
Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates

 ,
,  , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Epigenetic Regulation and Cancer
2.1. Cancer and DNA Methylation
2.2. Oncohistones and Histone Changes
2.2.1. Histone Methylation and Cancer
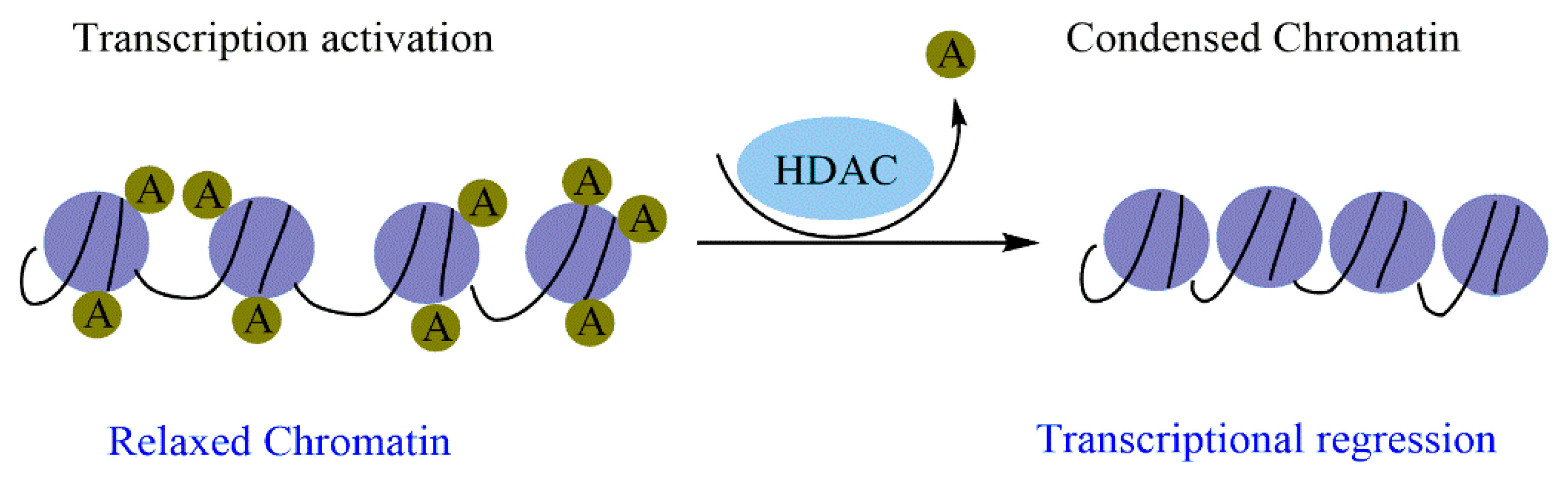
2.2.2. Histone Acetylation/Deacetylation and Cancer
2.2.3. Phosphorylation, Ubiquitination, SUMOylation, and Cancer
2.2.4. Epigenetic Regulation by miRNAs and Cancer
3. The Role of HDAC in Cancer
3.1. HDAC in Different Cancer Stages
3.1.1. Cell Cycle Progression and Apoptosis
3.1.2. Differentiation
3.1.3. DNA Damage Response
3.1.4. Metastasis
3.1.5. Angiogenesis
3.1.6. Autophagy
4. Natural Bioactive Compounds Targeting HDAC in Human Cancers
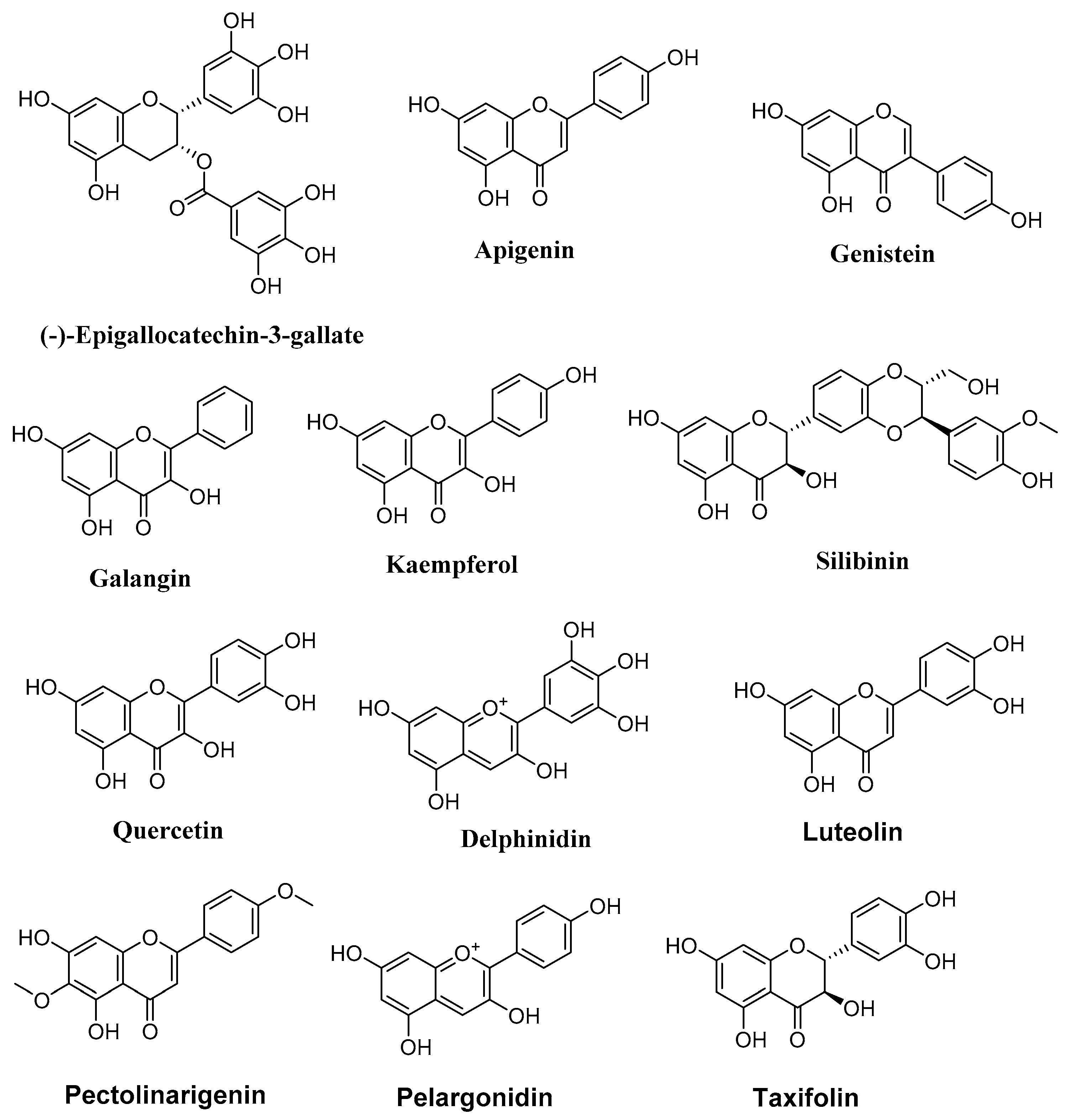
4.1. Flavonoids
4.1.1. Epigallocatechin Gallate
4.1.2. Apigenin
4.1.3. Galangin
4.1.4. Genistein
4.1.5. Silibinin and Kaempferol
4.1.6. Quercetin
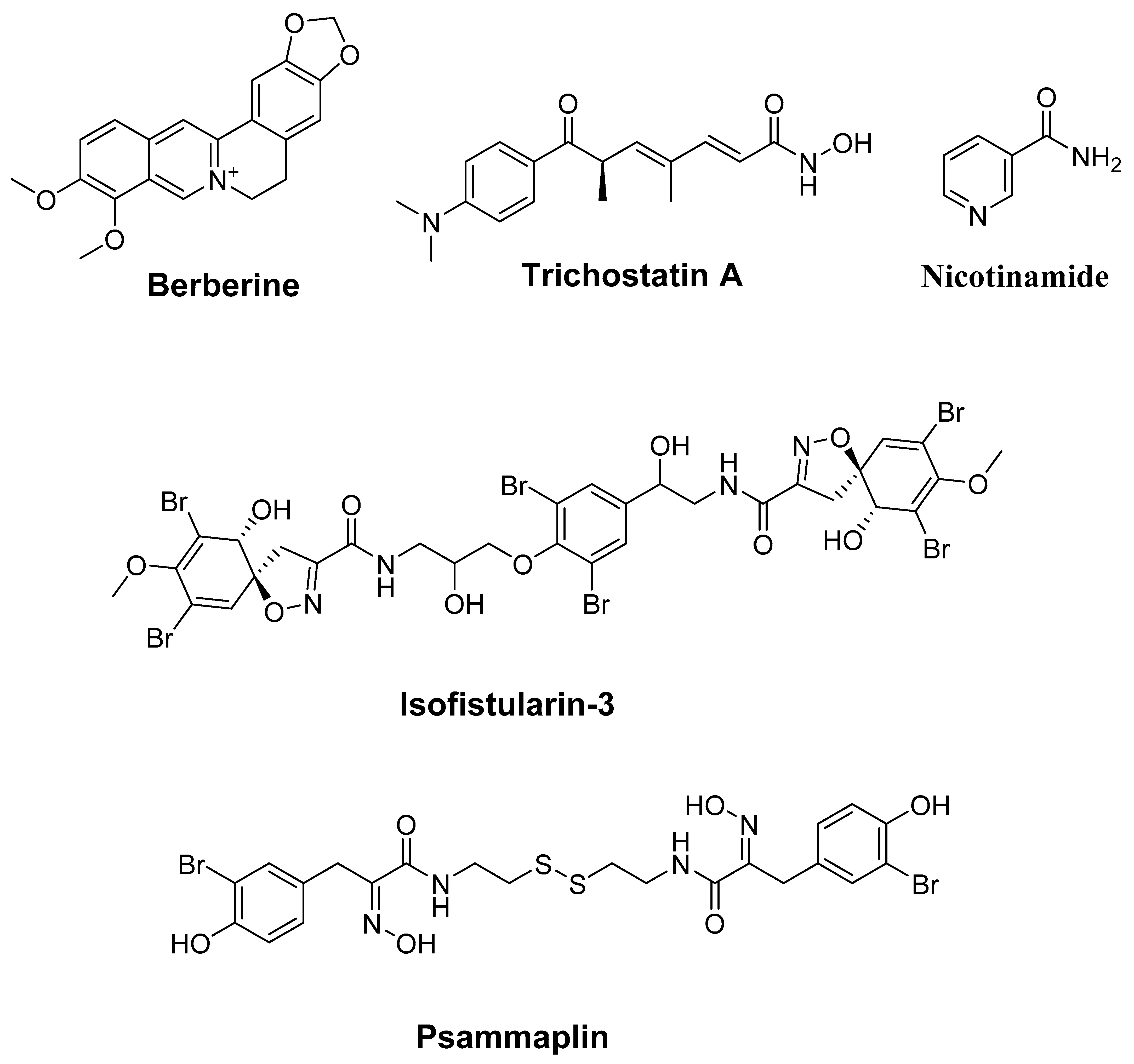
4.2. Alkaloids
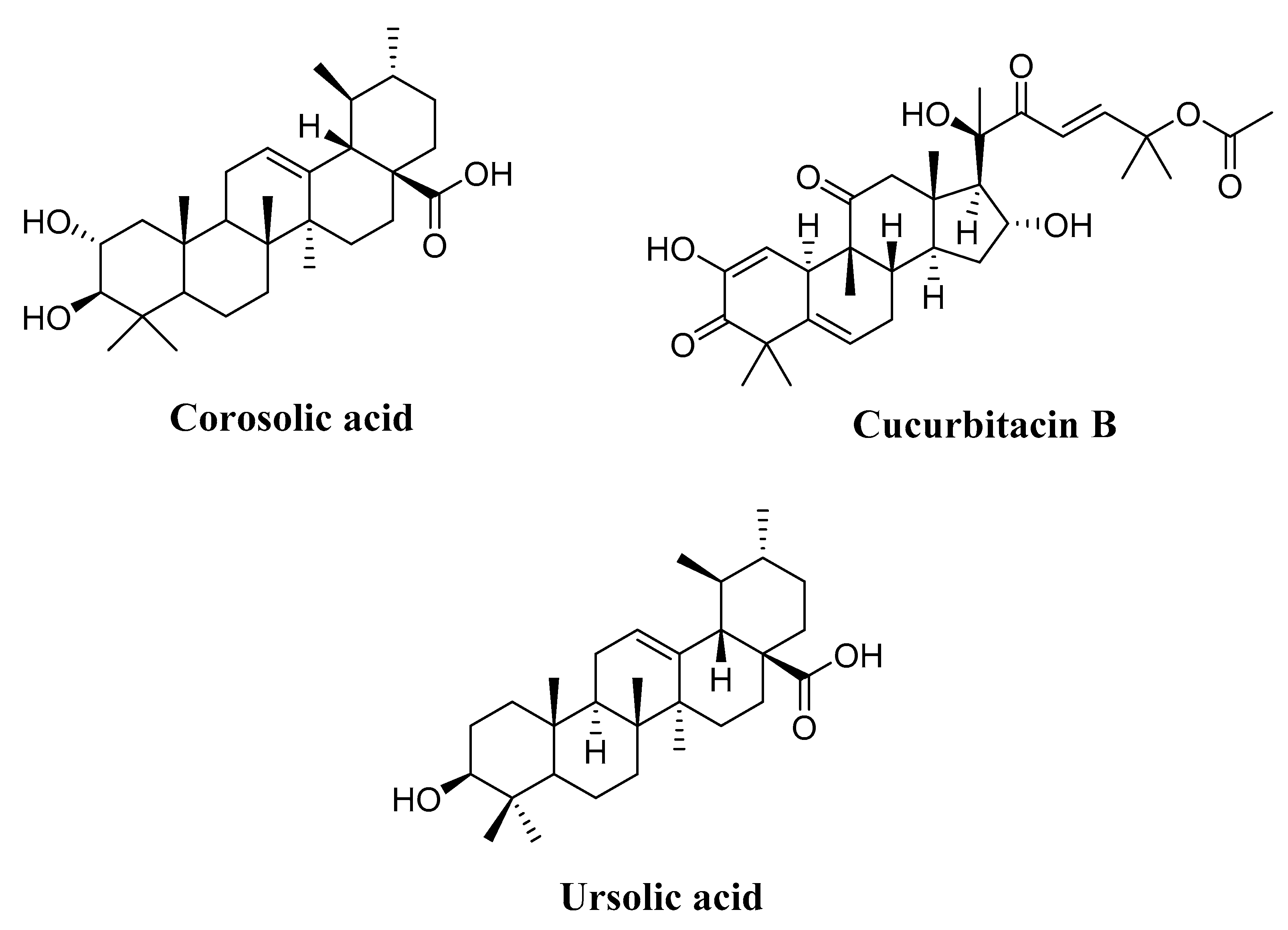
4.3. Terpenoids
4.3.1. Corosolic Acid
4.3.2. Cucurbitacin B
4.3.3. Ursolic Acid
4.4. Fatty Acids
4.4.1. Butyrate
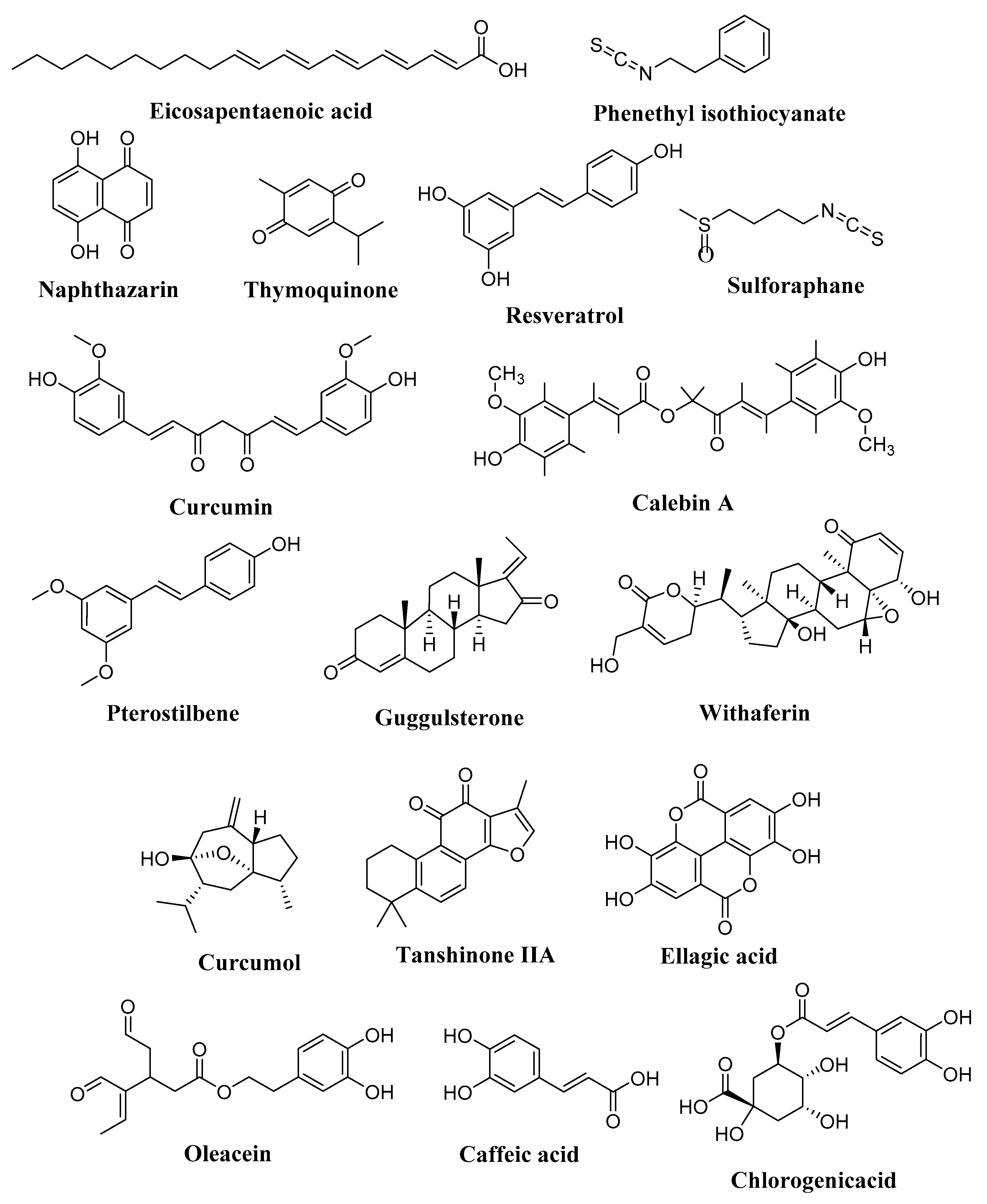
4.4.2. Butyric Acid and Eicosapentaenoic Acid
4.5. Isothiocyanate
4.5.1. Phenethyl Isothiocyanate (PEITC)
4.5.2. Sulforaphane (SFN)
4.6. Quinones
4.6.1. Naphthazarin
4.6.2. Thymoquinone
4.7. Stilbenes
4.7.1. Resveratrol
4.7.2. Curcumin
4.7.3. Calebin-A
4.7.4. Pterostilbene
4.8. Steroids
4.8.1. Guggulsterone
4.8.2. Withaferin A (WFA)
4.9. Phenolic Acids, Secoiridoids, Tannins, and Tanshinones
4.9.1. Phenolic Acids: Caffeic Acid Chlorogenic Acid
4.9.2. Secoiridoids: Oleacein
4.9.3. Secoiridoids: Ellagic Acid
4.9.4. Tanshinones: Tanshinone IIA
4.10. Other Molecules Targeting HDAC in Human Cancers
4.10.1. Arsenic Trioxide
4.10.2. Curcumol
4.10.3. Selenium
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; El Menyiy, N.; Oumeslakht, L.; El Allam, A.; Balahbib, A.; Rauf, A.; Muhammad, N.; Kuznetsova, E.; Derkho, M.; Thiruvengadam, M.; et al. Preclinical and Clinical Antioxidant Effects of Natural Compounds against Oxidative Stress-Induced Epigenetic Instability in Tumor Cells. Antioxidants 2021, 10, 1553. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Huang, S. Cancer Epigenetics: Mechanisms and Crosstalk of a HDAC Inhibitor, Vorinostat. Chemotherapy 2013, 2, 14934. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M. Cancer Biology: Mechanism of Antitumour Action of Vorinostat (Suberoylanilide Hydroxamic Acid), a Novel Histone Deacetylase Inhibitor. Br. J. Cancer 2006, 95, S2–S6. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma after Prior Systemic Therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Patra, S.; Praharaj, P.P.; Klionsky, D.J.; Bhutia, S.K. Vorinostat in Autophagic Cell Death: A Critical Insight into Autophagy-Mediated, -Associated and -Dependent Cell Death for Cancer Prevention. Drug Discov. Today 2022, 27, 269–279. [Google Scholar] [CrossRef]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Oki, Y.; Buglio, D.; Fanale, M.; Fayad, L.; Copeland, A.; Romaguera, J.; Kwak, L.W.; Pro, B.; de Castro Faria, S.; Neelapu, S.; et al. Phase I Study of Panobinostat plus Everolimus in Patients with Relapsed or Refractory Lymphoma. Clin. Cancer Res. 2013, 19, 6882–6890. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent Progress on HDAC Inhibitors with Dual Targeting Capabilities for Cancer Treatment. Eur. J. Med. Chem. 2020, 208, 112831. [Google Scholar] [CrossRef]
- Bouyahya, A.; El Omari, N.; Hakkur, M.; El Hachlafi, N.; Charfi, S.; Balahbib, A.; Guaouguaou, F.-E.; Rebezov, M.; Maksimiuk, N.; Shariati, M.A.; et al. Sources, Health Benefits, and Biological Properties of Zeaxanthin. Trends Food Sci. Technol. 2021, 118, 519–538. [Google Scholar] [CrossRef]
- Balahbib, A.; El Omari, N.; Hachlafi, N.E.; Lakhdar, F.; El Menyiy, N.; Salhi, N.; Mrabti, H.N.; Bakrim, S.; Zengin, G.; Bouyahya, A. Health Beneficial and Pharmacological Properties of P-Cymene. Food Chem. Toxicol. 2021, 153, 112259. [Google Scholar] [CrossRef] [PubMed]
- El Hachlafi, N.; Lakhdar, F.; Khouchlaa, A.; Bakrim, S.; El Omari, N.; Balahbib, A.; Shariati, M.A.; Zengin, G.; Fikri-Benbrahim, K.; Orlando, G.; et al. Health Benefits and Pharmacological Properties of Hinokitiol. Processes 2021, 9, 1680. [Google Scholar] [CrossRef]
- El Omari, N.; El Menyiy, N.; Zengin, G.; Goh, B.H.; Gallo, M.; Montesano, D.; Naviglio, D.; Bouyahya, A. Anticancer and Anti-Inflammatory Effects of Tomentosin: Cellular and Molecular Mechanisms. Separations 2021, 8, 207. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Bakha, M.; Lorenzo, J.M.; Rebezov, M.; Shariati, M.A.; Aboulaghras, S.; Balahbib, A.; Khayrullin, M.; Bouyahya, A. Natural Bioactive Compounds Targeting Epigenetic Pathways in Cancer: A Review on Alkaloids, Terpenoids, Quinones, and Isothiocyanates. Nutrients 2021, 13, 3714. [Google Scholar] [CrossRef]
- El Omari, N.; Bakha, M.; Imtara, H.; Guaouguaoua, F.-E.; Balahbib, A.; Zengin, G.; Bouyahya, A. Anticancer Mechanisms of Phytochemical Compounds: Focusing on Epigenetic Targets. Environ. Sci. Pollut. Res. 2021, 28, 47869–47903. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Sandoval, J.; Esteller, M. Cancer Epigenomics: Beyond Genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, S. Epigenetics and Cancer. J. Appl. Physiol. 2010, 109, 598–605. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Ashworth, A. Marked for Death: Targeting Epigenetic Changes in Cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. DNA Methylation and Epigenetic Inheritance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 326, 329–338. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Nielsen, H.M.; Hansen, L.L. Epigenetics and Cancer Treatment. Eur. J. Pharmacol. 2009, 625, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Karimzadeh, M.R.; Pourdavoud, P.; Ehtesham, N.; Qadbeigi, M.; Asl, M.M.; Alani, B.; Mosallaei, M.; Pakzad, B. Regulation of DNA Methylation Machinery by Epi-MiRNAs in Human Cancer: Emerging New Targets in Cancer Therapy. Cancer Gene Ther. 2021, 28, 157–174. [Google Scholar] [CrossRef]
- Ross, J.P.; Rand, K.N.; Molloy, P.L. Hypomethylation of Repeated DNA Sequences in Cancer. Epigenomics 2010, 2, 245–269. [Google Scholar] [CrossRef]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A Gene Hypermethylation Profile of Human Cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar]
- He, J.; Zhou, M.; Li, X.; Gu, S.; Cao, Y.; Xing, T.; Chen, W.; Chu, C.; Gu, F.; Zhou, J. SLC34A2 Simultaneously Promotes Papillary Thyroid Carcinoma Growth and Invasion through Distinct Mechanisms. Oncogene 2020, 39, 2658–2675. [Google Scholar] [CrossRef]
- Sastry, N.G.; Wan, X.; Huang, T.; Alvarez, A.A.; Pangeni, R.P.; Song, X.; James, C.D.; Horbinski, C.M.; Brennan, C.W.; Nakano, I. LY6K Promotes Glioblastoma Tumorigenicity via CAV-1–Mediated ERK1/2 Signaling Enhancement. Neuro-Oncol. 2020, 22, 1315–1326. [Google Scholar] [CrossRef]
- Xiao, C.; Wu, G.; Zhou, Z.; Zhang, X.; Wang, Y.; Song, G.; Ding, E.; Sun, X.; Zhong, L.; Li, S. RBBP6, a RING Finger-Domain E3 Ubiquitin Ligase, Induces Epithelial–Mesenchymal Transition and Promotes Metastasis of Colorectal Cancer. Cell Death Dis. 2019, 10, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eads, C.A.; Lord, R.V.; Kurumboor, S.K.; Wickramasinghe, K.; Skinner, M.L.; Long, T.I.; Peters, J.H.; DeMeester, T.R.; Danenberg, K.D.; Danenberg, P.V. Fields of Aberrant CpG Island Hypermethylation in Barrett’s Esophagus and Associated Adenocarcinoma. Cancer Res. 2000, 60, 5021–5026. [Google Scholar] [PubMed]
- Issa, J.-P.J.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated Age-Related CpG Island Methylation in Ulcerative Colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Greger, V.; Passarge, E.; Höpping, W.; Messmer, E.; Horsthemke, B. Epigenetic Changes May Contribute to the Formation and Spontaneous Regression of Retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL Tumor-Suppressor Gene by DNA Methylation in Renal Carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Zulueta, M.; Bender, C.M.; Yang, A.S.; Nguyen, T.; Beart, R.W.; Van Tornout, J.M.; Jones, P.A. Methylation of the 5′ CpG Island of the P16/CDKN2 Tumor Suppressor Gene in Normal and Transformed Human Tissues Correlates with Gene Silencing. Cancer Res. 1995, 55, 4531–4535. [Google Scholar]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the HMLH1 Promoter Correlates with Lack of Expression of HMLH1 in Sporadic Colon Tumors and Mismatch Repair-Defective Human Tumor Cell Lines. Cancer Res. 1997, 57, 808–811. [Google Scholar]
- Richter, A.M.; Woods, M.L.; Küster, M.M.; Walesch, S.K.; Braun, T.; Boettger, T.; Dammann, R.H. RASSF10 Is Frequently Epigenetically Inactivated in Kidney Cancer and Its Knockout Promotes Neoplasia in Cancer Prone Mice. Oncogene 2020, 39, 3114–3127. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Feng, J.; Wang, W.; Deng, Z.; Zhang, Y.; Xiao, L.; Wang, Z.; Liu, C.; Liu, Q.; Chen, S. The EGFR-ZNF263 Signaling Axis Silences SIX3 in Glioblastoma Epigenetically. Oncogene 2020, 39, 3163–3178. [Google Scholar] [CrossRef]
- Marini, A.; Mirmohammadsadegh, A.; Nambiar, S.; Gustrau, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Inactivation of Tumor Suppressor Genes in Serum of Patients with Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 422–431. [Google Scholar] [CrossRef]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, S.; Buckles, E.; Estrada, J.; Koochekpour, S. Aberrant DNA Methylation and Prostate Cancer. Curr. Genom. 2011, 12, 486–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, A.; Bell, D.; Weber, R.S.; El-Naggar, A.K. CpG Island Methylation Profiling in Human Salivary Gland Adenoid Cystic Carcinoma. Cancer 2011, 117, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, F.; Yang, L.; Guo, C.; Wan, R.; Ke, A.; Xu, L.; Hu, G.; Xu, X.; Shen, J. Expression of DNMT1 and DNMT3a Are Regulated by GLI1 in Human Pancreatic Cancer. PLoS ONE 2011, 6, e27684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Ohm, J.E. Epigenetic Gene Silencing in Cancer–a Mechanism for Early Oncogenic Pathway Addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef]
