
Design and Synthesis of New α-hydroxy β-fluoro/β-trifluoromethyl and Unsaturated Phosphonates from Carbohydrate-Derived Building Blocks via Pudovik and Horner–Wadsworth–Emmons Reactions †


Abstract
:
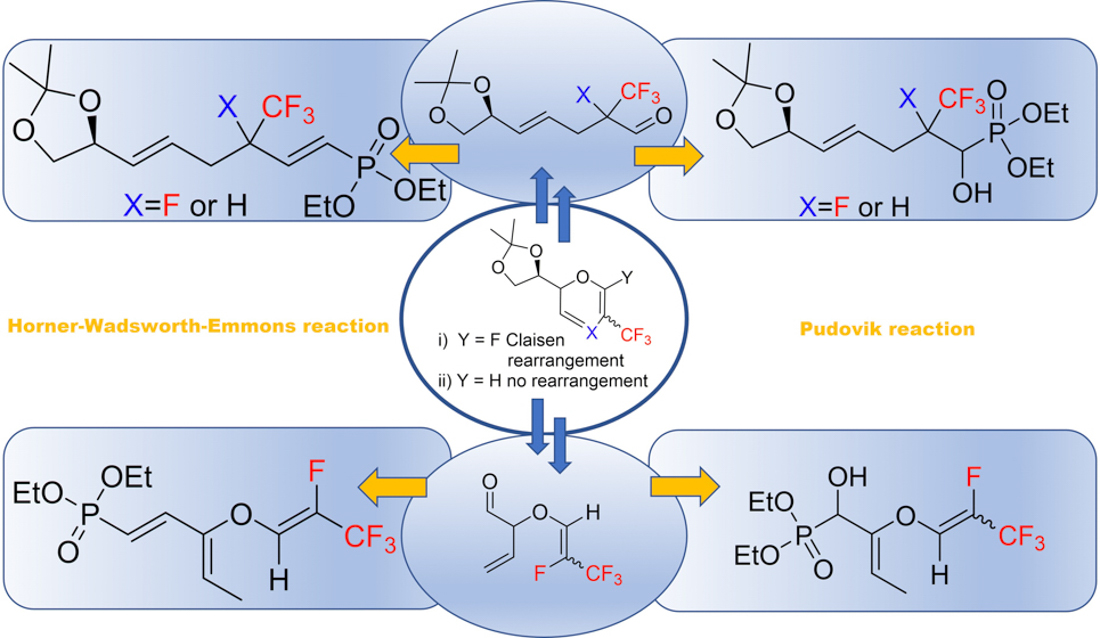
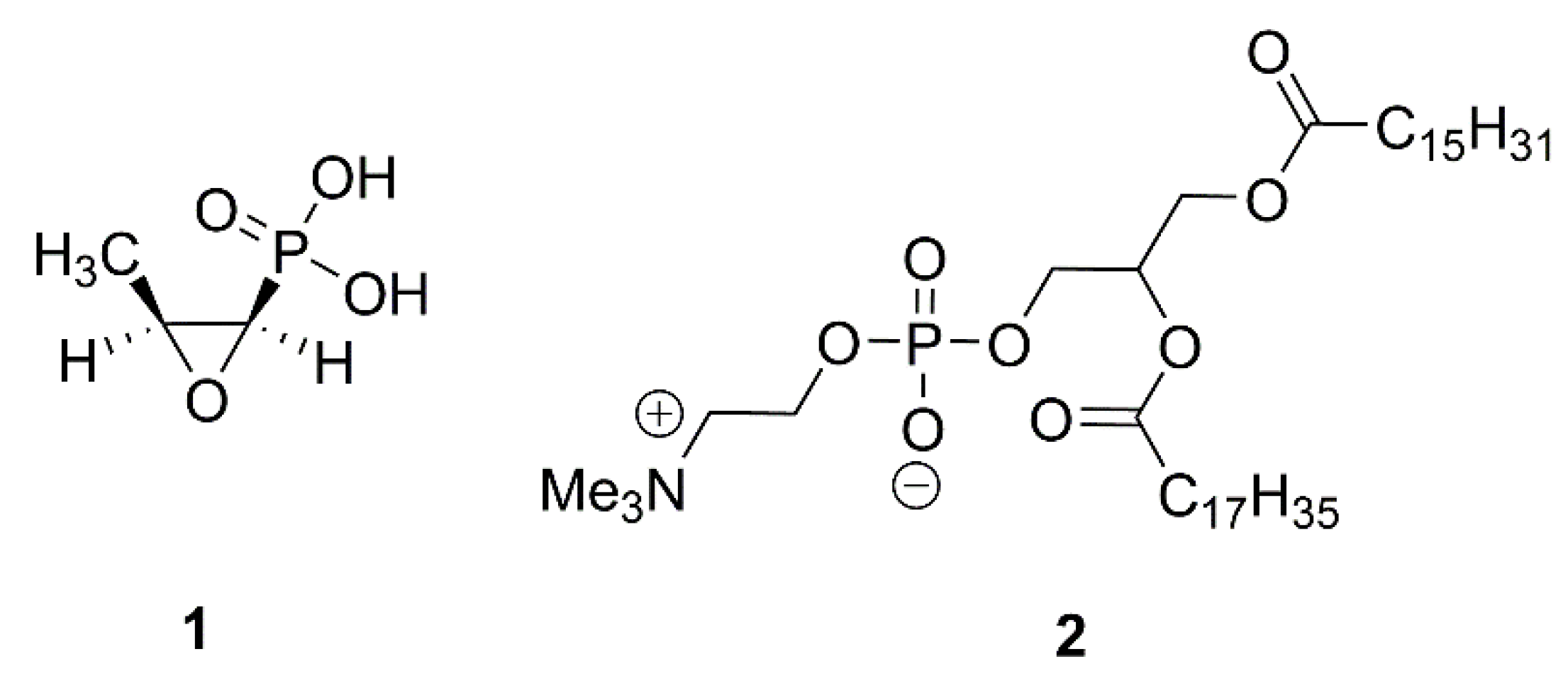
1. Introduction
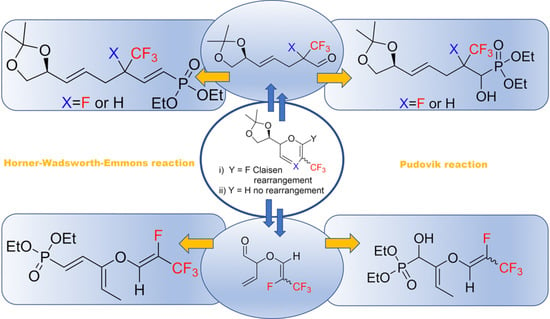
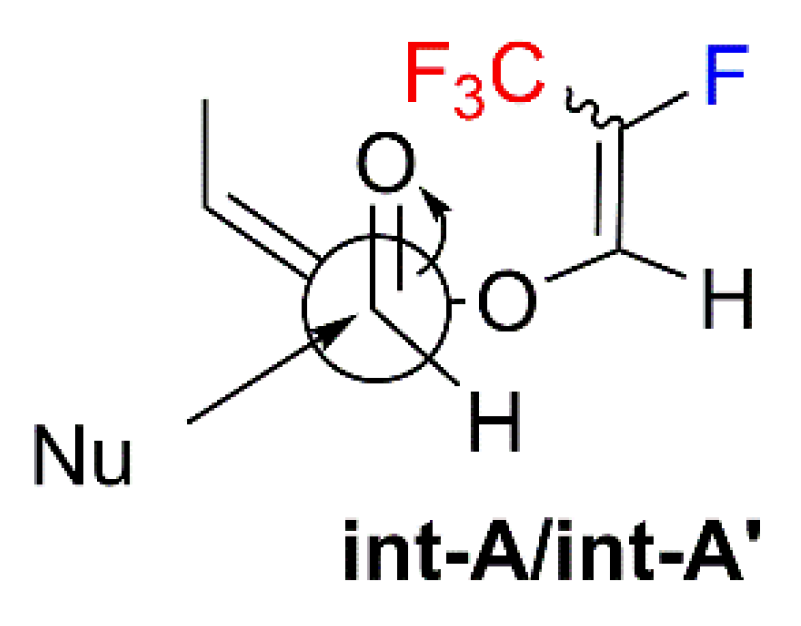
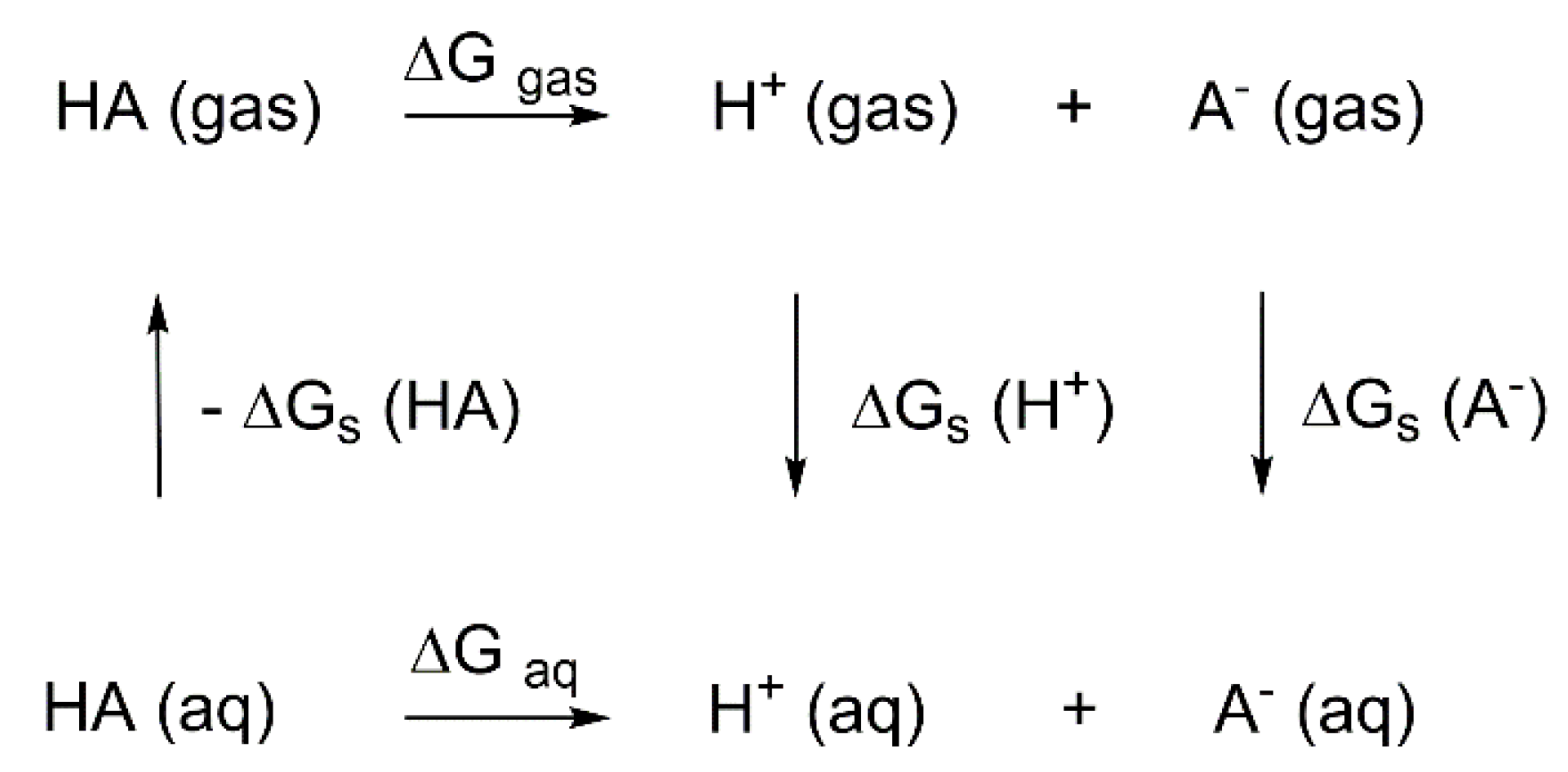
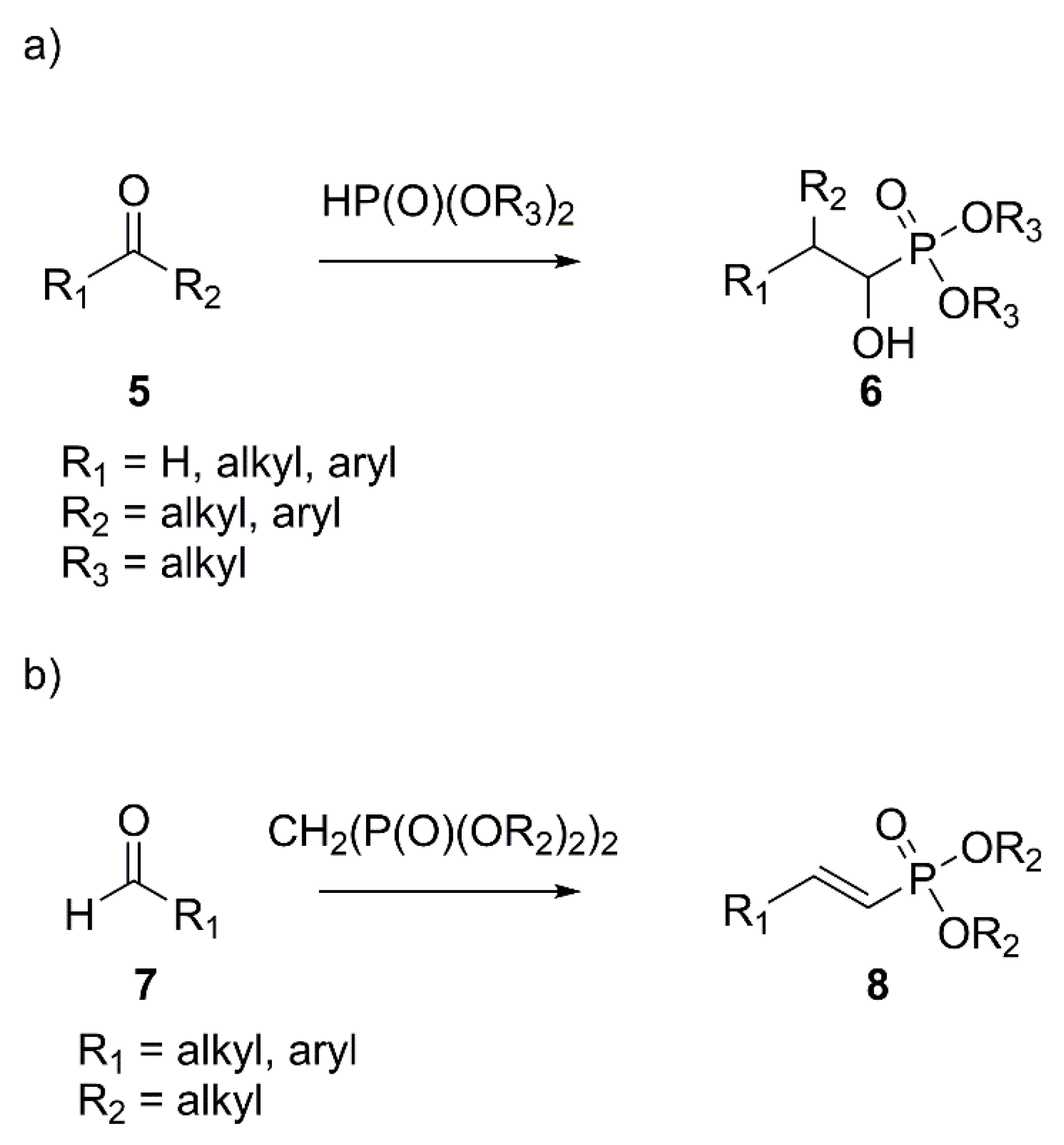
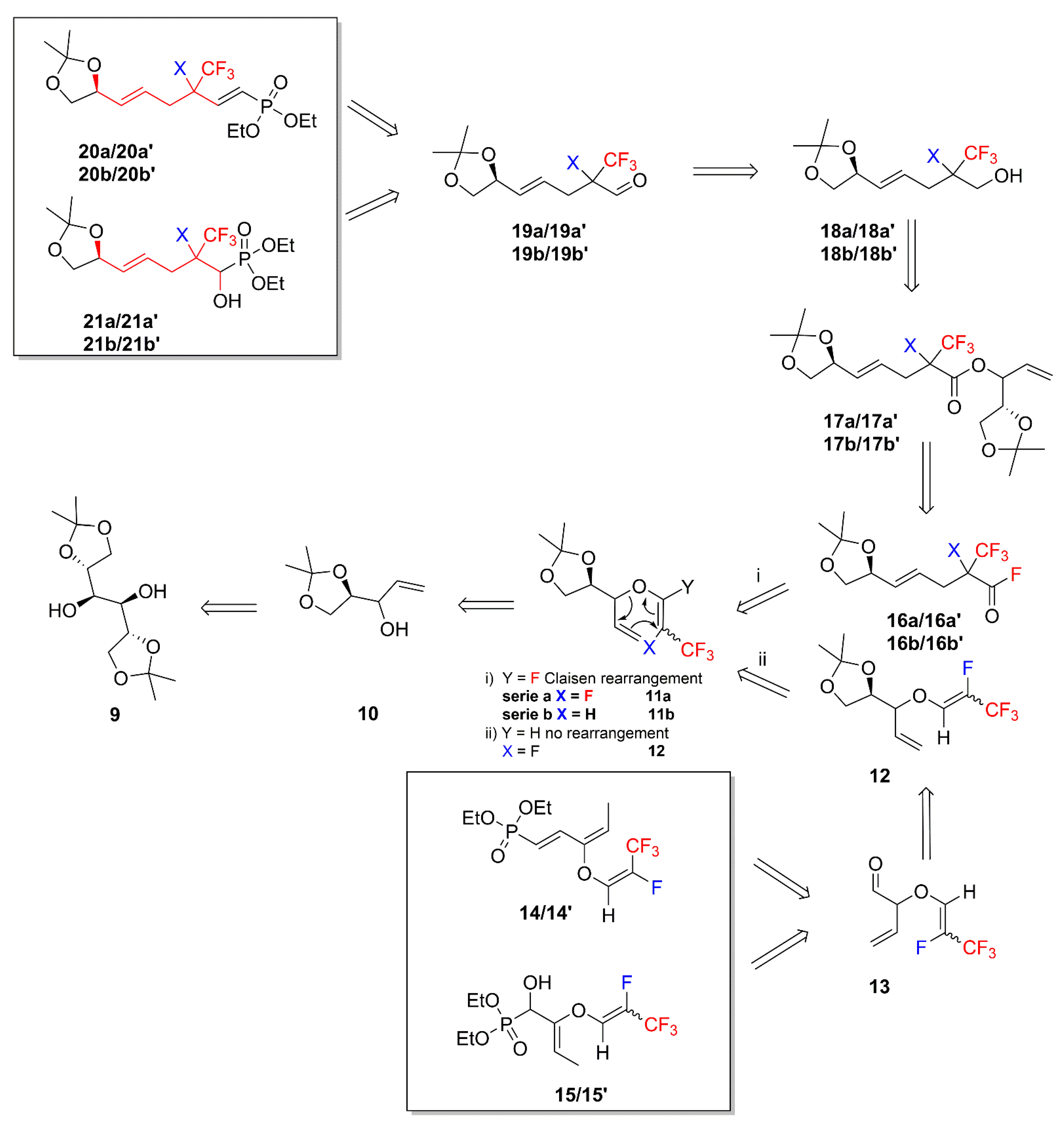
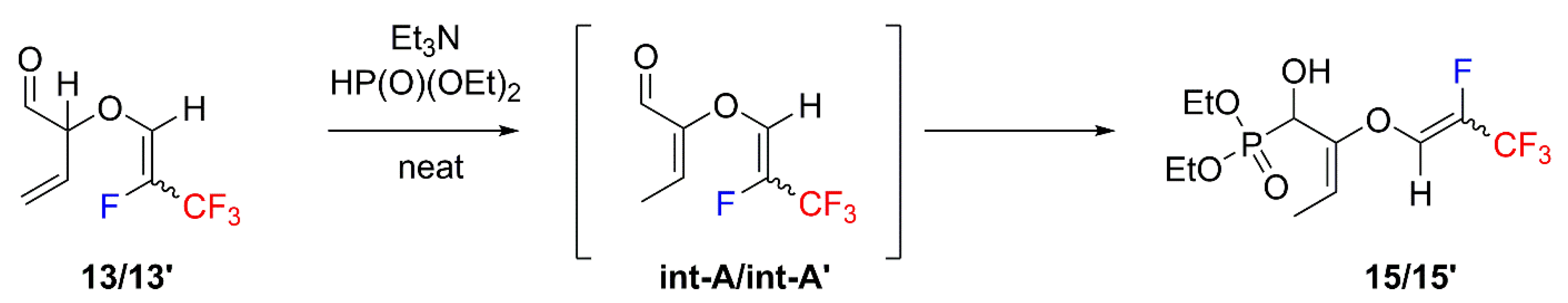
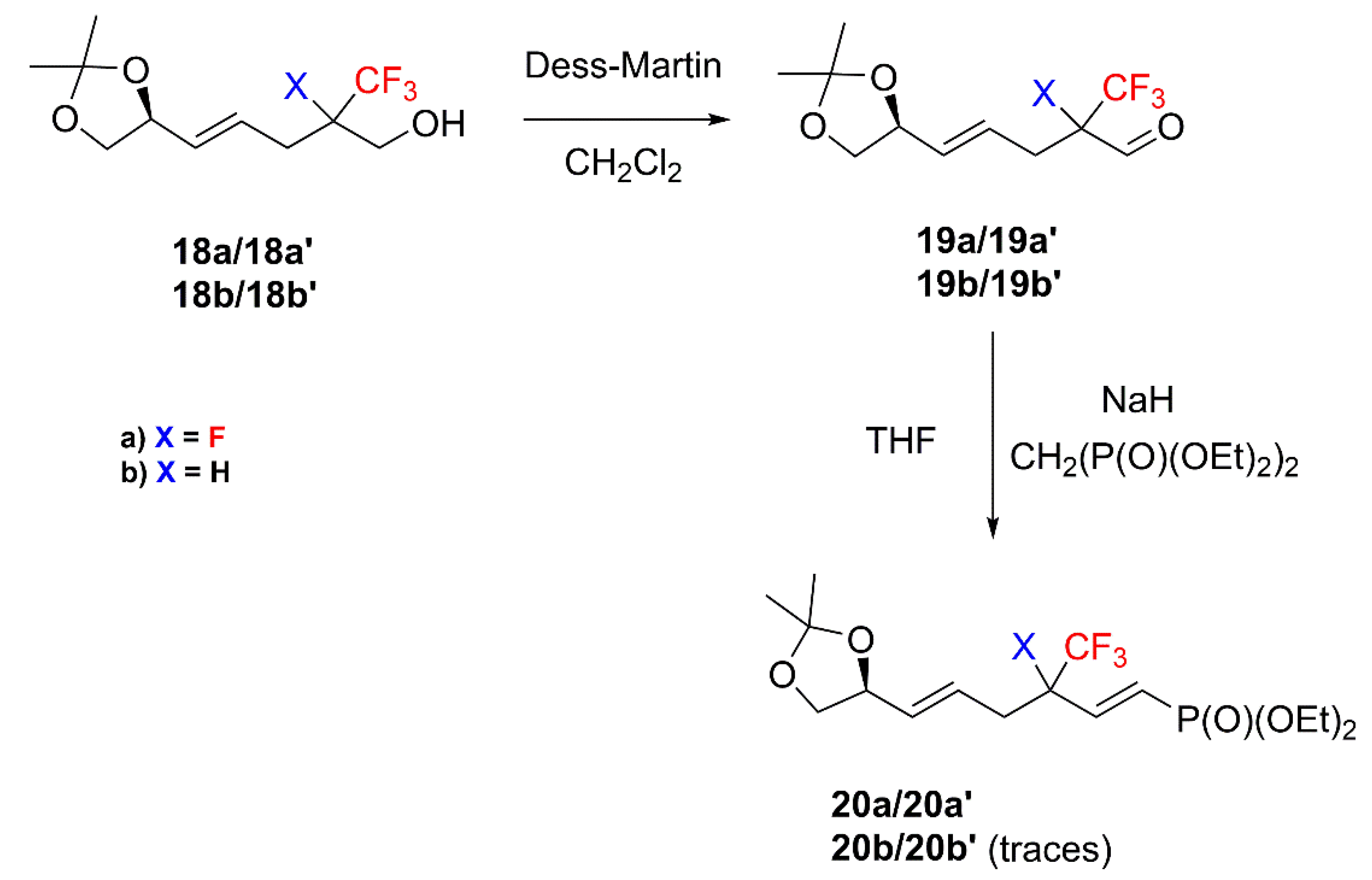
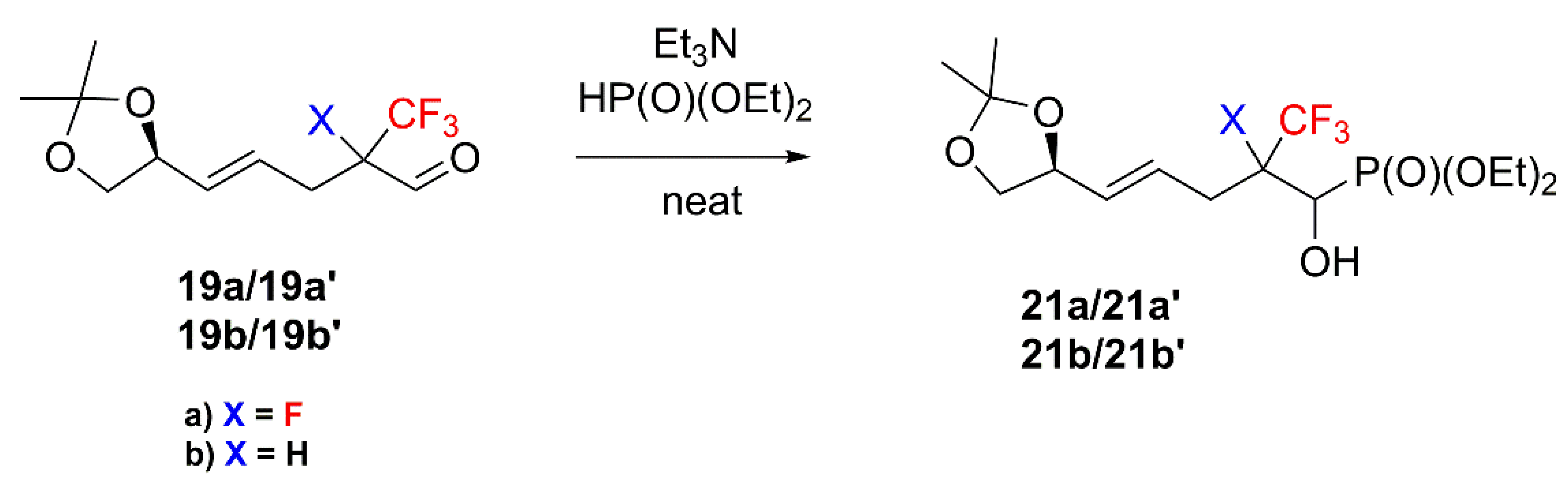
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Methods
4.2. General Procedure of Periodic Acid Oxidation Synthesis of 13
4.3. General procedure of Dess-Martin oxidation Synthesis of 19
4.4. General Procedure of HWE Reaction Synthesis of 14 and 20
4.5. General Procedure of Pudovik Reaction Synthesis of 15 and 21
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grygorenko, O.O.; Volochnyuk, D.M.; Vashchenko, B.V. Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances. Eur. J. Org. Chem. 2021, 2021, 6478–6510. [Google Scholar] [CrossRef]
- Zimmer, L.E.; Sparr, C.; Gilmour, R. Fluorine Conformational Effects in Organocatalysis: An Emerging Strategy for Molecular Design. Angew. Chem. Int. Ed. 2011, 50, 11860–11871. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Jäckel, C.; Koksch, B. Fluorine in Peptide Design and Protein Engineering. Eur. J. Org. Chem. 2005, 2005, 4483–4503. [Google Scholar] [CrossRef]
