
In Silico Screening of Natural Compounds for Candidates 5HT6 Receptor Antagonists against Alzheimer’s Disease


Abstract
:1. Introduction
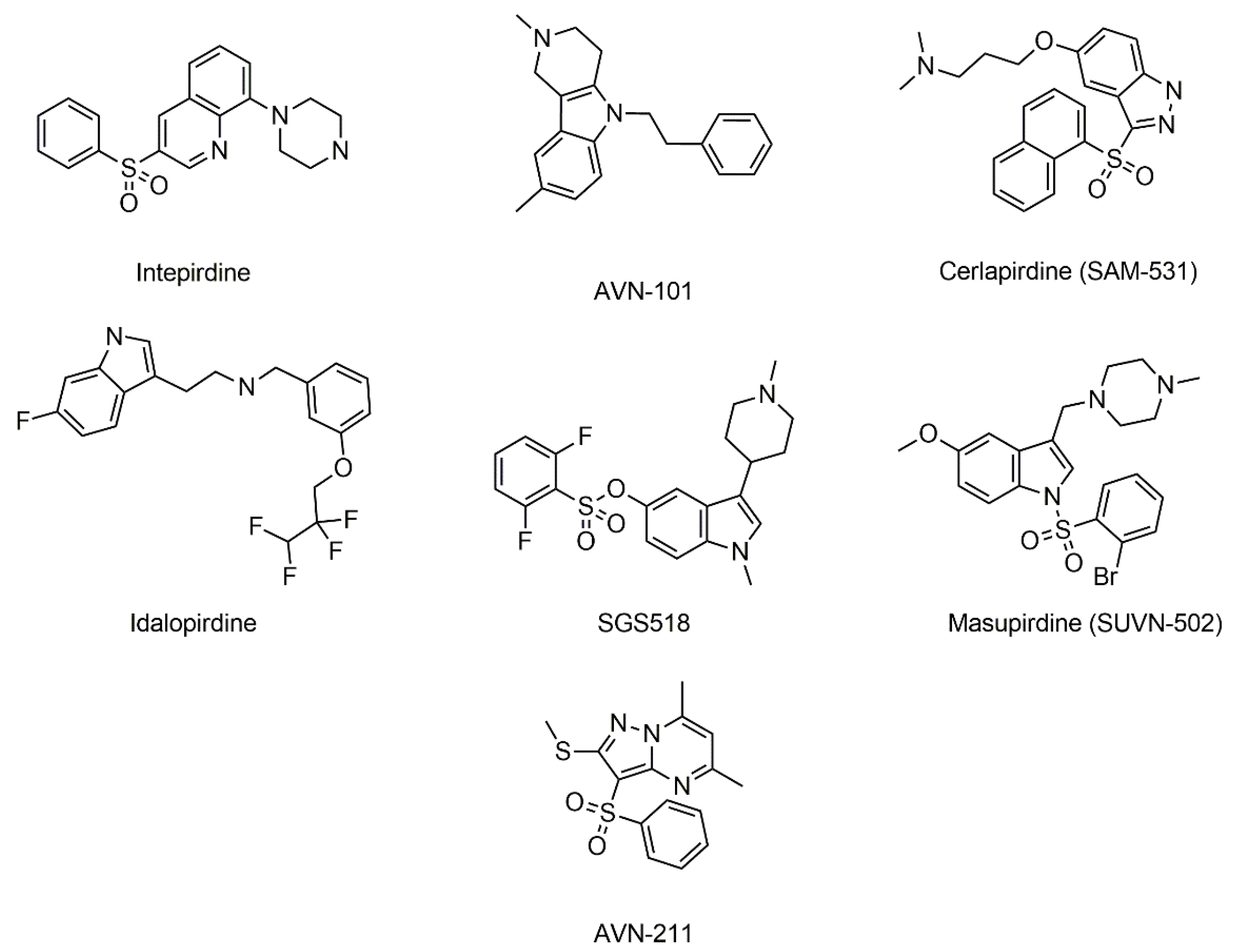
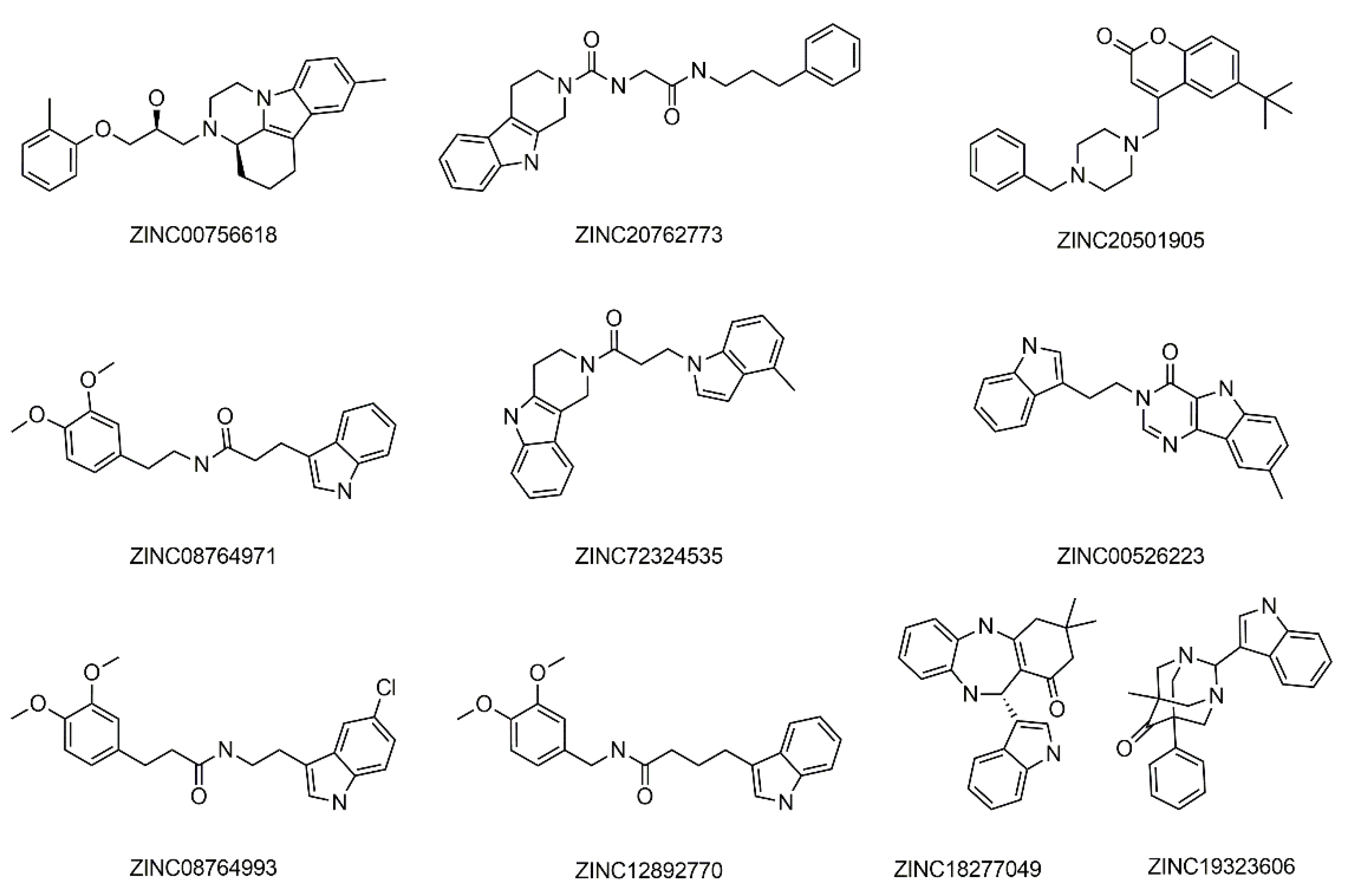
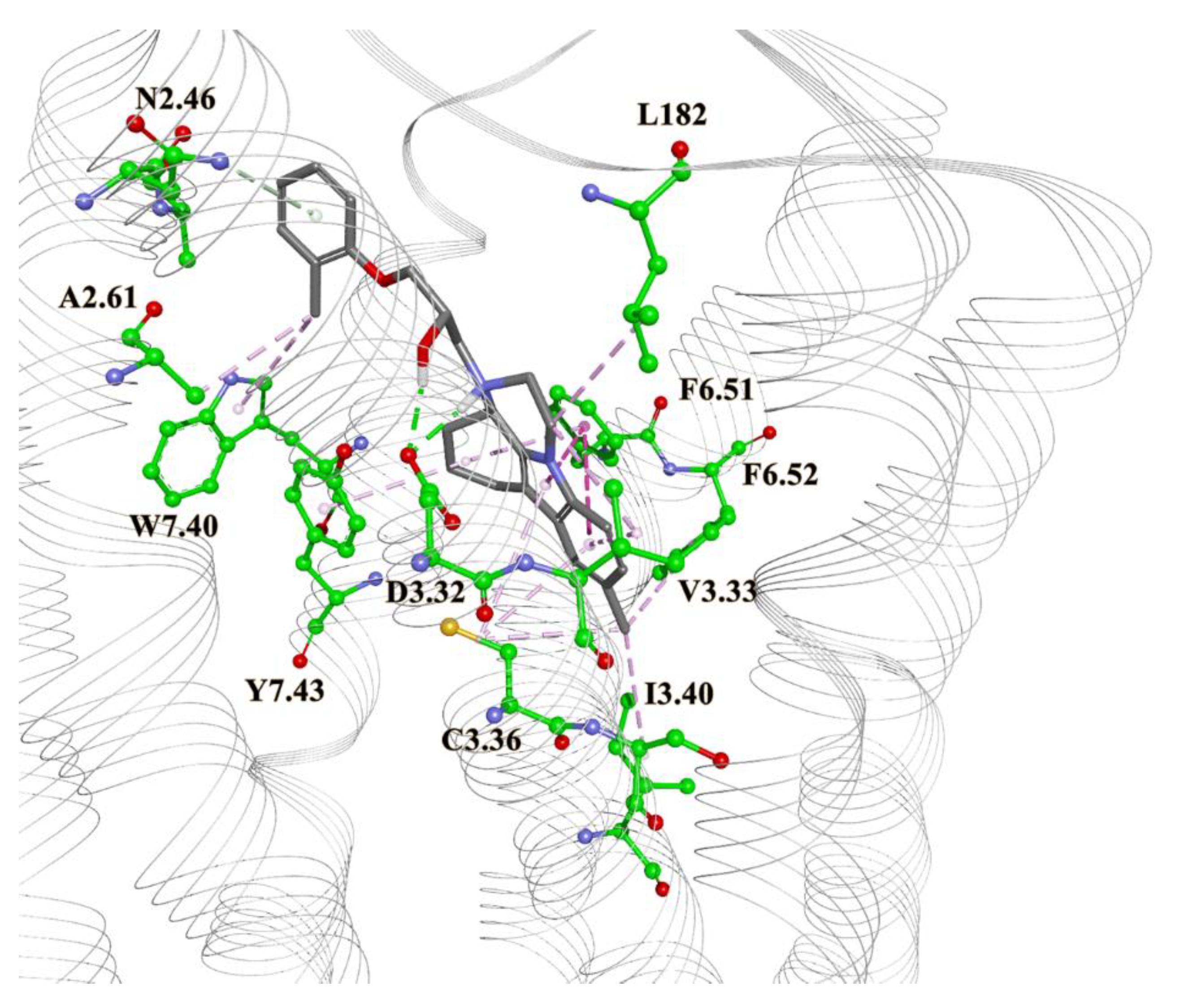
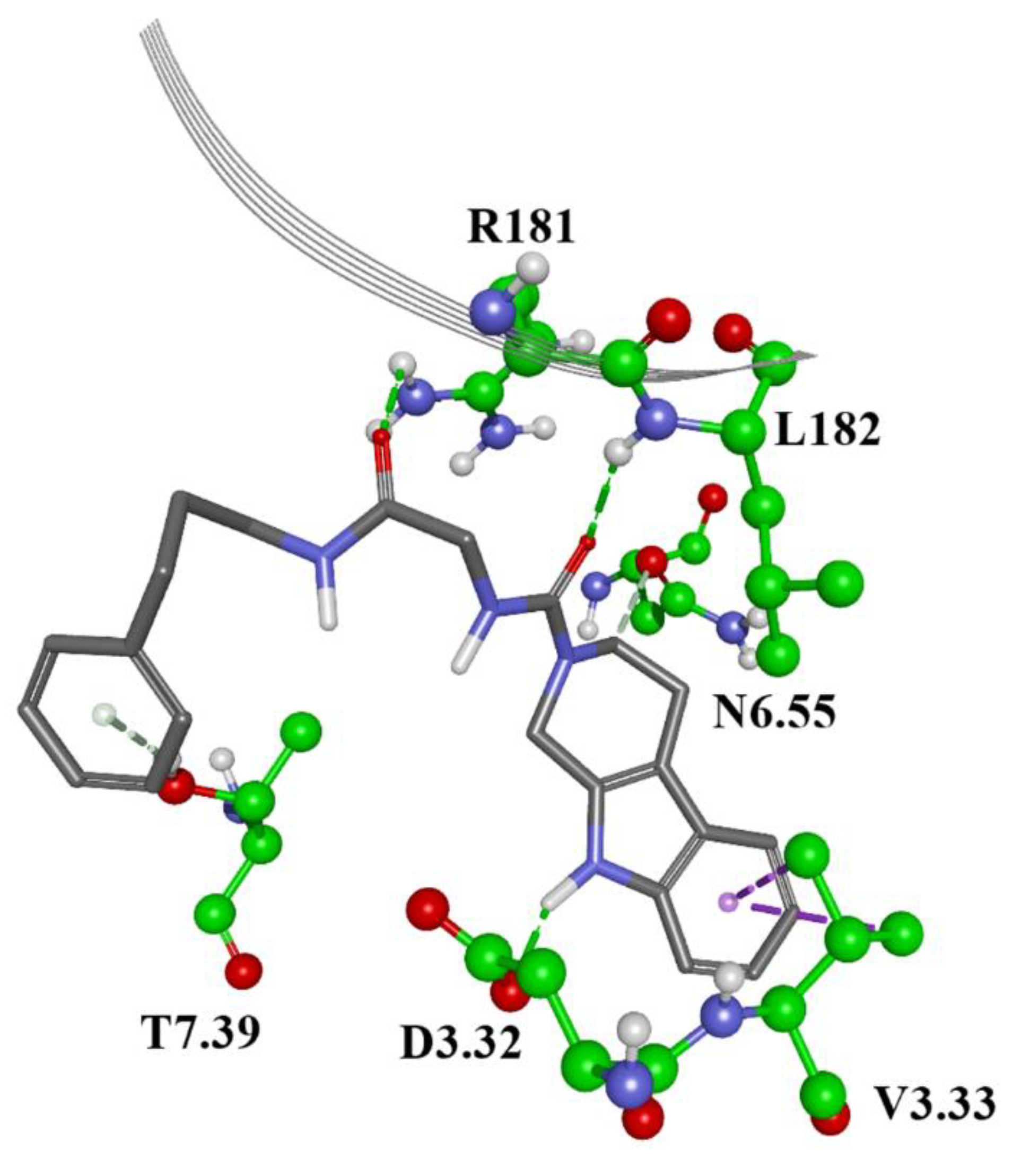
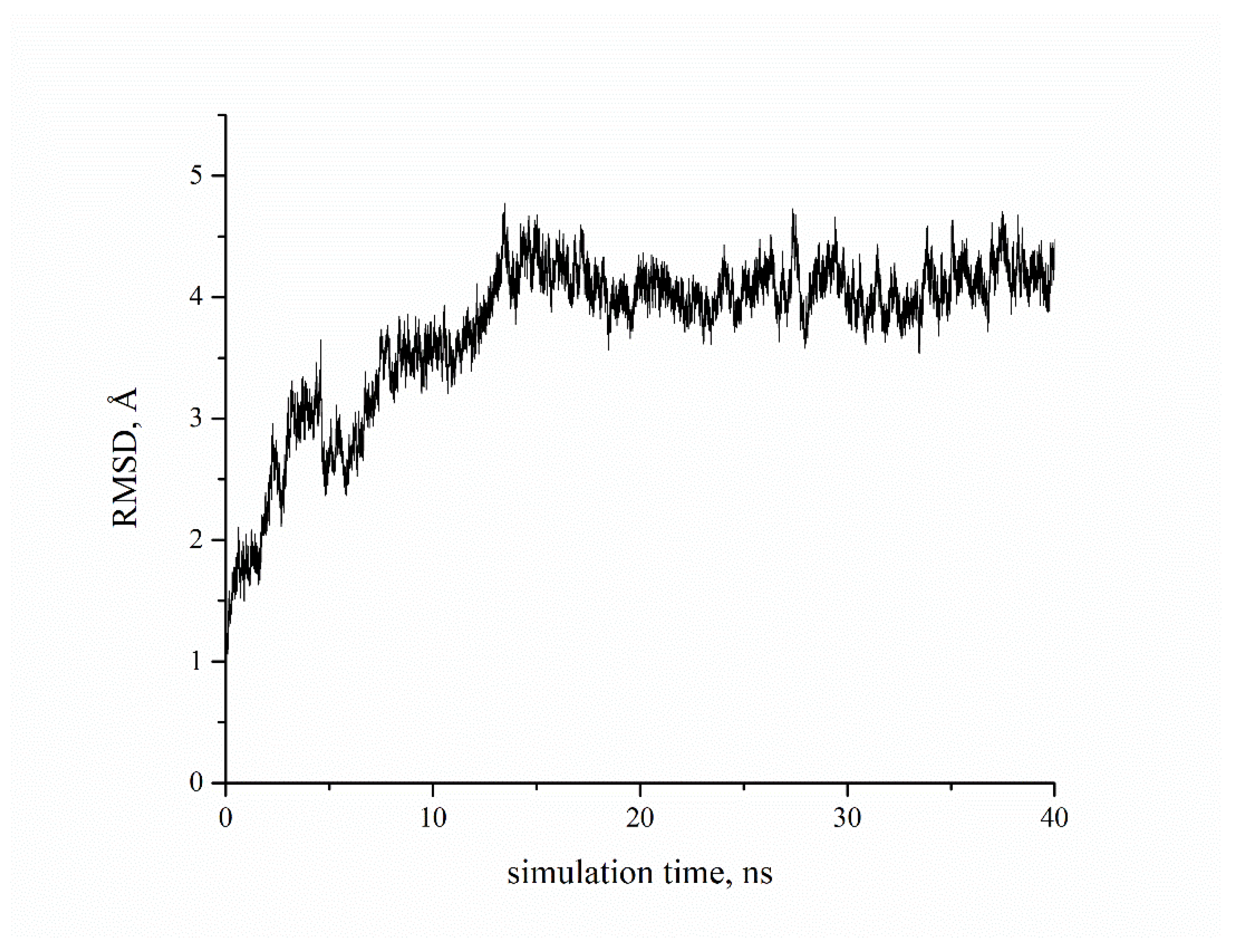
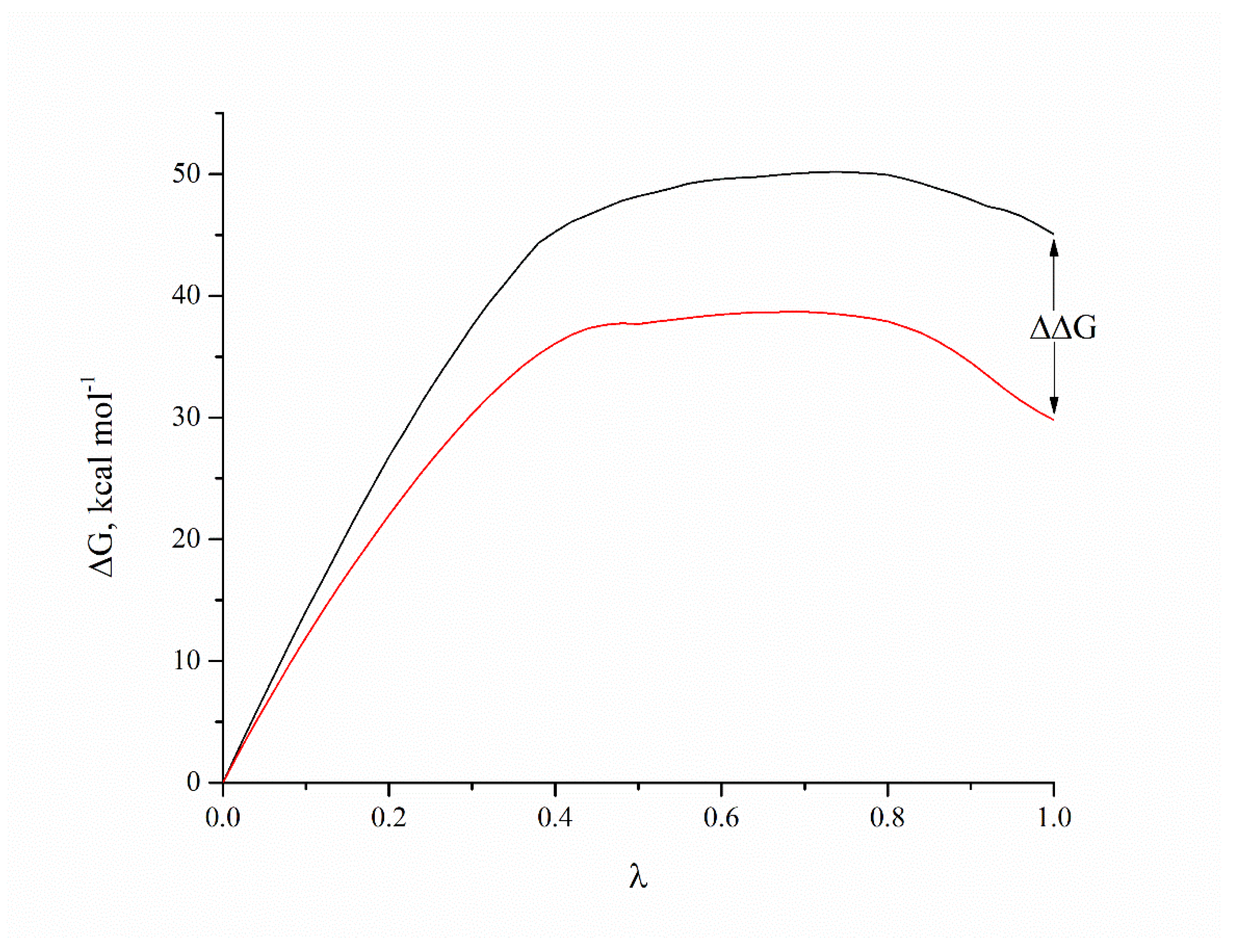
2. Results and Discussion
3. Materials and Methods
3.1. EIIP/AQVN
3.2. Zinc Ligand Database and Preparation
3.3. PCA Analysis of Virtual Screening
3.4. Receptor Preparation
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations and Binding Free Energy Calculations
3.7. ADME/Toxicity Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Andrews, M.; Tousi, B.; Sabbagh, M.N. 5HT6 Antagonists in the Treatment of Alzheimer’s Dementia: Current Progress. Neurol. Ther. 2018, 7, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero, H.; Solas, M.; Francis, P.T.; Ramirez, M.J. Serotonin 5-HT(6) Receptor Antagonists in Alzheimer’s Disease: Therapeutic Rationale and Current Development Status. CNS Drugs 2017, 31, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Kanagaratnam, L.; Dramé, M.; Trenque, T.; Oubaya, N.; Nazeyrollas, P.; Novella, J.