- Markl, I.D.; Cheng, J.; Liang, G.; Shibata, D.; Laird, P.W.; Jones, P.A. Global and Gene-Specific Epigenetic Patterns in Human Bladder Cancer Genomes Are Relatively Stable in Vivo and in Vitro over Time. Cancer Res. 2001, 61, 5875–5884. [Google Scholar]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The Expanding Landscape of ‘Oncohistone’Mutations in Human Cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L. Mutational Landscape of Uterine and Ovarian Carcinosarcomas Implicates Histone Genes in Epithelial–Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M. Driver Mutations in Histone H3. 3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.S. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar]
- Koelsche, C.; Schrimpf, D.; Tharun, L.; Roth, E.; Sturm, D.; Jones, D.T.; Renker, E.-K.; Sill, M.; Baude, A.; Sahm, F. Histone 3.3 Hotspot Mutations in Conventional Osteosarcomas: A Comprehensive Clinical and Molecular Characterization of Six H3F3A Mutated Cases. Clin. Sarcoma Res. 2017, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsik, P.; Baude, A.; Mancarella, D.; Öz, S.; Kühn, A.; Toth, R.; Hey, J.; Toprak, U.H.; Lim, J.; Nguyen, V.H. Globally Altered Epigenetic Landscape and Delayed Osteogenic Differentiation in H3. 3-G34W-Mutant Giant Cell Tumor of Bone. Nat. Commun. 2020, 11, 5414. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting Epigenetic Modifications in Cancer Therapy: Erasing the Roadmap to Cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of Acetylation at Lys16 and Trimethylation at Lys20 of Histone H4 Is a Common Hallmark of Human Cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prathipati, P.; Pratiwi, R.; Phanus-Umporn, C.; Malik, A.A.; Schaduangrat, N.; Seenprachawong, K.; Wongchitrat, P.; Supokawej, A.; Prachayasittikul, V. Exploring the Epigenetic Drug Discovery Landscape. Expert Opin. Drug Discov. 2017, 12, 345–362. [Google Scholar] [CrossRef]
- Kubicek, S.; Gilbert, J.C.; Fomina-Yadlin, D.; Gitlin, A.D.; Yuan, Y.; Wagner, F.F.; Holson, E.B.; Luo, T.; Lewis, T.A.; Taylor, B. Chromatin-Targeting Small Molecules Cause Class-Specific Transcriptional Changes in Pancreatic Endocrine Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 5364–5369. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone Deacetylases in Acute Myeloid Leukaemia Show a Distinctive Pattern of Expression That Changes Selectively in Response to Deacetylase Inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef]
- Fang, D.; Gan, H.; Cheng, L.; Lee, J.-H.; Zhou, H.; Sarkaria, J.N.; Daniels, D.J.; Zhang, Z. H3. 3K27M Mutant Proteins Reprogram Epigenome by Sequestering the PRC2 Complex to Poised Enhancers. Elife 2018, 7, e36696. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C. Histone H3. 3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in Cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Gan, H.; Lee, J.-H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J. The Histone H3. 3K36M Mutation Reprograms the Epigenome of Chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R. Histone H3K36 Mutations Promote Sarcomagenesis through Altered Histone Methylation Landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhoumaud, P.; Badri, S.; Rodriguez-Hernaez, J.; Sakellaropoulos, T.; Sethia, G.; Kloetgen, A.; Cornwell, M.; Bhattacharyya, S.; Ay, F.; Bonneau, R. NSD2 Overexpression Drives Clustered Chromatin and Transcriptional Changes in a Subset of Insulated Domains. Nat. Commun. 2019, 10, 4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-Affinity, Small-Molecule Peptidomimetic Inhibitors of MLL1/WDR5 Protein–Protein Interaction. J. Am. Chem. Soc. 2013, 135, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xhemalce, B.; Dawson, M.A.; Bannister, A.J. Histone Modifications. In Reviews in Cell Biology and Molecular Medicine; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Marmorstein, R. Structure of Histone Acetyltransferases. J. Mol. Biol. 2001, 311, 433–444. [Google Scholar] [CrossRef]
- Calcagno, D.Q.; Wisnieski, F.; Mota, E.R.; Maia de Sousa, S.B.; Costa da Silva, J.M.; Leal, M.F.; Gigek, C.O.; Santos, L.C.; Rasmussen, L.T.; Assumpção, P.P. Role of Histone Acetylation in Gastric Cancer: Implications of Dietetic Compounds and Clinical Perspectives. Epigenomics 2019, 11, 349–362. [Google Scholar] [CrossRef]
- Gruber, J.J.; Geller, B.; Lipchik, A.M.; Chen, J.; Salahudeen, A.A.; Ram, A.N.; Ford, J.M.; Kuo, C.J.; Snyder, M.P. HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Mol. Cell 2019, 75, 711–724. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Bian, J.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Correction: Role of Novel Histone Modifications in Cancer. Oncotarget 2018, 9, 19460. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 Pathway Facilitates Nuclear Accumulation of HIF-1α and Functional Activation of HIF-1 by Deacetylating Hsp70 in the Cytosol. Cell Cycle 2015, 14, 2520–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, C.; Xiong, T.; Zhuang, C.; Zhuang, C.; Li, Y.; Ye, J.; Gui, Y. A CRISPR Interference of CBP and P300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro. Int. J. Biol. Sci. 2019, 15, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.K.; Brachmann, R.K. Chromatin Remodeling and Cancer. Cancer Biol. Ther. 2003, 2, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P. The Origins of Protein Phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten Things You Should Know about Protein Kinases: IUPHAR R Eview 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Seeler, J.-S.; Dejean, A. SUMO and the Robustness of Cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic Variation in MicroRNA Networks: The Implications for Cancer Research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Kasinski, A.L.; Slack, F.J. MiRNA-34 Prevents Cancer Initiation and Progression in a Therapeutically Resistant K-Ras and P53-Induced Mouse Model of Lung Adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K. Frequent Deletions and Down-Regulation of Micro-RNA Genes MiR15 and MiR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasinski, A.L.; Slack, F.J. MicroRNAs En Route to the Clinic: Progress in Validating and Targeting MicroRNAs for Cancer Therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Liang, G.; Liu, C.-C.; Wolff, E.M.; Tsai, Y.C.; Ye, W.; Zhou, X.; Jones, P.A. The Putative Tumor Suppressor MicroRNA-101 Modulates the Cancer Epigenome by Repressing the Polycomb Group Protein EZH2. Cancer Res. 2009, 69, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Cao, Q.; Mani, R.-S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K. Genomic Loss of MicroRNA-101 Leads to Overexpression of Histone Methyltransferase EZH2 in Cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C. MicroRNA-29 Family Reverts Aberrant Methylation in Lung Cancer by Targeting DNA Methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [Green Version]
- Bandres, E.; Agirre, X.; Bitarte, N.; Ramirez, N.; Zarate, R.; Roman-Gomez, J.; Prosper, F.; Garcia-Foncillas, J. Epigenetic Regulation of MicroRNA Expression in Colorectal Cancer. Int. J. Cancer 2009, 125, 2737–2743. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, X.; Ma, L. Downregulation of MicroRNA-100 Correlates with Tumor Progression and Poor Prognosis in Hepatocellular Carcinoma. Mol. Cell. Biochem. 2013, 383, 49–58. [Google Scholar] [CrossRef]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Müller, M.; Römermann, D.; Länger, F.; Kreipe, H. Epigenetic Inactivation of MicroRNA Gene Hsa-Mir-9-1 in Human Breast Cancer. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2008, 214, 17–24. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific Activation of MicroRNA-127 with Downregulation of the Proto-Oncogene BCL6 by Chromatin-Modifying Drugs in Human Cancer Cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, T.; Amini, M.; Hashemi, Z.S.; Mansoori, B.; Rezaei, S.; Karami, H.; Mosafer, J.; Mokhtarzadeh, A.; Baradaran, B. MicroRNA-181 Serves as a Dual-Role Regulator in the Development of Human Cancers. Free. Radic. Biol. Med. 2020, 152, 432–454. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S. Dysregulation of TET2 in Hematologic Malignancies. Int. J. Hematol. 2017, 105, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Inomata, M.; Tagawa, H.; Guo, Y.-M.; Kameoka, Y.; Takahashi, N.; Sawada, K. MicroRNA-17-92 down-Regulates Expression of Distinct Targets in Different B-Cell Lymphoma Subtypes. Blood J. Am. Soc. Hematol. 2009, 113, 396–402. [Google Scholar] [CrossRef]