- Buer, B.C.; Marsh, E.N. Fluorine: A new element in protein design. Protein Sci. 2012, 21, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Sloand, J.N.; Miller, M.A.; Medina, S.H. Fluorinated peptide biomaterials. Pept. Sci. 2021, 113, e24184. [Google Scholar] [CrossRef]
- O’Hagan, D.; Schaffrath, C.; Cobb, S.L.; Hamilton, J.T.G.; Murphy, C.D. Biosynthesis of an organofluorine molecule. Nature 2002, 416, 279. [Google Scholar] [CrossRef]
- Hall, R.J. The distribution of organic fluorine in some toxic tropical plants. New Phytol. 1972, 71, 855–871. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Johnson, B.M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Filler, R.; Saha, R. Fluorine in medicinal chemistry: A century of progress and a 60-year retrospective of selected highlights. Future Med. Chem. 2009, 1, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Vulpetti, A.; Dalvit, C. Fluorine local environment: From screening to drug design. Drug Discov. Today 2012, 17, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar]
- Kafarski, P.; Lejczak, B. Biological Activity of Aminophosphonic Acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Engel, R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977, 77, 349–367. [Google Scholar] [CrossRef]
- Azéma, L.; Baron, R.; Ladame, S. Targeting Enzymes with Phosphonate-Based Inhibitors: Mimics of Tetrahedral Transition States and Stable Isosteric Analogues of Phosphates. Curr. Enzym. Inhib. 2006, 2, 61–72. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric synthesis of hydroxyphosphonates. Tetrahedron 2005, 16, 3295–3340. [Google Scholar] [CrossRef]
- Rádai, Z. α-Hydroxyphosphonates as versatile starting materials. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 425–437. [Google Scholar] [CrossRef]