-L.; Jolly, D.; Mahmoudi, R. Adverse drug reactions in elderly patients with cognitive disorders: A systematic review. Maturitas 2016, 85, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Haque, M.E.; Jakaria, M.; Jo, S.-H.; Kim, I.-S.; Choi, D.-K. G-Protein-Coupled Receptors in CNS: A Potential Therapeutic Target for Intervention in Neurodegenerative Disorders and Associated Cognitive Deficits. Cells 2020, 9, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudoł, S.; Cios, A.; Jastrzębska-Więsek, M.; Honkisz-Orzechowska, E.; Mordyl, B.; Wilczyńska-Zawal, N.; Satała, G.; Kucwaj-Brysz, K.; Partyka, A.; Latacz, G.; et al. The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. Int. J. Mol. Sci. 2021, 22, 10773. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, L.; Liu, J.; Zhang, H.; Chen, H.; Yang, L.; Wu, M.; Li, C.; Zhu, X.; Ding, Y.; et al. Safety, Tolerability and Pharmacokinetics of the Serotonin 5-HT6 Receptor Antagonist, HEC30654, in Healthy Chinese Subjects. Front. Pharmacol. 2021, 12, 726536. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, G.; De Deurwaerdère, P. Serotonin research: Crossing scales and boundaries. Neuropharmacology 2020, 181, 108340. [Google Scholar] [CrossRef]