- Wang, H.; An, X.; Yu, H.; Zhang, S.; Tang, B.; Zhang, X.; Li, Z. MiR-29b/TET1/ZEB2 Signaling Axis Regulates Metastatic Properties and Epithelial-Mesenchymal Transition in Breast Cancer Cells. Oncotarget 2017, 8, 102119. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chen, C.-S.; Lin, S.-P.; Weng, J.-R.; Chen, C.-S. Targeting Histone Deacetylase in Cancer Therapy. Med. Res. Rev. 2006, 26, 397–413. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of Histone Deacetylases in Epigenetic Regulation: Emerging Paradigms from Studies with Inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone Deacetylases and Epigenetic Therapies of Hematological Malignancies. Pharmacol. Res. 2010, 62, 18–34. [Google Scholar] [CrossRef]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and P21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 MRNA Expression in Invasive Carcinoma of the Breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 Expression upon Loss of APC in Colorectal Tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of Histone Deacetylase 2 Increases Apoptosis and P21 Cip1/WAF1 Expression, Independent of Histone Deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y. Increased Expression of Histone Deacetylase 2 Is Found in Human Gastric Cancer. Apmis 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F. A Truncating Mutation of HDAC2 in Human Cancers Confers Resistance to Histone Deacetylase Inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [CrossRef]
- Zimmermann, S.; Kiefer, F.; Prudenziati, M.; Spiller, C.; Hansen, J.; Floss, T.; Wurst, W.; Minucci, S.; Göttlicher, M. Reduced Body Size and Decreased Intestinal Tumor Rates in HDAC2-Mutant Mice. Cancer Res. 2007, 67, 9047–9054. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.-W.; Hiebert, S.W. Deletion of Histone Deacetylase 3 Reveals Critical Roles in S Phase Progression and DNA Damage Control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Glaser, K.B.; Li, J.; Staver, M.J.; Wei, R.-Q.; Albert, D.H.; Davidsen, S.K. Role of Class I and Class II Histone Deacetylases in Carcinoma Cells Using SiRNA. Biochem. Biophys. Res. Commun. 2003, 310, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Mai, A.; Dyer, M.J.; Cohen, G.M. Inhibition of Histone Deacetylase Class I but Not Class II Is Critical for the Sensitization of Leukemic Cells to Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Induced Apoptosis. Cancer Res. 2006, 66, 6785–6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.-C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I Histone Deacetylase Expression Has Independent Prognostic Impact in Human Colorectal Cancer: Specific Role of Class I Histone Deacetylases in Vitro and in Vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of P21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-H.; Kim, S.-H.; Choi, M.-C.; Lee, J.; Oh, D.-Y.; Im, S.-A.; Bang, Y.-J.; Kim, T.-Y. Class II Histone Deacetylases Play Pivotal Roles in Heat Shock Protein 90-Mediated Proteasomal Degradation of Vascular Endothelial Growth Factor Receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-ΚB-Dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P. Composition and Histone Substrates of Polycomb Repressive Group Complexes Change during Cellular Differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O. HDAC6 Rescues Neurodegeneration and Provides an Essential Link between Autophagy and the UPS. Nature 2007, 447, 860–864. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The Mammalian SIR2α Protein Has a Role in Embryogenesis and Gametogenesis. Mol. Cell. Biol. 2003, 23, 38–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.-H.; Sengupta, K.; Li, C.; Kim, H.-S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, L.R.; Verdin, E. Sirtuins: Critical Regulators at the Crossroads between Cancer and Aging. Oncogene 2007, 26, 5489–5504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A. Proteomics-Based Identification of Differentially Expressed Genes in Human Gliomas: Down-Regulation of SIRT2 Gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in Mammals: Insights into Their Biological Function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The Cytoplasmic Deacetylase HDAC6 Is Required for Efficient Oncogenic Tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [Green Version]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcene, A.; Verdin, E.; Castronovo, V. HDAC4 Represses P21 WAF1/Cip1 Expression in Human Cancer Cells through a Sp1-Dependent, P53-Independent Mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C. Role for Histone Deacetylase 1 in Human Tumor Cell Proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef] [Green Version]
- Draney, C.; Austin, M.C.; Leifer, A.H.; Smith, C.J.; Kener, K.B.; Aitken, T.J.; Hess, K.H.; Haines, A.C.; Lett, L.A.; Hernandez-Carretero, A. HDAC1 Overexpression Enhances β-Cell Proliferation by down-Regulating Cdkn1b/P27. Biochem. J. 2018, 475, 3997–4010. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Yang, C.; Zhou, J.; Wang, L.; Yi, K.; Li, Y.; Shi, J.; Kang, C.; Zeng, L. EGFR-VIII Downregulated H2AZK4/7AC Though the PI3K/AKT-HDAC2 Axis to Regulate Cell Cycle Progression. Clin. Transl. Med. 2020, 9, 10. [Google Scholar] [CrossRef]
- Wei, D.; Lu, T.; Ma, D.; Yu, K.; Zhang, T.; Xiong, J.; Wang, W.; Zhang, Z.; Fang, Q.; Wang, J. Synergistic Activity of Imatinib and AR-42 against Chronic Myeloid Leukemia Cells Mainly through HDAC1 Inhibition. Life Sci. 2018, 211, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Hsieh, J. HDAC3 Controls Gap 2/Mitosis Progression in Adult Neural Stem/Progenitor Cells by Regulating CDK1 Levels. Proc. Natl. Acad. Sci. USA 2014, 111, 13541–13546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.-L.; Lu, L.-L.; Dong, L.-L.; Liu, Y.; Bian, X.-Y.; Lian, B.-F.; Xie, L.; Wen, D.; Gao, D.-M.; Ke, A.-W. MiR-17-5p and MiR-20a-5p Suppress Postoperative Metastasis of Hepatocellular Carcinoma via Blocking HGF/ERBB3-NF-ΚB Positive Feedback Loop. Theranostics 2020, 10, 3668–3683. [Google Scholar] [CrossRef]
- Yang, J.; Li, B.; Zhao, S.; Du, H.; Du, Y. Exosomal MiR-638 Inhibits Hepatocellular Carcinoma Progression by Targeting SP1. OncoTargets Ther. 2020, 13, 6709–6720. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-K.; Cai, M.-Y.; Luo, R.-Z.; Tian, X.; Liao, Q.-M.; Zhang, X.-Y.; Han, J.-D. Overexpression of P300 Correlates with Poor Prognosis in Patients with Cutaneous Squamous Cell Carcinoma. Br. J. Dermatol. 2015, 172, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, Z.; Liao, S.; Li, E.; Wu, X.; Zeng, W. Elevated MicroRNA-7 Inhibits Proliferation and Tumor Angiogenesis and Promotes Apoptosis of Gastric Cancer Cells via Repression of Raf-1. Cell Cycle 2020, 19, 2496–2508. [Google Scholar] [CrossRef]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.-H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A. Histone Deacetylase 11 Inhibition Promotes Breast Cancer Metastasis from Lymph Nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, S.; Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Jena, M.; Bhutia, S.K. Dysregulation of Histone Deacetylases in Carcinogenesis and Tumor Progression: A Possible Link to Apoptosis and Autophagy. Cell. Mol. Life Sci. 2019, 76, 3263–3282. [Google Scholar] [CrossRef]
- Atsumi, A.; Tomita, A.; Kiyoi, H.; Naoe, T. Histone Deacetylase 3 (HDAC3) Is Recruited to Target Promoters by PML-RARα as a Component of the N-CoR Co-Repressor Complex to Repress Transcription in Vivo. Biochem. Biophys. Res. Commun. 2006, 345, 1471–1480. [Google Scholar] [CrossRef]
- Chauchereau, A.; Mathieu, M.; de Saintignon, J.; Ferreira, R.; Pritchard, L.L.; Mishal, Z.; Dejean, A.; Harel-Bellan, A. HDAC4 Mediates Transcriptional Repression by the Acute Promyelocytic Leukaemia-Associated Protein PLZF. Oncogene 2004, 23, 8777–8784. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, Fusion Partner in t (8; 21) Acute Myeloid Leukemia, Represses Transcription by Interaction with the Human N-CoR/MSin3/HDAC1 Complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, J.M.; Nip, J.; Strom, D.K.; Lutterbach, B.; Harada, H.; Lenny, N.; Downing, J.R.; Meyers, S.; Hiebert, S.W. ETO, a Target of t (8; 21) in Acute Leukemia, Makes Distinct Contacts with Multiple Histone Deacetylases and Binds MSin3A through Its Oligomerization Domain. Mol. Cell. Biol. 2001, 21, 6470–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K. The HDAC6/8/10 Inhibitor TH34 Induces DNA Damage-Mediated Cell Death in Human High-Grade Neuroblastoma Cell Lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Nishimoto, K.; Niida, H.; Uchida, C.; Ohhata, T.; Kitagawa, K.; Motegi, A.; Suda, T.; Kitagawa, M. HDAC3 Is Required for XPC Recruitment and Nucleotide Excision Repair of DNA Damage Induced by UV Irradiation. Mol. Cancer Res. 2020, 18, 1367–1378. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Zhang, R.; Jin, T.; Qu, L.; Jin, Q.; Zheng, J.; Sun, J.; Wu, Z.; Wang, L. HDAC10 Is Positively Associated with PD-L1 Expression and Poor Prognosis in Patients with NSCLC. Front. Oncol. 2020, 10, 485. [Google Scholar] [CrossRef]
- Yang, W.-C.; Bao, H.-Y.; Liu, Y.-Y.; Nie, Y.-Y.; Yang, J.-M.; Hong, P.-Z.; Zhang, Y. Depsidone Derivatives and a Cyclopeptide Produced by Marine Fungus Aspergillus Unguis under Chemical Induction and by Its Plasma Induced Mutant. Molecules 2018, 23, 2245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xiang, S.; Joo, H.-Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C. HDAC6 Deacetylates and Ubiquitinates MSH2 to Maintain Proper Levels of MutSα. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Lagunas-Rangel, F.A. Current Role of Mammalian Sirtuins in DNA Repair. DNA Repair 2019, 80, 85–92. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of P53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 Promotes DNA Repair under Stress by Activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meter, M.; Simon, M.; Tombline, G.; May, A.; Morello, T.D.; Hubbard, B.P.; Bredbenner, K.; Park, R.; Sinclair, D.A.; Bohr, V.A. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016, 16, 2641–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, S.; Jia, X.; Huang, Y.; Ji, R.; Zhao, L. Comparation of the Phytotoxicity between Chemically and Green Synthesized Silver Nanoparticles. Sci. Total Environ. 2021, 752, 142264. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Signaling Networks Guiding Epithelial–Mesenchymal Transitions during Embryogenesis and Cancer Progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.-H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ding, X.; Wang, S.; Xu, L.; Yin, T.; Han, S.; Geng, J.; Sun, W. Downregulation of Serum Exosomal MiR-320d Predicts Poor Prognosis in Hepatocellular Carcinoma. J. Clin. Lab. Anal. 2020, 34, e23239. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 Induces EMT by Cooperating with EMT Transcription Factors and Enhances Prostate Cancer Cell Migration and Metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Hung, M.-C. Breast Cancer Brain Metastases. Cancer Metastasis Rev. 2007, 26, 635–643. [Google Scholar] [CrossRef]
