.36 (2H, q, J = 7.2 Hz), 7.88 (1H, d, J = 6.4 Hz), 8.62 (1H, d, J = 6.4 Hz), 9.01 (1H, s), 9.77 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 14.8, 15.2 (CH3), 46.1 (CH2), 113.1, 136.3, 145.1, 151.8 (CHarom), 120.6, 124.8, 126.4, 128.5, 129.7, 139.8 (Carom). HRMS (ESI+) calcd for C13H13N4O2 (M + H+) 257.1033, found 257.1025. HPLC: purity > 96%, λ = 270 nm, tR = 20.2 min (Method A).
5.1.11. 1,4-Diethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (3b)
Compound 1c (150 mg, 0.619 mmol), ethylmagnesium bromide (0.33 M/THF, 5.63 mL, 1.858 mmol), −40 °C, 4 h. Then DDQ (141 mg, 0.619 mmol) in DCM/MeOH 3:2 mixture (60 mL). Purification by two flash chromatographies first with EtOAc/cyclohexane 1:1 + 0.5% Et3N then with DCM/MeOH 5% to give compound 3b (25 mg, 0.092 mmol, 15%) as a yellow powder. Rf = 0.50 (DCM/MeOH 9:1). Mp. 96–97 °C. IR (ATR): 2977, 1602, 1502, 1490, 1375, 1271 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.33 (3H, t, J = 7.2 Hz), 1.40 (3H, t, J = 7.6 Hz), 3.72 (2H, q, J = 7.6 Hz), 4.36 (2H, q, J = 7.2 Hz), 7.89 (1H, d, J = 6.4 Hz), 8.62 (1H, d, J = 6.0 Hz), 9.03 (1H, s), 9.82 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 14.8, 16.8 (CH3), 22.1, 46.0 (CH2), 113.3, 135.8, 145.0, 151.4 (CHarom), 119.6, 125.0, 126.6, 127.8, 129.7, 145.6 (Carom). HRMS (ESI+) calcd for C14H15N4O2 (M + H+) 271.1189, found 271.1195. HPLC: purity > 98%, λ = 270 nm, tR = 21.1 min (Method A).
5.1.12. 1-Ethyl-9-nitro-4-propyl-1H-pyrazolo[3,4-g]isoquinoline (3c)
Compound 1c (50 mg, 0.206 mmol), propylmagnesium bromide (0.36 M/THF, 1.72 mL, 0.619 mmol), 0 °C, 3 h. Then DDQ (47 mg, 0.206 mmol) in DCM/MeOH 3:2 mixture (20 mL). Purification by two flash chromatographies first with EtOAc/cyclohexane 1:1 + 0.5% Et3N then with DCM/Acetone 2% to give compound 3c (27 mg, 0.095 mmol, 46%) as a yellow powder. Rf = 0.55 (DCM/Acetone 9:1). Mp. 109–110 °C. IR (ATR): 3119, 1605, 1504, 1450, 1338, 1255 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.00 (3H, t, J = 7.2 Hz), 1.33 (3H, t, J = 7.2 Hz), 1.80 (2H, hex, J = 7.2 Hz), 3.68 (2H, t, J = 7.4 Hz), 4.36 (2H, q, J = 7.2 Hz), 7.89 (1H, d, J = 6.0 Hz), 8.62 (1H, d, J = 6.0 Hz), 9.04 (1H, s), 9.83 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 14.0, 14.8 (CH3), 25.5, 30.3, 46.1 (CH2), 113.3, 136.1, 145.0, 151.6 (CHarom), 120.1, 125.1, 126.6, 128.4, 129.6, 144.0 (Carom). HRMS (ESI+) calcd for C15H17N4O2 (M + H+) 285.1346, found 285.1339. HPLC: purity > 99%, λ = 270 nm, tR = 22.2 min (Method A).