- Grimaldi, B.; Bonnin, A.; Fillion, M.P.; Ruat, M.; Traiffort, E.; Fillion, G. Characterization of 5-ht6 receptor and expression of 5-ht6 mRNA in the rat brain during ontogenetic development. Naunyn Schmiedebergs Arch. Pharmacol. 1998, 357, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Grysman, N.; Gold, J.; Patel, K.; Grossberg, G.T. The role of 5 HT6-receptor antagonists in Alzheimer’s disease: An update. Expert Opin. Investig. Drugs 2018, 27, 523–533. [Google Scholar] [CrossRef]
- Ramírez, M.J. 5-HT6 receptors and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codony, X.; Vela, J.M.; Ramírez, M.J. 5-HT(6) receptor and cognition. Curr. Opin. Pharmacol. 2011, 11, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Shinde, A.; Kambhampati, R.S.; Mohammed, A.R.; Saraf, S.K.; Badange, R.K.; Bandyala, T.R.; Bhatta, V.; Bojja, K.; Reballi, V.; et al. Discovery and Development of 1-[(2-Bromophenyl)sulfonyl]-5-methoxy-3-[(4-methyl-1-piperazinyl)methyl]-1H-indole Dimesylate Monohydrate (SUVN-502): A Novel, Potent, Selective and Orally Active Serotonin 6 (5-HT(6)) Receptor Antagonist for Potential Treatment of Alzheimer’s Disease. J. Med. Chem. 2017, 60, 1843–1859. [Google Scholar] [CrossRef] [PubMed]
- Bourson, A.; Borroni, E.; Austin, R.H.; Monsma, F.J.J.; Sleight, A.J. Determination of the role of the 5-ht6 receptor in the rat brain: A study using antisense oligonucleotides. J. Pharmacol. Exp. Ther. 1995, 274, 173–180. [Google Scholar] [PubMed]