- Zhou, J.; Li, W.; Guo, J.; Li, G.; Chen, F.; Zhou, J. Downregulation of MiR-329 Promotes Cell Invasion by Regulating BRD4 and Predicts Poor Prognosis in Hepatocellular Carcinoma. Tumor Biol. 2016, 37, 3561–3569. [Google Scholar] [CrossRef]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone Deacetylase Inhibitors. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.-P. Disease-Causing Mutations in Parkin Impair Mitochondrial Ubiquitination, Aggregation, and HDAC6-Dependent Mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and BHLH Factors in Tumour Progression: An Alliance against the Epithelial Phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P. E-Cadherin Regulates Metastasis of Pancreatic Cancer in Vivo and Is Suppressed by a SNAIL/HDAC1/HDAC2 Repressor Complex. Gastroenterology 2009, 137, 361–371. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in Life, Disease and Medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Moehler, T.; Neben, K.; Ho, A.; Goldschmidt, H. Angiogenesis in Hematologic Malignancies. Ann. Hematol. 2001, 80, 695–705. [Google Scholar] [CrossRef]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y. Histone Deacetylases Induce Angiogenesis by Negative Regulation of Tumor Suppressor Genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef]
- Hrgovic, I.; Doll, M.; Pinter, A.; Kaufmann, R.; Kippenberger, S.; Meissner, M. Histone Deacetylase Inhibitors Interfere with Angiogenesis by Decreasing Endothelial VEGFR-2 Protein Half-Life in Part via a VE-Cadherin-Dependent Mechanism. Exp. Dermatol. 2017, 26, 194–201. [Google Scholar] [CrossRef]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs Mediate HIF-1α Stability Through PHD2-Dependent Mechanism, While HDAC6, a Class IIb Member, Promotes HIF-1α Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. J. Bone Miner. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; Rössig, L.; Kaluza, D.; Potente, M.; Boeckel, J.-N.; Knau, A.; Diehl, F.; Geng, J.-G.; Hofmann, W.-K.; Zeiher, A.M. HDAC5 Is a Repressor of Angiogenesis and Determines the Angiogenic Gene Expression Pattern of Endothelial Cells. Blood J. Am. Soc. Hematol. 2009, 113, 5669–5679. [Google Scholar] [CrossRef] [Green Version]
- Tsou, P.-S.; Khanna, D.; Sawalha, A.H. Identification of Cysteine-Rich Angiogenic Inducer 61 as a Potential Antifibrotic and Proangiogenic Mediator in Scleroderma. Arthritis Rheumatol. 2019, 71, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.-P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.G. Class IIb HDAC6 Regulates Endothelial Cell Migration and Angiogenesis by Deacetylation of Cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Levine, B. The Role of Autophagy in Mammalian Development: Cell Makeover Rather than Cell Death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylases 1 and 2 Regulate Autophagy Flux and Skeletal Muscle Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Majora, M.; Sondenheimer, K.; Knechten, M.; Uthe, I.; Esser, C.; Schiavi, A.; Ventura, N.; Krutmann, J. HDAC Inhibition Improves Autophagic and Lysosomal Function to Prevent Loss of Subcutaneous Fat in a Mouse Model of Cockayne Syndrome. Sci. Transl. Med. 2018, 10, eaam7510. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Lee, K.Y.; Lee, Y.M. Downregulation of a Tumor Suppressor RECK by Hypoxia through Recruitment of HDAC1 and HIF-1α to Reverse HRE Site in the Promoter. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M. Histone Deacetylase 10 Promotes Autophagy-Mediated Cell Survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [Green Version]
- Garva, R.; Thepmalee, C.; Yasamut, U.; Sudsaward, S.; Guazzelli, A.; Rajendran, R.; Tongmuang, N.; Khunchai, S.; Meysami, P.; Limjindaporn, T. Sirtuin Family Members Selectively Regulate Autophagy in Osteosarcoma and Mesothelioma Cells in Response to Cellular Stress. Front. Oncol. 2019, 9, 949. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 Positively Regulates Autophagy and Mitochondria Function in Embryonic Stem Cells under Oxidative Stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L. SIRT5 Regulation of Ammonia-Induced Autophagy and Mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT 5-Mediated Deacetylation of LDHB Promotes Autophagy and Tumorigenesis in Colorectal Cancer. Mol. Oncol. 2019, 13, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury through Targeting Nkx3. 2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef]
- Moseley, V.R.; Morris, J.; Knackstedt, R.W.; Wargovich, M.J. Green Tea Polyphenol Epigallocatechin 3-Gallate, Contributes to the Degradation of DNMT3A and HDAC3 in HCT 116 Human Colon Cancer Cells. Anticancer. Res. 2013, 33, 5325–5333. [Google Scholar]
- Groh, I.A.M.; Chen, C.; Lüske, C.; Cartus, A.T.; Esselen, M. Plant Polyphenols and Oxidative Metabolites of the Herbal Alkenylbenzene Methyleugenol Suppress Histone Deacetylase Activity in Human Colon Carcinoma Cells. J. Nutr. Metab. 2013, 2013, 821082. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Chan, T.-H.; Tollefsbol, T.O. A Novel Prodrug of Epigallocatechin-3-Gallate: Differential Epigenetic HTERT Repression in Human Breast Cancer Cells. Cancer Prev. Res. 2011, 4, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-Gallate Reactivates Silenced Tumor Suppressor Genes, Cip1/P21 and P16INK4a, by Reducing DNA Methylation and Increasing Histones Acetylation in Human Skin Cancer Cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Ciesielski, O.; Biesiekierska, M.; Balcerczyk, A. Epigallocatechin-3-Gallate (EGCG) Alters Histone Acetylation and Methylation and Impacts Chromatin Architecture Profile in Human Endothelial Cells. Molecules 2020, 25, 2326. [Google Scholar] [CrossRef]
- Pandey, M.; Kaur, P.; Shukla, S.; Abbas, A.; Fu, P.; Gupta, S. Plant Flavone Apigenin Inhibits HDAC and Remodels Chromatin to Induce Growth Arrest and Apoptosis in Human Prostate Cancer Cells: In Vitro and in Vivo Study. Mol. Carcinog. 2012, 51, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, T.-H.; Chien, M.-H.; Lin, W.-L.; Wen, Y.-C.; Chow, J.-M.; Chen, C.-K.; Kuo, T.-C.; Lee, W.-J. Inhibition of MDA-MB-231 Breast Cancer Cell Proliferation and Tumor Growth by Apigenin through Induction of G2/M Arrest and Histone H3 Acetylation-Mediated P21 WAF1/CIP1 Expression: APIGENIN INHIBITS MDA-MB-231 BREAST CANCER CELL. Environ. Toxicol. 2017, 32, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Huang, P.; Wang, X.; Wu, J.; Wu, M.; Huang, J. Galangin-Induced down-Regulation of BACE1 by Epigenetic Mechanisms in SH-SY5Y Cells. Neuroscience 2015, 294, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of Hypermethylation and Reactivation of P16INK4a, RARβ, and MGMT Genes by Genistein and Other Isoflavones from Soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein Reverses Hypermethylation and Induces Active Histone Modifications in Tumor Suppressor Gene B-Cell Translocation Gene 3 in Prostate Cancer. Cancer 2009, 116, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic Reactivation of Estrogen Receptor-α (ERα) by Genistein Enhances Hormonal Therapy Sensitivity in ERα-Negative Breast Cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Q.; Chen, H. DNA Methylation and Histone Modifications of Wnt Genes by Genistein during Colon Cancer Development. Carcinogenesis 2013, 34, 1756–1763. [Google Scholar] [CrossRef] [Green Version]
- Mateen, S.; Tyagi, A.; Agarwal, C.; Singh, R.P.; Agarwal, R. Silibinin Inhibits Human Nonsmall Cell Lung Cancer Cell Growth through Cell-Cycle Arrest by Modulating Expression and Function of Key Cell-Cycle Regulators. Mol. Carcinog. 2010, 49, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Anestopoulos, I.; Sfakianos, A.; Franco, R.; Chlichlia, K.; Panayiotidis, M.; Kroll, D.; Pappa, A. A Novel Role of Silibinin as a Putative Epigenetic Modulator in Human Prostate Carcinoma. Molecules 2016, 22, 62. [Google Scholar] [CrossRef]
- Berger, A.; Venturelli, S.; Kallnischkies, M.; Böcker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Weiss, T.S.; Lauer, U.M.; et al. Kaempferol, a New Nutrition-Derived Pan-Inhibitor of Human Histone Deacetylases. J. Nutr. Biochem. 2013, 24, 977–985. [Google Scholar] [CrossRef]
- Lee, W.-J.; Chen, Y.-R.; Tseng, T.-H. Quercetin Induces FasL-Related Apoptosis, in Part, through Promotion of Histone H3 Acetylation in Human Leukemia HL-60 Cells. Oncol. Rep. 2011, 25, 583–591. [Google Scholar] [PubMed]