5.1.13. 4-Cyclopropyl-1-ethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (3d)
Compound 1c (100 mg, 0.413 mmol), cyclopropylmagnesium bromide (0.44 M/THF, 2.81 mL, 1.236 mmol), 0 °C, 6 h. Then DDQ (94 mg, 0.413 mmol) in DCM/MeOH 3:2 mixture (40 mL). Purification by three chromatographies first by flash column with DCM/Acetone 10% then by a first preparative TLC eluted with DCM/Acetone 10% then a second one using EtOAc to give compound 3d (4.3 mg, 0.015 mmol, 3%) as a yellow powder. Rf = 0.60 (DCM/Acetone 9:1). Mp. 97 °C. IR (ATR): 2927, 1604, 1501, 1450, 1331, 1271 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.03-1.09 (2H, m), 1.33 (3H, t, J = 7.2 Hz), 1.43–1.50 (2H, m), 2.78–2.87 (1H, m), 4.36 (2H, q, J = 7.2 Hz), 7.87 (1H, d, J = 6.4 Hz), 8.64 (1H, d, J = 6.0 Hz), 8.94 (1H, s), 10.12 (1H, s). 13C NMR (100 MHz, CDCl3) δ 11.3 (CH3), 7.8 (2CH2), 46.7 (CH2), 15.2 (CH), 113.9, 135.3, 144.8, 152.3 (CHarom), 122.9, 127.1, 128.4, 129.3, 130.4, 142.0 (Carom). HRMS (ESI+) calcd for C15H15N4O2 (M + H+) 283.1189, found 283.1182. HPLC: purity > 98%, λ = 270 nm, tR = 21.3 min (Method A).
5.1.14. 4-Butyl-1-ethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (3e)
Compound 1c (100 mg, 0.413 mmol), butylmagnesium bromide (0.17 M/THF, 7.16 mL, 1.239 mmol), 0 °C, 3 h. Then DDQ (94 mg, 0.413 mmol) in DCM/MeOH 3:2 mixture (40 mL). Purification by flash chromatography with DCM/Acetone 3% to give compound 3e (63 mg, 0.211 mmol, 51%) as a yellow powder. Rf = 0.60 (DCM/Acetone 9:1). Mp. 120–121 °C. IR (ATR): 2970, 1612, 1506, 1456, 1336, 1300 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 0.92 (3H, t, J = 7.2 Hz), 1.34 (3H, t, J = 7.2 Hz), 1.43 (2H, hex, J = 7.2 Hz), 1.75 (2H, quint, J = 7.2 Hz), 3.71 (2H, t, J = 7.6 Hz), 4.36 (2H, q, J = 7.2 Hz), 7.89 (1H, d, J = 6.4 Hz), 8.62 (1H, d, J = 6.0 Hz), 9.03 (1H, s), 9.82 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 13.8, 14.8 (CH3), 22.3, 28.4, 34.3, 46.0 (CH2), 113.3, 136.0, 144.9, 151.5 (CHarom), 120.0, 125.1, 126.6, 128.3, 129.6, 144.2 (Carom). HRMS (ESI+) calcd for C16H19N4O2 (M + H+) 299.1502, found 299.1498. HPLC: purity > 95%, λ = 270 nm, tR = 24.1 min (Method A).
5.1.15. 8-Bromo-1-ethyl-9-nitro-1H-pyrazolo[3,4-g]isoquinoline (4)
To a solution of compound 1c (30 mg, 0.124 mmol) in DMF (2 mL) at room temperature under argon was added freshly recrystallized N-bromosuccinimide (22 mg, 0.124 mmol). The reaction mixture was stirred overnight at room temperature. Two other portions of NBS (6 mg each, 0.037 mmol) were successively added after 24 and 48 h, but no complete conversion was reached. EtOAc was added, and the organic phase was washed with saturated aqueous Na2S2O3 and NaHCO3 solutions, dried over MgSO4, filtered and evaporated under reduced pressure. Residue was purified by flash chromatography (EtOAc/cyclohexane 1:1) yielding compound 4 (10.8 mg, 0.034 mmol, 27%) as an orange powder, Rf = 0.35 (EtOAc/cyclohexane 1:1). Mp = 172–173 °C; IR (ATR): 2932, 1623, 1525, 1456, 1286 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 1.37 (3H, t, J = 7.2 Hz), 4.31 (2H, q, J = 7.2 Hz), 8.82 (1H, s), 8.91 (1H, s), 9.21 (1H, s), 9.62 (1H, s). 13C NMR (100 MHz, DMSO-d6) δ 15.6 (CH3), 44.9 (CH2), 127.9, 136.5, 148.3, 155.4 (CHarom), 110.3, 121.2, 124.0, 126.2, 128.3, 130.2 (Carom). HRMS (ESI+) calcd for C12H1079BrN4O2 (M + H+) 320.9982, found 320.9976. HRMS (ESI+) calcd for C12H1081BrN4O2 (M + H+) 322.9961, found 322.9953. HPLC: purity > 95%, λ = 240 nm, tR = 30.3 min (Method A).

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}