- Nikiforuk, A. The procognitive effects of 5-HT6 receptor ligands in animal models of schizophrenia. Rev. Neurosci. 2014, 25, 367–382. [Google Scholar] [CrossRef] [PubMed]
- De Jong, I.E.M.; Mørk, A. Antagonism of the 5-HT(6) receptor—Preclinical rationale for the treatment of Alzheimer’s disease. Neuropharmacology 2017, 125, 50–63. [Google Scholar] [CrossRef] [PubMed]
- West, P.J.; Marcy, V.R.; Marino, M.J.; Schaffhauser, H. Activation of the 5-HT(6) receptor attenuates long-term potentiation and facilitates GABAergic neurotransmission in rat hippocampus. Neuroscience 2009, 164, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.M.; Mo, Y.; Sabbagh, M.; Solomon, P.; Boada, M.; Jones, R.W.; Frisoni, G.B.; Grimmer, T.; Dubois, B.; Harnett, M.; et al. Intepirdine as adjunctive therapy to donepezil for mild-to-moderate Alzheimer’s disease: A randomized, placebo-controlled, phase 3 clinical trial (MINDSET). Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12136. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Baltrukevich, H.; Czarnota, K.; Handzlik, J. Chemical update on the potential for serotonin 5-HT(6) and 5-HT(7) receptor agents in the treatment of Alzheimer’s disease. Bioorganic Med. Chem. Lett. 2021, 49, 128275. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Abraham, R.; Benade, V.; Medapati, R.B.; Jayarajan, P.; Bhyrapuneni, G.; Muddana, N.; Mekala, V.R.; Subramanian, R.; Shinde, A.; et al. SUVN-502, a novel, potent, pure, and orally active 5-HT6 receptor antagonist: Pharmacological, behavioral, and neurochemical characterization. Behav. Pharmacol. 2019, 30, 16–35. [Google Scholar] [CrossRef] [PubMed]
- NIH SUVN-502 with Donepezil and Memantine for the Treatment of Moderate Alzheimer’s Disease-Phase 2a Study 2019, Identifier: NCT02580305. Available online: https://clinicaltrials.gov/ct2/show/NCT02580305 (accessed on 31 March 2022).
- Nirogi, R.; Shinde, A.; Jayarajan, P.; Goyal, V.; Bhyrapuneni, G.; Subramanian, R.; Jeta, S.; Ravula, J.; Jasti, V. Potential benefits of Masupirdine (SUVN-502) on Behavioral and Psychological symptoms in patients with moderate Alzheimer’s Disease (5090). Neurology 2020, 94, 5090. [Google Scholar]
- Suven Life Sciences Suven Life Sciences Announces Phase 3 Clinical Trial of SUVN-502 (Masupirdine), a 5-HT6 Antagonist for Treatment of Agitation and Aggression in Alzheimer’s Type Dementias. News Release 2021. Available online: http://www.suven.com/pdf/NewRelease16Aug2021.pdf (accessed on 31 March 2022).