- Kedhari Sundaram, M.; Hussain, A.; Haque, S.; Raina, R.; Afroze, N. Quercetin Modifies 5′CpG Promoter Methylation and Reactivates Various Tumor Suppressor Genes by Modulating Epigenetic Marks in Human Cervical Cancer Cells. J. Cell. Biochem. 2019, 120, 18357–18369. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA Methyltransferases in Breast Cancer Patients and to Analyze the Effect of Natural Compounds on DNA Methyltransferases and Associated Proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, G.; Thakur, V.S.; Limaye, A.M.; Gupta, S. Epigenetic Induction of Tissue Inhibitor of Matrix Metalloproteinase-3 by Green Tea Polyphenols in Breast Cancer Cells. Mol. Carcinog. 2015, 54, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, S.N.; Kala, R.; Tollefsbol, T.O. Molecular Mechanisms for Inhibition of Colon Cancer Cells by Combined Epigenetic-Modulating Epigallocatechin Gallate and Sodium Butyrate. Exp. Cell Res. 2014, 324, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-Epigallocatechin-3-Gallate Reverses the Expression of Various Tumor-Suppressor Genes by Inhibiting DNA Methyltransferases and Histone Deacetylases in Human Cervical Cancer Cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green Tea Polyphenol EGCG Reverse Cisplatin Resistance of A549/DDP Cell Line through Candidate Genes Demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef]
- Borutinskaitė, V.; Virkšaitė, A.; Gudelytė, G.; Navakauskienė, R. Green Tea Polyphenol EGCG Causes Anti-Cancerous Epigenetic Modulations in Acute Promyelocytic Leukemia Cells. Leuk. Lymphoma 2018, 59, 469–478. [Google Scholar] [CrossRef]
- Deb, G.; Shankar, E.; Thakur, V.S.; Ponsky, L.E.; Bodner, D.R.; Fu, P.; Gupta, S. Green Tea-Induced Epigenetic Reactivation of Tissue Inhibitor of Matrix Metalloproteinase-3 Suppresses Prostate Cancer Progression through Histone-Modifying Enzymes. Mol. Carcinog. 2019, 58, 1194–1207. [Google Scholar] [CrossRef]
- Steed, K.L.; Jordan, H.R.; Tollefsbol, T.O. SAHA and EGCG Promote Apoptosis in Triple-Negative Breast Cancer Cells, Possibly Through the Modulation of CIAP2. Anticancer Res. 2020, 40, 9–26. [Google Scholar] [CrossRef]
- Kuo, H.-C.D.; Wu, R.; Li, S.; Yang, A.Y.; Kong, A.-N. Anthocyanin Delphinidin Prevents Neoplastic Transformation of Mouse Skin JB6 P+ Cells: Epigenetic Re-Activation of Nrf2-ARE Pathway. AAPS J. 2019, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Gonzalez, X.; Fuentes, F.; Su, Z.-Y.; Kong, A.-N.T. Apigenin Reactivates Nrf2 Anti-Oxidative Stress Signaling in Mouse Skin Epidermal JB6 P + Cells Through Epigenetics Modifications. AAPS J. 2014, 16, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.; Li, Y.; Tollefsbol, T. The Effects of Combinatorial Genistein and Sulforaphane in Breast Tumor Inhibition: Role in Epigenetic Regulation. Int. J. Mol. Sci. 2018, 19, 1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, M.; Mz, A.; Aa, M. Genistein Induces Alterations of Epigenetic Modulatory Signatures in Human Cervical Cancer Cells. Anticancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef]
- Attoub, S.; Hassan, A.H.; Vanhoecke, B.; Iratni, R.; Takahashi, T.; Gaben, A.-M.; Bracke, M.; Awad, S.; John, A.; Kamalboor, H.A.; et al. Inhibition of Cell Survival, Invasion, Tumor Growth and Histone Deacetylase Activity by the Dietary Flavonoid Luteolin in Human Epithelioid Cancer Cells. Eur. J. Pharmacol. 2011, 651, 18–25. [Google Scholar] [CrossRef]
- Zuo, Q.; Wu, R.; Xiao, X.; Yang, C.; Yang, Y.; Wang, C.; Lin, L.; Kong, A. The Dietary Flavone Luteolin Epigenetically Activates the Nrf2 Pathway and Blocks Cell Transformation in Human Colorectal Cancer HCT116 Cells. J. Cell. Biochem. 2018, 119, 9573–9582. [Google Scholar] [CrossRef]
- Zhang, T.; Li, S.; Li, J.; Yin, F.; Hua, Y.; Wang, Z.; Lin, B.; Wang, H.; Zou, D.; Zhou, Z.; et al. Natural Product Pectolinarigenin Inhibits Osteosarcoma Growth and Metastasis via SHP-1-Mediated STAT3 Signaling Inhibition. Cell Death Dis. 2016, 7, e2421. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, W.; Wang, C.; Wu, R.; Yin, R.; Kuo, H.-C.; Wang, L.; Kong, A.-N. Pelargonidin Reduces the TPA Induced Transformation of Mouse Epidermal Cells –Potential Involvement of Nrf2 Promoter Demethylation. Chem. Biol. Interact. 2019, 309, 108701. [Google Scholar] [CrossRef]
- Kauntz, H.; Bousserouel, S.; Gossé, F.; Raul, F. Epigenetic Effects of the Natural Flavonolignan Silibinin on Colon Adenocarcinoma Cells and Their Derived Metastatic Cells. Oncol. Lett. 2013, 5, 1273–1277. [Google Scholar] [CrossRef] [Green Version]
- Kuang, H.; Tang, Z.; Zhang, C.; Wang, Z.; Li, W.; Yang, C.; Wang, Q.; Yang, B.; Kong, A.-N. Taxifolin Activates the Nrf2 Anti-Oxidative Stress Pathway in Mouse Skin Epidermal JB6 P+ Cells through Epigenetic Modifications. Int. J. Mol. Sci. 2017, 18, 1546. [Google Scholar] [CrossRef] [Green Version]
- Priyadarsini, R.V.; Vinothini, G.; Murugan, R.S.; Manikandan, P.; Nagini, S. The Flavonoid Quercetin Modulates the Hallmark Capabilities of Hamster Buccal Pouch Tumors. Nutr. Cancer 2011, 63, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.-G.; Wang, J.-L.; Yang, S.-L.; Wu, J.-L. Aberrant Epigenetic Alteration in Eca9706 Cells Modulated by Nanoliposomal Quercetin Combined with Butyrate Mediated via Epigenetic-NF-ΚB Signaling. Asian Pac. J. Cancer Prev. 2014, 15, 4539–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, Y.; Xue, Y.; Hu, H.; Ye, J.; Li, X.; Lu, Z.; Meng, F.; Liang, S. Berberine Acts as a Putative Epigenetic Modulator by Affecting the Histone Code. Toxicol. Vitr. 2016, 36, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florean, C.; Schnekenburger, M.; Lee, J.-Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.-M.; Grandjenette, C.; Kim, J.-G.; Yoon, A.-Y.; et al. Discovery and Characterization of Isofistularin-3, a Marine Brominated Alkaloid, as a New DNA Demethylating Agent Inducing Cell Cycle Arrest and Sensitization to TRAIL in Cancer Cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Gupta, K.P. Modulation of MiR-203 and Its Regulators as a Function of Time during the Development of 7, 12 Dimethylbenz [a] Anthracene Induced Mouse Skin Tumors in Presence or Absence of the Antitumor Agents. Toxicol. Appl. Pharmacol. 2014, 278, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Ducourouble, M.; Van Seuningen, I. Epigenetic Regulation of the Human Mucin Gene MUC4 in Epithelial Cancer Cell Lines Involves Both DNA Methylation and Histone Modifications Mediated by DNA Methyltransferases and Histone Deacetylases. FASEB J. 2008, 22, 3035–3045. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A Natural Histone Deacetylase Inhibitor, Psammaplin A, Induces Cell Cycle Arrest and Apoptosis in Human Endometrial Cancer Cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the Mechanism of Action and Enzymatic Selectivity of Psammaplin A against Its Epigenetic Targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef]
- Piña, I.C.; Gautschi, J.T.; Wang, G.-Y.-S.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the Sponge Pseudoceratina p Urpurea: Inhibition of Both Histone Deacetylase and DNA Methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Inhibition of DNA Methyltransferases, Histone Deacetylases and Lysine-Specific Demethylase-1 Suppresses the Tumorigenicity of the Ovarian Cancer Ascites Cell Line SKOV3. Int. J. Oncol. 2013, 43, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, C.; Li, W.; Wu, R.; Guo, Y.; Cheng, D.; Yang, Y.; Androulakis, I.P.; Kong, A.-N. Pharmacokinetics and Pharmacodynamics of the Triterpenoid Ursolic Acid in Regulating the Antioxidant, Anti-Inflammatory, and Epigenetic Gene Responses in Rat Leukocytes. Mol. Pharm. 2017, 14, 3709–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Khan, S.; Kumar, S.; Sinha, S.; Farhan, M.; Bora, H.K.; Maurya, R.; Meeran, S.M. Cucurbitacin B Alters the Expression of Tumor-Related Genes by Epigenetic Modifications in NSCLC and Inhibits NNK-Induced Lung Tumorigenesis. Cancer Prev. Res. 2015, 8, 552–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, D.G.; Neis, V.B.; Balen, G.O.; Colla, A.; Cunha, M.P.; Dalmarco, J.B.; Pizzolatti, M.G.; Prediger, R.D.; Rodrigues, A.L.S. Antidepressant-like Effect of Ursolic Acid Isolated from Rosmarinus officinalis L. in Mice: Evidence for the Involvement of the Dopaminergic System. Pharmacol. Biochem. Behav. 2012, 103, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; MacKinnon, S.L.; Craft, C.C.; Matchett, M.D.; Hurta, R.A.; Neto, C.C. Ursolic Acid and Its Esters: Occurrence in Cranberries and Other Vaccinium Fruit and Effects on Matrix Metalloproteinase Activity in DU145 Prostate Tumor Cells. J. Sci. Food Agric. 2011, 91, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Gopal, Y.N.V.; Arora, T.S.; Van Dyke, M.W. Parthenolide Specifically Depletes Histone Deacetylase 1 Protein and Induces Cell Death through Ataxia Telangiectasia Mutated. Chem. Biol. 2007, 14, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Guzman, M.L.; Chen, S.; Wang, L.; Yeung, S.-K.; Pei, X.-Y.; Dent, P.; Jordan, C.T.; Grant, S. The NF (Nuclear Factor)-ΚB Inhibitor Parthenolide Interacts with Histone Deacetylase Inhibitors to Induce MKK7/JNK1-Dependent Apoptosis in Human Acute Myeloid Leukaemia Cells: Parthenolide Potentiates HDAC Inhibitor Lethality in AML. Br. J. Haematol. 2010, 151, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghantous, A.; Saikali, M.; Rau, T.; Gali-Muhtasib, H.; Schneider-Stock, R.; Darwiche, N. Inhibition of Tumor Promotion by Parthenolide: Epigenetic Modulation of P21. Cancer Prev. Res. 2012, 5, 1298–1309. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Buttitta, G.; Emanuele, S.; di Fiore, R.; Martinez, R.; Rolfo, C.; Vento, R.; Tesoriere, G. The Synergistic Effect of SAHA and Parthenolide in MDA-MB231 Breast Cancer Cells: THE EFFECT OF PN/SAHA COMBINATION IN MDA-MB231 CELLS. J. Cell. Physiol. 2015, 230, 1276–1289. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Ramirez, C.N.; Su, Z.-Y.; Kong, A.-N.T. Epigenetic Modifications of Triterpenoid Ursolic Acid in Activating Nrf2 and Blocking Cellular Transformation of Mouse Epidermal Cells. J. Nutr. Biochem. 2016, 33, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Li, L.; Dou, G.; Wang, C.; Li, J.; He, H.; Wu, M.; Qi, H. Z-Ligustilide Restores Tamoxifen Sensitivity of ERα Negative Breast Cancer Cells by Reversing MTA1/IFI16/HDACs Complex Mediated Epigenetic Repression of ERα. Oncotarget 2017, 8, 29328–29345. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Gao, J.; Rizzo, M.; Harrison, T.; Tiedje, J.M. Diet is a major factor governing the fecal butyrate-producing community structure across Mammalia, Aves and Reptilia. ISME J. 2015, 9, 832–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, V.; Ronchetti, S.; Marchetti, M.C.; Calvitti, M.; Riccardi, C.; Grignani, F.; Vecchini, A. Molecular Mechanisms Underlying Eicosapentaenoic Acid Inhibition of HDAC1 and DNMT Expression and Activity in Carcinoma Cells. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2020, 1863, 194481. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Sun, Y.; Lim, S.K.; Tam, J.P.; Dekker, M.; Chen, H.; Sze, S.K. Dietary Phytochemical PEITC Restricts Tumor Development via Modulation of Epigenetic Writers and Erasers. Sci. Rep. 2017, 7, 40569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Bhatt, L.K.; Momin, M. Potent Antitumor Activity of Laccaic Acid and Phenethyl Isothiocyanate Combination in Colorectal Cancer via Dual Inhibition of DNA Methyltransferase-1 and Histone Deacetylase-1. Toxicol. Appl. Pharmacol. 2019, 377, 114631. [Google Scholar] [CrossRef]