- Romanenko, V.D.; Kukhar, V.P. Fluorinated Phosphonates: Synthesis and Biomedical Application. Chem. Rev. 2006, 106, 3868–3935. [Google Scholar] [CrossRef] [PubMed]
- Cytlak, T.; Kaźmierczak, M.; Skibińska, M.; Koroniak, H. Latest achievements in the preparation of fluorinated aminophosphonates and aminophosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 602–620. [Google Scholar] [CrossRef]
- Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Regioselective Fluorination of α-Hydroxy-β-aminophosphonates by Using PyFluor. Eur. J. Org. Chem. 2018, 2018, 3844–3852. [Google Scholar] [CrossRef]
- Kaźmierczak, M.; Dutkiewicz, G.; Cytlak, T. Application of α-Amino-β-fluorophosphonates in Construction of their Dipeptide Analogues. Synthesis 2020, 52, 2364–2372. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Dutkiewicz, G.; Koroniak, H. Deoxyfluorinating reagents as tools for γ-amino-α-hydroxyphosphonates modification. Org. Biomol. Chem. 2022, 20, 5615–5623. [Google Scholar] [CrossRef]
- Kaźmierczak, M.; Koroniak, H. Efficient synthesis of dipeptide analogues of α-fluorinated β-aminophosphonates. Beilstein J. Org. Chem. 2020, 16, 756–762. [Google Scholar] [CrossRef]
- Elliott, T.S.; Slowey, A.; Ye, Y.; Conway, S.J. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. MedChemComm 2012, 3, 735–751. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Röschenthaler, G.-V.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Cui, P.; McCalmont, W.F.; Tomsig, J.L.; Lynch, K.R.; Macdonald, T.L. alpha- and beta-substituted phosphonate analogs of LPA as autotaxin inhibitors. Bioorg. Med. Chem. 2008, 16, 2212–2225. [Google Scholar] [CrossRef] [Green Version]