- Ivachtchenko, A.V.; Lavrovsky, Y.; Okun, I. AVN-101: A Multi-Target Drug Candidate for the Treatment of CNS Disorders. J. Alzheimer’s Dis. 2016, 53, 583–620. [Google Scholar] [CrossRef] [Green Version]
- Hesselink, J.M.K. Idalopirdine (LY483518, SGS518, Lu AE 58054) in Alzheimer disease: Never change a winning team and do not build exclusively on surrogates. Lessons Learned from Drug Development Trials. J. Pharmacol. Clin. Res. 2016, 2, 001–004. [Google Scholar] [CrossRef]
- Ivachtchenko, A.V.; Lavrovsky, Y.; Ivanenkov, Y.A. AVN-211, Novel and Highly Selective 5-HT6 Receptor Small Molecule Antagonist, for the Treatment of Alzheimer’s Disease. Mol. Pharm. 2016, 13, 945–963. [Google Scholar] [CrossRef]
- Piñeiro-Núñez, M.M.; Bauzon, D.D.; Bymaster, F.P.; Chen, Z.; Chernet, M.P.; Clay, M.P.; Crile, R.; Delapp, N.W.; Denny, C.P.; Falcone, J.F.; et al. Discovery and SAR studies of 2,6-difluorobenzenesulfonic acid 1-methyl-3-(1-methylpiperidin-4-yl)-1H-indol-5-yl ester, a novel and potent 5-HT6 antagonist for the treatment of cognitive deficit. In Proceedings of the 229th ACS National Meeting, San Diego, CA, USA, 13–17 March 2005. [Google Scholar]
- Filla, S.A.; Flaugh, M.E.; Gillig, J.R.; Heinz, L.J.; Krushinski, J.H.J.; Liu, B.; Pineiro-Nunez, M.M.; Schaus, J.M.; Ward, J.S. Benzenesulfonic Acid Indol-5-yl Esters as Antagonists of the 5-ht6 Receptor. Patent WO2002060871A2, 8 August 2002. [Google Scholar]
- Sunshine Lake Pharma Co., Ltd. The Study of a Selective 5-ht6 Receptor Antagonist, HEC30654AcOH, in Healthy Subjects. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03655873 (accessed on 31 March 2022).
- Pfizer. Scopolamine Challenge Study. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT01213355 (accessed on 31 March 2022).
- GlaxoSmithKline. A Study to Assess the Pharmacokinetics of SB-742457 Formulated as a Capsule and a Tablet in Healthy Elderly Volunteers. 2008. Available online: https://clinicaltrials.gov/ct2/show/NCT00551772 (accessed on 31 March 2022).
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimer’s Dement. 2017, 3, 367–384. [Google Scholar] [CrossRef]
- Tewari, D.; Stankiewicz, A.M.; Mocan, A.; Sah, A.N.; Tzvetkov, N.T.; Huminiecki, L.; Horbańczuk, J.O.; Atanasov, A.G. Ethnopharmacological Approaches for Dementia Therapy and Significance of Natural Products and Herbal Drugs. Front. Aging Neurosci. 2018, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Barker, A.; Kettle, J.G.; Nowak, T.; Pease, J.E. Expanding medicinal chemistry space. Drug Discov. Today 2013, 18, 298–304. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Modern Natural Products Drug Discovery and Its Relevance to Biodiversity Conservation. J. Nat. Prod. 2011, 74, 496–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nisbet, R.M.; Polanco, J.-C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glisic, S.; Sencanski, M.; Perovic, V.; Stevanovic, S.; García-Sosa, A.T. Arginase Flavonoid Anti-Leishmanial in Silico Inhibitors Flagged against Anti-Targets. Molecules 2016, 21, 589. [Google Scholar] [CrossRef] [Green Version]
- Kelemen, A.A.; Mordalski, S.; Bojarski, A.J.; Keseru, G.M. Computational modeling of drugs for Alzheimer’s disease: Design of serotonin 5-HT 6 antagonists. In Computational Modeling of Drugs against Alzheimer’s Disease; Springer: New York, NY, USA; Humana Press: Totowa, NJ, USA, 2018; pp. 419–461. [Google Scholar]
- Da Silva, A.P.; de Angelo, R.M.; de Paula, H.; Honório, K.M.; da Silva, A.B.F. Drug design of new 5-HT6 antagonists: A QSAR study of arylsulfonamide derivatives. Struct. Chem. 2020, 31, 1585–1597. [Google Scholar] [CrossRef]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilhelm, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.-C. TCM Database@Taiwan: The World’s Largest Traditional Chinese Medicine Database for Drug Screening In Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CHembl CHEMBL3371. Available online: https://www.ebi.ac.uk/chembl/target_report_card/CHEMBL3371/ (accessed on 31 March 2022).