- de Figueiredo, S.M.; Binda, N.S.; Nogueira-Machado, J.A.; Vieira-Filho, S.A.; Caligiorne, R.B. The Antioxidant Properties of Organosulfur Compounds (Sulforaphane). Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 24–39. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane Causes Epigenetic Repression of HTERT Expression in Human Breast Cancer Cell Lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Zhou, S.-J.; Zhang, X.-M.; Chen, H.-Q.; Liu, W. Sulforaphane Suppresses in Vitro and in Vivo Lung Tumorigenesis through Downregulation of HDAC Activity. Biomed. Pharmacother. 2016, 78, 74–80. [Google Scholar] [CrossRef]
- dos Santos, P.W.; Machado, A.R.T.; De Grandis, R.A.; Ribeiro, D.L.; Tuttis, K.; Morselli, M.; Aissa, A.F.; Pellegrini, M.; Antunes, L.M.G. Transcriptome and DNA Methylation Changes Modulated by Sulforaphane Induce Cell Cycle Arrest, Apoptosis, DNA Damage, and Suppression of Proliferation in Human Liver Cancer Cells. Food Chem. Toxicol. 2020, 136, 111047. [Google Scholar] [CrossRef]
- Royston, K.; Udayakumar, N.; Lewis, K.; Tollefsbol, T. A Novel Combination of Withaferin A and Sulforaphane Inhibits Epigenetic Machinery, Cellular Viability and Induces Apoptosis of Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1092. [Google Scholar] [CrossRef]
- Gao, P.; Jin, J.; Liu, R.; Jin, Q.; Wang, X. Chemical Compositions of Walnut (Juglans regia L.) Oils from Different Cultivated Regions in China. J. Am. Oil Chem. Soc. 2018, 95, 825–834. [Google Scholar] [CrossRef]
- Li, Y.; Buckhaults, P.; Li, S.; Tollefsbol, T. Temporal Efficacy of a Sulforaphane-Based Broccoli Sprout Diet in Prevention of Breast Cancer through Modulation of Epigenetic Mechanisms. Cancer Prev. Res. 2018, 11, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.L.; Kala, R.; Tollefsbol, T.O. Mechanisms for the Inhibition of Colon Cancer Cells by Sulforaphane through Epigenetic Modulation of MicroRNA-21 and Human Telomerase Reverse Transcriptase (HTERT) down-Regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef] [PubMed]
- da Santos, P.W. Influência do Sulforafano, um Inibidor de Histonas Desacetilases, Sobre a Instabilidade genômica e Mecanismos Epigenéticos em Linhagens Celulares Humanas. text. Master’s Thesis, Universidade de São Paulo, São Paulo, Spain, 2019. [Google Scholar]
- Gupta, A.; Behl, T.; Panichayupakaranan, P. A Review of Phytochemistry and Pharmacology Profile of Juglans Regia. Obes. Med. 2019, 16, 100142. [Google Scholar] [CrossRef]
- Kim, M.Y.; Park, S.-J.; Shim, J.W.; Yang, K.; Kang, H.S.; Heo, K. Naphthazarin Enhances Ionizing Radiation-Induced Cell Cycle Arrest and Apoptosis in Human Breast Cancer Cells. Int. J. Oncol. 2015, 46, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Qadi, S.A.; Hassan, M.A.; Sheikh, R.A.; Baothman, O.A.; Zamzami, M.A.; Choudhry, H.; Al-Malki, A.L.; Albukhari, A.; Alhosin, M. Thymoquinone-Induced Reactivation of Tumor Suppressor Genes in Cancer Cells Involves Epigenetic Mechanisms. Genet. Epigenet 2019, 12, 251686571983901. [Google Scholar] [CrossRef]
- Yang, Y.; Bae, M.; Kim, B.; Park, Y.-K.; Koo, S.I.; Lee, J.-Y. Astaxanthin Prevents and Reverses the Activation of Mouse Primary Hepatic Stellate Cells. J. Nutr. Biochem. 2016, 29, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parashar, G.; Capalash, N. Promoter Methylation-Independent Reactivation of PAX1 by Curcumin and Resveratrol Is Mediated by UHRF1. Clin. Exp. Med. 2016, 16, 471–478. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Xu, C.; Wang, J.; Zhu, B.; Huang, Q.; Chen, D.; Sheng, J.; Zou, Y.; Lee, Y.M.; et al. Comparative Profiling of Analog Targets: A Case Study on Resveratrol for Mouse Melanoma Metastasis Suppression. Theranostics 2018, 8, 3504–3516. [Google Scholar] [CrossRef]
- Gao, Y.; Tollefsbol, T. Combinational Proanthocyanidins and Resveratrol Synergistically Inhibit Human Breast Cancer Cells and Impact Epigenetic–Mediating Machinery. Int. J. Mol. Sci. 2018, 19, 2204. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Chen, J.; Shi, Y.; Jia, J.; Zhang, Y. Curcumin-Induced Histone Hypoacetylation: The Role of Reactive Oxygen Species. Biochem. Pharmacol. 2005, 69, 1205–1213. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Cui, G.; Zhou, J. Curcumin, a Potent Anti-Tumor Reagent, Is a Novel Histone Deacetylase Inhibitor Regulating B-NHL Cell Line Raji Proliferation. Acta Pharmacol. Sin. 2005, 26, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, Both Histone Deacetylase and P300/CBP-Specific Inhibitor, Represses the Activity of Nuclear Factor Kappa B and Notch 1 in Raji Cells. Basic Clin. Pharmacol. Toxicol. 2007, 101, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Bora-Tatar, G.; Dayangaç-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekçi, K.; Erdem-Yurter, H. Molecular Modifications on Carboxylic Acid Derivatives as Potent Histone Deacetylase Inhibitors: Activity and Docking Studies. Bioorganic Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Krauthauser, C.; Maduskuie, V.; Fawcett, P.T.; Olson, J.M.; Rajasekaran, S.A. Curcumin-Induced HDAC Inhibition and Attenuation of Medulloblastoma Growth in Vitro and in Vivo. BMC Cancer 2011, 11, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, L.; Khor, T.O.; Lee, J.-H.; Boyanapalli, S.S.S.; Huang, Y.; Wu, T.-Y.; Saw, C.L.-L.; Cheung, K.-L.; Kong, A.-N.T. Epigenetic CpG Demethylation of the Promoter and Reactivation of the Expression of Neurog1 by Curcumin in Prostate LNCaP Cells. AAPS J. 2011, 13, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-J.; Tsai, Y.-J.; Lin, M.-Y.; You, H.-L.; Kalyanam, N.; Ho, C.-T.; Pan, M.-H. Calebin-A Induced Death of Malignant Peripheral Nerve Sheath Tumor Cells by Activation of Histone Acetyltransferase. Phytomedicine 2019, 57, 377–384. [Google Scholar] [CrossRef]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-Based Combinatorial Resveratrol and Pterostilbene Alters DNA Damage Response by Affecting SIRT1 and DNMT Enzyme Expression, Including SIRT1-Dependent γ-H2AX and Telomerase Regulation in Triple-Negative Breast Cancer. BMC Cancer 2015, 15, 672. [Google Scholar] [CrossRef] [Green Version]
- Juli, G.; Oliverio, M.; Bellizzi, D.; Gallo Cantafio, M.E.; Grillone, K.; Passarino, G.; Colica, C.; Nardi, M.; Rossi, M.; Procopio, A.; et al. Anti-Tumor Activity and Epigenetic Impact of the Polyphenol Oleacein in Multiple Myeloma. Cancers 2019, 11, 990. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.; Okla, M.; Chung, S. Ellagic Acid Inhibits Adipocyte Differentiation through Coactivator-Associated Arginine Methyltransferase 1-Mediated Chromatin Modification. J. Nutr. Biochem. 2014, 25, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, C.; Guo, Y.; Su, Z.-Y.; Yang, Y.; Shu, L.; Kong, A.-N.T. Blocking of JB6 Cell Transformation by Tanshinone IIA: Epigenetic Reactivation of Nrf2 Antioxidative Stress Pathway. AAPS J. 2014, 16, 1214–1225. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wu, D.; Zhao, L.; Yang, Y.; Ding, J.; Dong, L.; Hu, L.; Wang, F.; Zhao, X.; Cai, Y.; et al. Arsenic Trioxide Reduces Global Histone H4 Acetylation at Lysine 16 through Direct Binding to Histone Acetyltransferase HMOF in Human Cells. PLoS ONE 2015, 10, e0141014. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhou, W.; Zhang, C.; Liu, H.; Zhang, Y. Curcumol Controls Choriocarcinoma Stem-Like Cells Self-Renewal via Repression of DNA Methyltransferase (DNMT)- and Histone Deacetylase (HDAC)-Mediated Epigenetic Regulation. Med. Sci. Monit. 2018, 24, 461–472. [Google Scholar] [CrossRef]
- Xiang, N.; Zhao, R.; Song, G.; Zhong, W. Selenium Modifies GSTP1 and Histone Methylation and Acetylation in Human Prostate Cancer Cells. AACR J. 2008, 68, 749. [Google Scholar]
- Saiko, P.; Pemberger, M.; Horvath, Z.; Savinc, I.; Grusch, M.; Handler, N.; Erker, T.; Jaeger, W.; Fritzer-Szekeres, M.; Szekeres, T. Novel Resveratrol Analogs Induce Apoptosis and Cause Cell Cycle Arrest in HT29 Human Colon Cancer Cells: Inhibition of Ribonucleotide Reductase Activity. Oncol. Rep. 2008, 19, 1621–1626. [Google Scholar] [PubMed] [Green Version]
- Koushki, M.; Amiri-Dashatan, N.; Ahmadi, N.; Abbaszadeh, H.-A.; Rezaei-Tavirani, M. Resveratrol: A Miraculous Natural Compound for Diseases Treatment. Food Sci. Nutr. 2018, 6, 2473–2490. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Torres, E.; Hernández-Oliveras, A.; Meneses-Morales, I.; Rodríguez, G.; Fuentes-García, G.; Zarain-Herzberg, Á. Resveratrol Up-Regulates ATP2A3 Gene Expression in Breast Cancer Cell Lines through Epigenetic Mechanisms. Int. J. Biochem. Cell Biol. 2019, 113, 37–47. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Yuan, W.; Li, S.; Gupta, S.C. Curcumin-Free Turmeric Exhibits Anti-Inflammatory and Anticancer Activities: Identification of Novel Components of Turmeric. Mol. Nutr. Food Res. 2013, 57, 1529–1542. [Google Scholar] [CrossRef]