- Kaźmierczak, M.; Koroniak, M. DAST mediated preparation of β-fluoro-α-aminophosphonates. J. Fluor. Chem. 2012, 139, 23–27. [Google Scholar] [CrossRef]
- Harnden, M.R.; Parkin, A.; Parratt, M.J.; Perkins, R.M. Novel acyclonucleotides: Synthesis and antiviral activity of alkenylphosphonic acid derivatives of purines and a pyrimidine. J. Med. Chem. 1993, 36, 1343–1355. [Google Scholar] [CrossRef]
- Lazrek, H.B.; Rochdi, A.; Khaider, H.; Barascut, J.L.; Imbach, J.L.; Balzarini, J.; Witvrouw, M.; Pannecouque, C.; De Clercq, E. Synthesis of (Z) and (E) α-alkenyl phosphonic acid derivatives of purines and pyrimidines. Tetrahedron 1998, 54, 3807–3816. [Google Scholar] [CrossRef]
- Thomas, A.A.; Sharpless, K.B. The Catalytic Asymmetric Aminohydroxylation of Unsaturated Phosphonates. J. Org. Chem. 1999, 64, 8379–8385. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.B.; De Mendonca, D.J. An efficient synthesis of (Z)-γ-fluoroallylphosphonates using a base-promoted deconjugation of (E)-γ-fluorovinylphosphonates, and its utility as fluoroolefin-containing building block. J. Fluor. Chem. 2000, 102, 189–197. [Google Scholar] [CrossRef]
- Pajkert, R.; Koroniak, H. Simple synthesis of some unsaturated 1,1-difluoro-2-hydroxyethylphosphonates. J. Fluor. Chem. 2007, 128, 1260–1263. [Google Scholar] [CrossRef]
- Cherkupally, P.; Slazhnev, A.; Beier, P. Stereoselective Synthesis of (E)-alpha-Fluorovinylphosphonates from alpha, alpha-Difluorophosphonates. Synlett 2011, 2011, 331–334. [Google Scholar]
- Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Preparation and characterization of α-fluorinated-γ-aminophosphonates. J. Fluor. Chem. 2014, 167, 128–134. [Google Scholar] [CrossRef]
- Panigrahi, K.; Applegate, G.A.; Malik, G.; Berkowitz, D.B. Combining a Clostridial Enzyme Exhibiting Unusual Active Site Plasticity with a Remarkably Facile Sigmatropic Rearrangement: Rapid, Stereocontrolled Entry into Densely Functionalized Fluorinated Phosphonates for Chemical Biology. J. Am. Chem. Soc. 2015, 137, 3600–3609. [Google Scholar] [CrossRef] [Green Version]
- Rádai, Z.; Keglevich, G. Synthesis and Reactions of α-Hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef] [Green Version]
- Szalai, Z.; Keglevich, G. Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products. Molecules 2021, 26, 7575. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tanaka, K.; Kogen, H. Recent topics of the natural product synthesis by Horner–Wadsworth–Emmons reaction. Tetrahedron Lett. 2018, 59, 568–582. [Google Scholar] [CrossRef]
- Roman, D.; Sauer, M.; Beemelmanns, C. Applications of the Horner–Wadsworth–Emmons Olefination in Modern Natural Product Synthesis. Synthesis 2021, 53, 2713–2739. [Google Scholar]
- Nakao, M.; Tanaka, K.; Kitaike, S.; Sano, S. Synthesis of Fluorine-Containing Analogues of 1-Lysoglycerophospholipids via Horner–Wadsworth–Emmons Reaction. Synthesis 2017, 49, 3654–3661. [Google Scholar]
- Wójtowicz-Rajchel, H. Synthesis and applications of fluorinated nucleoside analogues. J. Fluor. Chem. 2012, 143, 11–48. [Google Scholar] [CrossRef]
- Edmondson, S.D.; Wei, L.; Xu, J.; Shang, J.; Xu, S.; Pang, J.; Chaudhary, A.; Dean, D.C.; He, H.; Leiting, B.; et al. Fluoroolefins as amide bond mimics in dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2409–2413. [Google Scholar] [CrossRef]