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, T.; Martín-Fontecha, M.; Sallander, J.; Benhamú, B.; Campillo, M.; Medina, R.A.; Pellissier, L.P.; Claeysen, S.; Dumuis, A.; Pardo, L.; et al. Benzimidazole derivatives as new serotonin 5-HT6 receptor antagonists. Molecular mechanisms of receptor inactivation. J. Med. Chem. 2010, 53, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Mierzejewski, P.; Bienkowski, P. Novel Arylsulfonamide Derivatives with 5-HT6/5-HT7 Receptor Antagonism Targeting Behavioral and Psychological Symptoms of Dementia. J. Med. Chem. 2014, 57, 4543–4557. [Google Scholar]
- Pullagurla, M.R.; Westkaemper, R.B.; Glennon, R.A. Possible differences in modes of agonist and antagonist binding at human 5-HT6 receptors. Bioorganic Med. Chem. Lett. 2004, 14, 4569–4573. [Google Scholar] [CrossRef] [PubMed]
- Gordillo-Cruz, R.E.; Rentería-Gómez, A.; Islas-Jácome, A.; Cortes-García, C.J.; Díaz-Cervantes, E.; Robles, J.; Gámez-Montaño, R. Synthesis of 3-tetrazolylmethyl-azepino[4,5-b]indol-4-ones in two reaction steps: (Ugi-azide/N-acylation/SN2)/free radical cyclization and docking studies to a 5-Ht6 model. Org. Biomol. Chem. 2013, 11, 6470–6476. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Kamińska, K.; Satała, G.; Jastrzębska-Więsek, M.; Partyka, A.; Bojarski, A.J.; Wesołowska, A.; Kieć-Kononowicz, K.; et al. The computer-aided discovery of novel family of the 5-HT6 serotonin receptor ligands among derivatives of 4-benzyl-1,3,5-triazine. Eur. J. Med. Chem. 2017, 135, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Staroń, J.; Kurczab, R.; Warszycki, D.; Satała, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2019, 185. [Google Scholar] [CrossRef]
- Dukat, M.; Mosier, P.D.; Kolanos, R.; Roth, B.L.; Glennon, R.A. Binding of serotonin and N1-benzenesulfonyltryptamine-related analogs at human 5-HT6 serotonin receptors: Receptor modeling studies. J. Med. Chem. 2008, 51, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Vera, G.; Lagos, C.F.; Almendras, S.; Hebel, D.; Flores, F.; Valle-Corvalán, G.; Pessoa-Mahana, C.D.; Mella-Raipán, J.; Montecinos, R.; Recabarren-Gajardo, G. Extended N-Arylsulfonylindoles as 5-HT6 Receptor Antagonists: Design, Synthesis & Biological Evaluation. Molecules 2016, 21, 1070. [Google Scholar]
- Harris, R.N.; Stabler, R.S.; Repke, D.B.; Kress, J.M.; Walker, K.A.; Martin, R.S.; Brothers, J.M.; Ilnicka, M.; Lee, S.W.; Mirzadegan, T. Highly potent, non-basic 5-HT6 ligands. Site mutagenesis evidence for a second binding mode at 5-HT6 for antagonism. Bioorg. Med. Chem. Lett. 2010, 20, 3436–3440. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, M.; Bucki, A.; Panek, D.; Siwek, A.; Fajkis, N.; Bednarski, M.; Zygmunt, M.; Godyń, J.; Del Rio Valdivieso, A.; Kotańska, M.; et al. Anti-Alzheimer’s multitarget-directed ligands with serotonin 5-HT6 antagonist, butyrylcholinesterase inhibitory, and antioxidant activity. Arch. Pharm. (Weinh.) 2019, 352, 1900041. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.; Bayada, D.M.; Delbressine, L.P.; Ploemen, J.P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Vera, J.A.; Medina, R.A.; Martín-Fontecha, M.; Gonzalez, A.; de la Fuente, T.; Vázquez-Villa, H.; García-Cárceles, J.; Botta, J.; McCormick, P.J.; Benhamú, B.; et al. A new serotonin 5-HT6 receptor antagonist with procognitive activity–Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, 41293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, L.; Scotti, M.T. In Silico Studies Applied to Natural Products with Potential Activity against Alzheimer’s Disease. In Computational Modeling of Drugs against Alzheimer’s Disease; Roy, K., Ed.; Springer: New York, NY, USA, 2018; pp. 513–531. ISBN 978-1-4939-7404-7. [Google Scholar]
- Veljkovic, N.; Glisic, S.; Perovic, V.; Veljkovic, V. The role of long-range intermolecular interactions in discovery of new drugs. Expert Opin. Drug Discov. 2011, 6, 1263–1270. [Google Scholar] [CrossRef]
- Veljkovic, N.; Glisic, S.; Prljic, J.; Perovic, V.; Veljkovic, V. Simple and general criterion for “in silico” screening of candidate HIV drugs. Curr. Pharm. Biotechnol. 2013, 14, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Manetti, F.; Veljkovic, N.; Perovic, V.; Vercammen, J.; Hayes, S.; Massa, S.; Witvrouw, M.; Debyser, Z.; Veljkovic, V.; et al. Novel Virtual Screening Protocol Based on the Combined Use of Molecular Modeling and Electron-Ion Interaction Potential Techniques To Design HIV-1 Integrase Inhibitors. J. Chem. Inf. Model. 2007, 47, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Goeijenbier, M.; Glisic, S.; Veljkovic, N.; Perovic, V.R.; Sencanski, M.; Branch, D.R.; Paessler, S. In silico analysis suggests repurposing of ibuprofen for prevention and treatment of EBOLA virus disease. F1000Research 2015, 4, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paessler, S.; Huang, C.; Sencanski, M.; Veljkovic, N.; Perovic, V.; Glisic, S.; Veljkovic, V. Ibuprofen as a template molecule for drug design against Ebola virus. Front. Biosci. (Landmark Ed.) 2018, 23, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojić, T.; Perović, V.R.; Senćanski, M.; Glišić, S. Identification of Candidate Allosteric Modulators of the M1 Muscarinic Acetylcholine Receptor Which May Improve Vagus Nerve Stimulation in Chronic Tinnitus. Front. Neurosci. 2017, 11, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojić, T.; Perović, V.R.; Glišić, S. In silico Therapeutics for Neurogenic Hypertension and Vasovagal Syncope. Front. Neurosci. 2016, 9, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Chen, W.; Zhou, J.; Dai, J.; Li, Y.; Zhao, Y. NPBS database: A chemical data resource with relational data between natural products and biological sources. Database 2020, 2020, baaa102. [Google Scholar] [CrossRef] [PubMed]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK Family Databases: Integrated Metabolite–Plant Species Databases for Multifaceted Plant Research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Durán, Á.; Zamora, I.; Pastor, M. Suitability of GRIND-Based Principal Properties for the Description of Molecular Similarity and Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2009, 49, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Pastor, M.; Cruciani, G.; McLay, I.; Pickett, S.; Clementi, S. GRid-INdependent Descriptors (GRIND): A Novel Class of Alignment-Independent Three-Dimensional Molecular Descriptors. J. Med. Chem. 2000, 43, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.; Martínez, G.C.; Pastor, M. Development and validation of AMANDA, a new algorithm for selecting highly relevant regions in Molecular Interaction Fields. J. Chem. Inf. Model. 2008, 48, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Receptor Molecular Biology; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. ISBN 1043-9471. [Google Scholar]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D.J. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Maia, J.D.C.; Radak, B.K.; Hardy, D.J.; Cai, W.; Chipot, C.; Tajkhorshid, E. Boosting Free-Energy Perturbation Calculations with GPU-Accelerated NAMD. J. Chem. Inf. Model. 2020, 60, 5301–5307. [Google Scholar] [CrossRef]
- Schrödinger LLC. QikProp. 2021. Available online: https://www.schrodinger.com/products/qikprop (accessed on 31 March 2022).