- Kala, R.; Tollefsbol, T.O.; Li, Y. Potential of Resveratrol in Inhibiting Cancer and Slowing Aging. J. Nutr. Food Sci. S 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The Critical Role of the Class III Histone Deacetylase SIRT1 in Cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Gore, S.D. Arsenic trioxide—An old drug rediscovered. Blood Rev. 2010, 24, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashem, S.; Nisar, S.; Sageena, G.; Macha, M.A.; Yadav, S.K.; Krishnankutty, R.; Uddin, S.; Haris, M.; Bhat, A.A. Therapeutic Effects of Curcumol in Several Diseases; An Overview. Nutr. Cancer 2021, 73, 181–195. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules (Origins) | Used Models | Methods | Key Findings | Ref. |
|---|---|---|---|---|
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | MCF-7 and MDA-MB-231 (Breast cancer cells) | Flow cytometry (apoptosis assay) RT-PCR and real-time PCR (Quantification of hTERT expression) DNMTs, HDACs, and HATs activity assays ChIP assay Western blot analysis | Inhibited the transcription of hTERT through epigenetic mechanisms in estrogen receptor (ER)-positive MCF-7 and ER-negative MDA-MB-231 cells. Inhibited the activities of DNMT and histone acetyltransferase (HAT). Remodelled the chromatin structures of the hTERT promoter by decreasing the level of acetyl-H3, acetyl-H3K9, and acetyl-H4 in the hTERT promoter. Induced chromatin alterations that facilitated the binding of many hTERT repressors such as MAD1 and E2F-1 to the hTERT regulatory region. | [189] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | A431 (Human skin cancer) | DNA methylation assay HDAC activity assay Western blot analysis Cell lysates | Decreased global DNA methylation levels in A431 cells in a dose-dependent manner. Decreased the HDAC activity. Increased the levels of acetylated lysine 9 and 14 on histone H3 (H3-Lys 9 and 14) and acetylated lysine 5, 12, and 16 on histone H4. Re-expressed the mRNA and proteins of silenced tumor suppressor genes, p16INK4a and Cip1/p21. | [190] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | HT-29 and HCT 116 (Human colon cancer cell lines) | Western blot analysis RNA extraction Real-time PCR | Reduced HDAC and DNMT protein expression. Decreased HDAC2 and HDAC3 expressions. Decreased association between UHRF1 and DNMT3. Decreased association between UHRF1 and HDAC3 in only the HCT 116 cell line. | [187] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | MCF7 and MDA MB 231 (Breast cancer cells) | RT-PCR Western blot analysis | Lowered the protein levels of DNMT1, HDAC1, and MeCP2. | [204] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | HT29 (Human colon cancer cells) | HDAC enzyme activity Western blot | Inhibited the HDAC activity in intact HT29 cells. Decreased the HDAC1 protein level. | [188] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Not reported) | MCF-7 and MDA-MB-231 (Breast cancer cells) | RNA extraction RT PCR ChIP method Western blot analysis | Reduced levels of the enhancer of zeste homolog 2 (EZH2) and Class I HDAC proteins. | [205] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | CRL-2577, HCT-116 and HT-29HTB-38 cells | Cell viability and apoptosis Real-time quantitative PCR HDAC activity assessment ChIP assay | Combinatorial effects of EGCG and NaB. Increased HDAC1 in RKO CRC cells. Inhibited the HDAC1, DNMT1, and survivin in all the three CRC cells tested. Affected the global DNA methylation. | [206] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | HeLa cells | HDAC activity assessment Bisulfite modification MS-PCR RT-PCR | Decreased the HDAC activity time-dependently. Zn ion was taken to be the substrate-binding site of HDAC1. | [207] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | A549/DDP cell line | Cell viability assay MTT assay RT- PCR Total HDAC activity and DNMT activity in vivo experiments | In vitro EGCG + cisplatin (DDP) treatment caused: Inhibition of DNMT and HDAC activities, reversal of hypermethylated status, and downregulation of the expression of GAS1, TIMP4, ICAM1, and WISP2 gene in A549/DDP cells. In vivo EGCG + DDP pre-treatment caused: Inhibition of tumors, a decrease in methylation levels of GAS1, TIMP4, ICAM, and WISP2, and an increase in their expression levels. | [208] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Not reported) | APL NB4 and HL-60 cells | RT-qPCR ChIP assay MSP and sequencing Western blot analysis | Downregulated the epigenetic modifiers HDAC1 and HDAC2 Downregulated the polycomb repressive complex 2 (PRC2) core components in gene and protein levels. Reduced gene-binding effect of core components of EZH2, SUZ12, and EED. | [209] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Not reported) | DUPRO and LNCaP (prostate cells) | RNA extraction RT– semi q-PCR Western blot analysis ChIP assay HDAC enzyme activity | Reduced the expression of both EZH2 enhancers and its catalytic product trimethylation of H3. Increased acetylation of histone H3K9/18. Reduced the activity of Class I HDACs, as well as EZH2 and H3K27me3 levels. | [210] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Not reported) | HMEC-1 and HUVECs cells | Cell proliferation assay RNA isolation Reverse transcription and RT- PCR Western blot analysis HDAC Activity Assay | Increased histone acetylation (H3K9/14ac, H3ac), as well as methylation of both active (H3K4me3) and repressive (H3K9me3) chromatin. Inhibited HDAC activity in both cellular and cell-free models. Altered epigenome modulator expression and activity (HDAC5 and 7, p300, CREBP, LSD1 or KMT2A). | [191] |
| (−)-Epigallocatechin-3-gallate (EGCG) (Purchased) | Breast cancer cells | RNA extraction Protein extraction qRT-PCR Western blot analysis ChIP assay HDAC activity HMT (H3K27me3) activity | Induced changes in histone modifications. Resulted in an increased apoptosis. | [211] |
| Delphinidin (Purchased) | JB6 P+ (Mouse epidermal cells) HepG2–C8 (hepatocellular cancer cells) | Western blot Quantitative real-time PCR Bisulfite genomic sequencing | Reduced DNMTs and HDACs protein expression. | [212] |
| Apigenin (Purchased) | Human prostate cancer cell lines 22Rv1 and PC-3 (prostate cancer cells) | HDAC activity assessment RNA isolation RT-PCR and q-PCR Western blot analysis ChIP assay Tumor xenograft studies | Inhibited Class I HDACs in prostate cancer cells. Inhibited HDAC enzyme activity, particularly HDAC1 and HDAC3. Induced histone acetylation. Increased p21/waf1 protein and mRNA expression, as well as p21/waf1 mRNA expression. Reduced (in vivo) tumor development, HDAC activity, HDAC1, and HDAC3 protein expression. | [192] |
| Apigenin (Purchased) | JB6 P+ (Skin cells) | RT-PCR Western blot analysis | Reduced some HDACs (1–8) and their expression levels. | [213] |
| Apigenin (Purchased) | MDA-MB-231 (breast cancer cells) | Immunoblot analysis HDAC assay Nuclear extract preparation ChIP assay | Inhibited HDAC activity. Induced acetylation of histone H3. Increased the acetylation of histone. | [193] |
| Galangin (Purchased) | Neuroblastoma cells | Flow cytometry ELISA RT-PCR and qPCR Western blot analysis ChIP assay DNA methylation analysis | Reduced the BACE1 at mRNA and protein levels. Reduced acetylated H3 in BACE1 promoter areas by increasing endogenous HDAC1-mediated deacetylation. | [194] |
| Genistein (Purchased) | Human esophageal squamous cell carcinoma cell lines KYSE 510 and KYSE 150 cancer cells | Modification by bisulfite Methylation-specific PCR RT-PCR | Reversed DNA hypermethylation. Reactivated RARβ, p16INK4a, and MGMT in KYSE 510 cells. Reversed DNA hypermethylation and reactivated RARβ in KYSE 150 cells and prostate cancer LNCaP and PC3 cells. Activated RARβ. Inhibited the activity of HDAC. | [195] |
| Genistein (Purchased) | MCF7 and MDA MB 231 (Breast cells) | Bisulfite conversion RT-PCR Western blot | Reduced HDAC1 and MeCP2 protein levels. | [204] |
| Genistein (Purchased) | A498, ACHN, HEK-293 and HK-2 cells | RT-PCR Sodium bisulfite modification and sequencing HAT and HDAC analysis | Increased HAT activity and reduced HDAC activity. | [205] |
| Genistein (Purchased) | LNCaP, PC3, and RWPE-1 (Prostate cells) | Quantitative RT-PCR Sodium bisulfite modification and sequencing ChIP analysis HAT and HDAC analysis | Increased the levels of acetylated histones 3, 4, histone three dimethylated at lysine 4, histone 3 trimethylated at lysine 4, and RNA polymerase II. Decreased DNA methyltransferase and methyl-binding domain protein 2 activity. Increased HAT activity. | [196] |
| Genistein (Purchased) | HT29 (colon cancer cells) | HDAC enzyme activity Western blot analysis | Inhibited HDAC activity in intact HT29 cells. Reduced HDAC1 protein levels. | [188] |
| Genistein (Purchased) | MCF-7, MDA-MB-231, and MDA-MB-157, HMECs cells, and two mouse models | MTT assay RT-PCR ChIP assay | Reduced the activity of HDAC, alone or in combination with TSA. Reduced binding to the ERα promoter, as well as gene expression for HDACs. Reduced HDAC1 protein and mRNA expression in both animal models studied. | [197] |
| Genistein (Purchased) | Rat colon tissues | RNA isolation qPCR analysis ChIP analysis Bisulfite sequencing | In the post-AOM phase, there was a decrease in H3Ac at the promoter of Wnt5a, Sfrp5, and Sfrp2. Repressed histone H3 lysine 9 methylation and serine 10 phosphorylation at the promoters of Sfrp2, Sfrp5, and Wnt5a in the post-AOM period. In the post-AOM period, the nuclear level of HDAC3 protein was increased. After AOM induction, H3Ac was reduced in the same region of the Sfrp5 promoter. | [198] |