- Bilska-Markowska, M.; Patyk-Kaźmierczak, E.; Lusina, A. Synthesis of Fluorinated Amides Starting from Carbohydrates Based on the Claisen Rearrangement. Eur. J. Org. Chem. 2022, 2022, e202101378. [Google Scholar] [CrossRef]
- Boysen, M.M.K. Carbohydrates as Synthetic Tools in Organic Chemistry. Chem. Eur. J. 2007, 13, 8648–8659. [Google Scholar] [CrossRef]
- Marciniak, B.; Bilska-Markowska, M.; Grzeszczuk, M.; Rapp, M.; Koroniak, H. A convenient synthesis of fluorinated vinyl ethers derived from di-O-isopropylidenehexofuranose and O-isopropylidenepentofuranose. J. Fluor. Chem. 2014, 167, 143–151. [Google Scholar] [CrossRef]
- Bilska-Markowska, M.; Szwajca, A.; Marciniak, B. Design, properties and applications of fluorinated and fluoroalkylated N-containing monosaccharides and their analogues. J. Fluor. Chem. 2019, 227, 109364. [Google Scholar] [CrossRef]
- Uhrig, M.L.; Lantaño, B.; Postigo, A. Synthetic strategies for fluorination of carbohydrates. Org. Biomol. Chem. 2019, 17, 5173–5189. [Google Scholar] [CrossRef]
- Linclau, B.; Ardá, A.; Reichardt, N.-C.; Sollogoub, M.; Unione, L.; Vincent, S.P.; Jiménez-Barbero, J. Fluorinated carbohydrates as chemical probes for molecular recognition studies. Current status and perspectives. Chem. Soc. Rev. 2020, 49, 3863–3888. [Google Scholar] [CrossRef] [PubMed]
- Shivatare, S.S.; Wong, C.-H. Synthetic Carbohydrate Chemistry and Translational Medicine. J. Org. Chem. 2020, 85, 15780–15800. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.-Y.; Lin, M.-C.; Wang, S.-H.; Lin, L.-S. A Mild Isomerization Reaction for β,γ-Unsaturated Ketone to α,β-Unsaturated Ketone. J. Chin. Chem. Soc. 2004, 51, 371–376. [Google Scholar] [CrossRef]