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- ChemAxon Ltd. MarvinSketch. 2022. Available online: https://chemaxon.com/products/marvin (accessed on 31 March 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Name | Gold Score | Autodock Vina Binding Energy (kcal/mol) | AQVN | EIIP |
|---|---|---|---|---|---|
| 1 | Intepirdine | 42.36 | −5.94 | 2.909 | 0.009 |
| 2 | Cerlapirdine (SAM-531) | 36.63 | −8.13 | 2.88 | 0.000 |
| 3 | Idalopirdine | 34.59 | −9.193 | 2.55 | 0.089 |
| 4 | SGS518 | 33.92 | −9.366 | 2.78 | 0.035 |
| 5 | Masupirdine (SUVN-502) | 35.55 | −10.52 | 2.79 | 0.032 |
| 6 | AVN-211 | 32.30 | −5.335 | 3.08 | 0.073 |
| No | Name | Gold Score | Autodock Vina Binding Energy (kcal/mol) | AQVN | EIIP |
|---|---|---|---|---|---|
| 1 | ZINC00756618 | 43.17 | −13.13 | 2.55 | 0.090 |
| 2 | ZINC20762773 | 39.05 | −11.57 | 2.55 | 0.090 |
| 3 | ZINC20501905 | 35.01 | −11.47 | 2.55 | 0.090 |
| 4 | ZINC08764971 | 34.65 | −9.11 | 2.72 | 0.055 |
| 5 | ZINC72324535 | 34.48 | −10.7 | 2.72 | 0.055 |
| 6 | ZINC00526223 | 34.38 | −9.135 | 2.99 | 0.009 |
| 7 | ZINC08764993 | 33.48 | −9.371 | 2.72 | 0.055 |
| 8 | ZINC12892770 | 32.94 | −8.995 | 2.72 | 0.055 |
| 9 | ZINC18277049 | 32.65 | −9.072 | 2.72 | 0.055 |
| 10 | ZINC19323606 | 32.08 | −10.04 | 2.72 | 0.055 |
| Compound | MW | RB | DM | MV | DHB | AHB | PSA | logP | logS | PCaco | PM | %HOA | VRF | VRT | QPlogBB | CNS MPO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ZINC00756618 | 390.5 | 6 | 3.2 | 1304.9 | 1 | 4.45 | 31.5 | 5.24 | −5.68 | 1416 | 8 | 100 | 1 | 1 | 0.33 | 3.22 |
| ZINC20762773 | 390.5 | 6 | 8.1 | 1287.3 | 2.25 | 3.75 | 86.8 | 3.98 | −4.91 | 348 | 4 | 95 | 0 | 0 | −0.92 | 4.81 |
| ZINC20501905 | 390.5 | 5 | 6.7 | 1317.7 | 0 | 6.5 | 46.9 | 3.48 | −3.34 | 125 | 4 | 85 | 0 | 0 | 0.29 | 3.22 |
| ZINC08764971 | 352.4 | 8 | 5.9 | 1133.5 | 2 | 4 | 65.4 | 3.74 | −3.92 | 1475 | 5 | 100 | 0 | 0 | −0.55 | 4.73 |
| ZINC72324535 | 357.5 | 3 | 6.3 | 1182.2 | 1 | 3 | 45.3 | 4.82 | −5.97 | 1440 | 4 | 100 | 0 | 1 | −0.28 | 4.53 |
| ZINC00526223 | 342.4 | 3 | 2.6 | 1102.3 | 2 | 4 | 69.6 | 3.99 | −5.73 | 839 | 3 | 100 | 0 | 1 | −0.75 | 4.41 |
| ZINC08764993 | 386.8 | 8 | 8.7 | 1202.5 | 2 | 4 | 63.3 | 4.46 | −5.39 | 1724 | 5 | 100 | 0 | 0 | −0.39 | 3.94 |
| ZINC12892770 | 352.4 | 8 | 5.1 | 1179.7 | 2 | 4 | 68.2 | 4.03 | −4.73 | 1159 | 5 | 100 | 0 | 0 | −0.69 | 4.58 |
| ZINC18277049 | 357.4 | 0 | 6.8 | 1087.1 | 2 | 2.5 | 62.2 | 4.61 | −5.69 | 1541 | 8 | 100 | 0 | 1 | −0.16 | 4.08 |
| ZINC19323606 | 357.4 | 0 | 4.7 | 1082.3 | 1 | 6 | 43.7 | 2.55 | −2.48 | 175 | 2 | 82 | 0 | 0 | 0.84 | 4.14 |
| No | Name | QPlogBB | CNS MPO |
|---|---|---|---|
| 1 | Intepirdine | −0.25 | 5.36 |
| 2 | Cerlapirdine (SAM-531) | −0.74 | 4.89 |
| 3 | Idalopirdine | 0.53 | 3.07 |
| 4 | SGS518 | 0.13 | 3.41 |
| 5 | Masupirdine (SUVN-502) | 0.46 | 4.52 |
| 6 | AVN-211 | −0.29 | 5.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojić, T.; Sencanski, M.; Perovic, V.; Milicevic, J.; Glisic, S. In Silico Screening of Natural Compounds for Candidates 5HT6 Receptor Antagonists against Alzheimer’s Disease. Molecules 2022, 27, 2626. https://doi.org/10.3390/molecules27092626
Bojić T, Sencanski M, Perovic V, Milicevic J, Glisic S. In Silico Screening of Natural Compounds for Candidates 5HT6 Receptor Antagonists against Alzheimer’s Disease. Molecules. 2022; 27(9):2626. https://doi.org/10.3390/molecules27092626
Chicago/Turabian StyleBojić, Tijana, Milan Sencanski, Vladimir Perovic, Jelena Milicevic, and Sanja Glisic. 2022. "In Silico Screening of Natural Compounds for Candidates 5HT6 Receptor Antagonists against Alzheimer’s Disease" Molecules 27, no. 9: 2626. https://doi.org/10.3390/molecules27092626