- Fustero, S.; Moscardó, J.; Sánchez-Roselló, M.; Flores, S.; Guerola, M.; Pozo, C.D. Organocatalytic enantioselective synthesis of quinolizidine alkaloids (+)-myrtine, (−)-lupinine, and (+)-epiepiquinamide. Tetrahedron 2011, 67, 7412–7417. [Google Scholar] [CrossRef]
- Li, Y.; Li, K.; Wu, Y.; Ma, Q.; Lei, X. Facile synthesis of fluorovinyl-containing lactams via ring-closing metathesis of N-substituted 2-fluoroallylamides. Tetrahedron 2016, 72, 4845–4853. [Google Scholar] [CrossRef]
- Wang, H. Horner-Wadsworth-Emmons Olefination. In Comprehensive Organic Name Reactions and Reagents; Wiley: Hoboken, NJ, USA, 2010; pp. 1484–1490. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Zeng, Q.; Jones, M.R.; Brooks, B.R. Absolute and relative pKa predictions via a DFT approach applied to the SAMPL6 blind challenge. J. Comput.-Aided Mol. Des. 2018, 32, 1179–1189. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, A.E. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian, Inc.: Wallingford, CT, USA, 2015. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Frisch, M.J.T.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Liptak, M.D.; Shields, G.C. Accurate pKa Calculations for Carboxylic Acids Using Complete Basis Set and Gaussian-n Models Combined with CPCM Continuum Solvation Methods. J. Am. Chem. Soc. 2001, 123, 7314–7319. [Google Scholar] [CrossRef]
- Bordwell pKa Table. Available online: https://organicchemistrydata.org/hansreich/resources/pka/#ka-water (accessed on 21 July 2022).
- Leung, P.S.-W.; Teng, Y.; Toy, P.H. Rasta Resin-PPh3 and its Use in Chromatography-Free Wittig Reactions. Synlett 2010, 2010, 1997–2001. [Google Scholar]
- Payne, J.E.; Bonnefous, C.; Hassig, C.A.; Symons, K.T.; Guo, X.; Nguyen, P.-M.; Annable, T.; Wash, P.L.; Hoffman, T.Z.; Rao, T.S.; et al. Identification of KD5170: A novel mercaptoketone-based histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2008, 18, 6093–6096. [Google Scholar] [CrossRef]
- Song, H.; Wu, D.; Mazunin, D.; Liu, S.M.; Sato, Y.; Broguiere, N.; Zenobi-Wong, M.; Bode, J.W. Post-Assembly Photomasking of Potassium Acyltrifluoroborates (KATs) for Two-Photon 3D Patterning of PEG-Hydrogels. Helv. Chim. Acta 2020, 103, e2000172. [Google Scholar] [CrossRef]
- Ortuzar, N.; Karu, K.; Presa, D.; Morais, G.R.; Sheldrake, H.M.; Shnyder, S.D.; Barnieh, F.M.; Loadman, P.M.; Patterson, L.H.; Pors, K.; et al. Probing cytochrome P450 (CYP) bioactivation with chloromethylindoline bioprecursors derived from the duocarmycin family of compounds. Bioorg. Med. Chem. 2021, 40, 116167. [Google Scholar] [CrossRef]
- Xie, M.; Berges, D.A.; Robins, M.J. Efficient “Dehomologation” of Di-O-isopropylidenehexofuranose Derivatives To Give O-Isopropylidenepentofuranoses by Sequential Treatment with Periodic Acid in Ethyl Acetate and Sodium Borohydride. J. Org. Chem. 1996, 61, 5178–5179. [Google Scholar] [CrossRef]
- Davis, F.A.; Kasu, P.V.N.; Sundarababu, G.; Qi, H. Nonracemic α-Fluoro Aldehydes: Asymmetric Synthesis of 4-Deoxy-4-fluoro-d-arabinopyranose. J. Org. Chem. 1997, 62, 7546–7547. [Google Scholar] [CrossRef]
- Allan, A.L.; Gladstone, P.L.; Price, M.L.P.; Hopkins, S.A.; Juarez, J.C.; Doñate, F.; Ternansky, R.J.; Shaw, D.E.; Ganem, B.; Li, Y.; et al. Synthesis and Evaluation of Multisubstrate Bicyclic Pyrimidine Nucleoside Inhibitors of Human Thymidine Phosphorylase. J. Med. Chem. 2006, 49, 7807–7815. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. pKa [64] | Calc. pKa | |
|---|---|---|
| Formic Acid | 3.8 | 3.7 |
| Trifluoroacetic Acid | 0.2 | 0.8 |
| Dimethyl Malonate | 13.5 | 11.9 |
| Acetone | 20.0 | 19.7 |
| Chloroacetone | 16.5 | 17.9 |
| 2-((2,3,3,3-tetrafluoroprop-1-en-1-yl)oxy)but-3-enal 13 | - | 13.3 |
| α-trifluoromethyl-γ,δ-unsaturated aldehyde 19b | - | 15.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilska-Markowska, M.; Jankowski, W.; Hoffmann, M.; Kaźmierczak, M. Design and Synthesis of New α-hydroxy β-fluoro/β-trifluoromethyl and Unsaturated Phosphonates from Carbohydrate-Derived Building Blocks via Pudovik and Horner–Wadsworth–Emmons Reactions. Molecules 2022, 27, 5404. https://doi.org/10.3390/molecules27175404
Bilska-Markowska M, Jankowski W, Hoffmann M, Kaźmierczak M. Design and Synthesis of New α-hydroxy β-fluoro/β-trifluoromethyl and Unsaturated Phosphonates from Carbohydrate-Derived Building Blocks via Pudovik and Horner–Wadsworth–Emmons Reactions. Molecules. 2022; 27(17):5404. https://doi.org/10.3390/molecules27175404
Chicago/Turabian StyleBilska-Markowska, Monika, Wojciech Jankowski, Marcin Hoffmann, and Marcin Kaźmierczak. 2022. "Design and Synthesis of New α-hydroxy β-fluoro/β-trifluoromethyl and Unsaturated Phosphonates from Carbohydrate-Derived Building Blocks via Pudovik and Horner–Wadsworth–Emmons Reactions" Molecules 27, no. 17: 5404. https://doi.org/10.3390/molecules27175404