| Genistein (Purchased) | MCF-7 and MDA-MB-231 cells | HDAC activity assay HMT activity | Inhibition of HDAC and HMT by GEN + SFN. Downregulation of HDAC2 and HDAC3 levels at the mRNA and protein levels. GEN + SFN downregulated the hTERT levels. | [214] |
| Genistein (Purchased) | Human cervical cancer cells | DNMT and HDAC activity analysis In silico studies in the post-AOM period | Reduced the expression of HDAC and enzymatic activity in a time-dependent manner. Interacted with members of the DNMT and HDAC families. Reversed the tumor suppressor genes’ promoter region methylation, and their expression was restored. | [215] |
| Genistein (Purchased) | HeLa cells | qPCR HDAC activity assay HMT-H3K9 activity Global DNA methylation | Altered HDACs, HMTs, demethylases, and histone phosphorylases’ expression. Reduced HDAC and HMT activity, as well as global DNA methylation levels. | [203] |
| Kaempferol (Not reported) | HepG2, Hep3B, and HCT-116 cells | In silico docking analysis HDACi screening assay HDAC inhibition profiling Immunoblotting Real-time cell monitoring | Inhibited the activity of HDAC. Induced hyperacetylation of histone complex H3. Reduced cell viability and proliferation rate. | [201] |
| Luteolin (Purchased) | MDA-MB231-1833, LNM35, HT29, HepG2, and MCF7/6 cells | HDAC assay and histone acetylation levels | Inhibited HDAC activity. | [216] |
| Luteolin (Purchased) | HCT116 (Colorectal cells) | HDAC activity assay | Reduced levels of HDAC protein and enzyme activity. | [217] |
| Pectolinarigenin (Purchased) | 143B, HOS, and MG63 (Osteosarcoma) | Western blot analysis RT-PCR ChIP assay MTS cell viability assay | Disrupted the development of the STAT3/DNMT1/HDAC1 complex. Caused an increase in SHP-1 expression in osteosarcoma. | [218] |
| Pelargonidin (Purchased) | Skin epidermal JB6 (JB6 P+) cells | Western blot analysis RT-PCR Bisulfite genomic sequencing | Reduced protein levels of genes encoding HDACs. | [219] |
| Silibinin (Not reported) | H1299 cells | HDAC activity RT- RT-PCR assay | Reduced the activity of HDAC in a dose-dependent manner. Reduced HDAC1, HDAC2, and HDAC3 protein levels, whereas HDAC1, HDAC6, SET domain proteins (SETD1A, D4, D6) and lysine-specific demethylases were upregulated (KDM 5B, 5C, 4A). | [199] |
| Silibinin (Purchased) | SW480 and SW620 (Colon cells) | HDAC activity | No effect on the activity of HDACs. | [220] |
| Silibinin (Purchased) | DU145 and PC3 (Prostate cells) | Western blot analysis RT-PCR HDACs assay | Reduced HDAC1-2 expression levels in a concentration-dependent manner. | [200] |
| Taxifolin (Purchased) | HepG2 cells, skin epidermal JB6 P+ cells, and HepG2-C8 cells | Western blot analysis RNA extraction qRT-PCR assay Bisulfite genomic sequencing | Inhibited DNMT and HDAC protein expression. | [221] |
| Quercetin (Purchased) | HL-60 (leukemia cells) | Western blot analysis RT-PCR HDAC assay HAT assay ChIP assay | Increased histone H3 acetylation, which promoted the production of FasL Activated HAT and inhibited HDAC. | [202] |
| Quercetin (Purchased) | Hamster buccal pouch (HBP) carcinomas | Immunohistochemistry Western blot analysis RT-PCR | Inhibited HDAC-1 and DNMT1 activities. | [222] |
| Quercetin (Purchased) | Eca9706 cells | MTT assay Immunoblotting MSP of p16INK4a gene promoter | Reduced the reverse expressions of global HDAC1. Quercetin + butyrate displayed a reverse effect targeting both altered DNA methylation and histone acetylation, acting as HDAC inhibitor mediated via epigenetic-NF-κB cascade signaling. | [223] |
| Quercetin (Purchased) | HeLa cells | DNMT and HDAC activity assay HMT-H3K9 activity assay Molecular modeling qRT-PCR | Reduced the activity of HDAC activity. Reduced the activation of HMT-H3K9. Modified the expression of many chromatin modifiers, and lowered the activity of HDACs and HMTs. Several DNMTs and HDACs interacted with residues in their catalytic cavities (as a competitive inhibitor). | [203] |
| Molecules (Origins) | Used Models | Methods | Key Findings | Ref. |
|---|---|---|---|---|
| Berberine (BBR) (Purchased) | U266, KG1-α, and HL-60 cells | Molecular Docking Growth inhibition assessment RT-PCR Western blot analysis | Affected the enzymes involved in histone acetylation and methylation. Induced cytotoxicity and apoptosis in HL-60/ADR and KG1-α cells. Upregulated histone acetyltransferase CREBBP and EP300, histone deacetylase SIRT3, histone demethylase KDM6A, as well as histone methyltransferase SETD7. Downregulated histone acetyltransferase HDAC8, histone methyltransferase WHSC1I, WHSC1II, and SMYD3. | [224] |
| Isofistularin-3 (Iso-3) (Aplysina aerophoba) | RAJI, U-937, JURKAT, K-562, HL-60, MEG-01, cells | HDAC activity assessment Molecular docking study Western blot analysis | Modified the aryl hydrocarbon receptor (AHR) promoter methylation and increased the AHR expression in RAJI cells. | [225] |
| Nicotinamide (NA) (Purchased) | Female mice skin | RT-PCR DNA bisulfite modification Western blot analysis | Downregulated the miR-203 levels at 16 weeks. Upregulated the HDAC, DNMT, and promoter methylation of miR-203 at 4 or 16 weeks. Prevented changed gene expression. | [226] |
| Trichostatin A (TSA) (Purchased) | PANC-1, CAPAN (1 and 2), and KATO-III cells | RT-PCR siRNA Western blot analysis ChIP assay | TSA + 5-aza increased MUC4 expression in a cell-specific manner. MUC4 silencing was directly engaged by HDAC3, HDAC1, DNMT3B, and DNMT3A binding to its 5′-UTR in a cell-specific manner. Inhibited the histone deacetylation, associated with strong MUC4 repression in high-expressing cells | [227] |
| Psammaplin (Jaspis sp. and Poecillastra wondoensis) | Ishikawa cancer cells | RT-PCR Western blot analysis | Induced accumulation of acetylated histones and reduced HDAC levels. Upregulated cyclin-dependent kinase inhibitor expression. | [228] |
| Psammaplin (Pseudoceratina purpurea) | A549, MCF7, and W138 cells | HDAC Assay Immunoblotting | Inhibited HDAC1 activity. Exhibited significant cytotoxicity in A549, MCF7, and W138 cells. Increased acetylation of histones. | [229] |
| Psammaplin (Pseudoceratina purpurea) | In vitro enzymatic assays | Cell proliferation assessment HDAC enzyme activity | Inhibited HDAC effectively. Induced weak cytotoxicity. | [230] |
| TSA (Purchased) | MCF-7, MDA-MB-231, and MDA-MB-157 cells and mouse models | RT-PCR assay Western blot analysis ChIP assay | Reduced the activity of HDAC, either alone or in conjunction with GEN. | [197] |
| TSA (Purchased) | SKOV3 (Ovarian cells) | Western blot analysis Histone immunoblots assay | TSA + decitabine reduced the DNMT and HDAC activities. TSA + decitabine increased acetylation of histone H3 and H4. TSA + decitabine inhibited the expression of lysine-specific demethylase-1. TSA + decitabine increased the transcription activity marker dimethylated-H3K4, while suppressing dimethylated-H3K9. | [231] |
| Molecules (Origins) | Used Models | Methods | Key Findings | Ref. |
|---|---|---|---|---|
| Corosolic acid (Purchased) | TRAMP-C1 cells | RT-PCR DNA extraction HDAC activity assessment ChIP assay Western blot analysis | Increased the acetylation of histone H3 lysine 27 (H3K27ac). Reduced trimethylation of histone H3 lysine 27 (H3K27Me3) in the Nrf2 promoter region. Reduced the expression and activity of epigenetically modifying enzymes in TRAMP-C1 cells. | [232] |
| Cucurbitacin B (L. graveolense Roxb.) | NSCLC cells | RT-PCR Western blot analysis HDAC activity Immunohistochemical staining | Altered histone modifications. | [233] |
| Parthenolide (PTL) (Purchased) | ZR-75-1 and 293T cells | RT-PCR HDAC activity Western blot analysis ChIP assay | Depleted HDAC1 protein, without affecting Class I/II HDAC proteins. HDAC1 depletion and cell death were promoted by the mutant DNA-damage transducer ataxia telangiectasia. | [236] |
| Parthenolide (PTL) (Purchased) | AML cells | Methylcellulose colony-forming Western blot analysis Flow cytometry | HDAC induced apoptosis was prevented in multiple human AML cells. Increased HDAC-induced lethality. PTL + HDACis caused apoptosis in MLL-ENL cells. PTL + HDACis triggered SAPK/JNK activation. | [237] |
| Parthenolide (PTL) (Not reported) | JB6 cells | Western blot analysis ChIP method RT-PCR siRNA transfection HDAC activity | Modulated gene expression. | [238] |
| Parthenolide (PTL) (Purchased) | MDA-MB231 cell line | Cell viability assessment ROS and GSH analysis Western blot analysis | PTL + HDACi induced GSH depletion, Cyt c release, activation of caspase 3 and apoptosis. PTL + HDACi sustained both HDACi-induced hyperacetylation of histones H3 and H4 and PTL-induced downregulation of DNMT1 expression PTL + HDACi boosted PTL’s cytotoxic impact. | [239] |
| Ursolic acid (Purchased) | JB6 P+ cells | qPCR Western blot analysis HDAC activity assay | Reduced the expression of HDAC1, 2, 3, and 8 (Class I) enzymes and the activity of HDAC6 and 7 (Class II). | [240] |
| Ursolic acid (Purchased) | Male Sprague-Dawley rats | mRNA expression measurement qPCR Development of PK/PD model | Inhibited the induction of epigenetic markers in leukocytes. | [232] |
| Z-Ligustilide (LIG) (Purchased) | HS578t, MDA-MB-231 and MDA-MB-453 cells | Western blot analysis Luciferase assay ChIP assay Immunoprecipitation | Increased Ace-H3 (lys9/14) enrichment in the ER promoter. Downregulated the HDAC activity. | [241] |
| Molecules (Origins) | Used Models | Methods | Key Findings | Ref. |
|---|---|---|---|---|
| Butyrate (Purchased) | Eca9706 cells | MTT assay Immunoblotting Methylation-specific PCR | Inhibited the development of human oesophageal cancer cells. Reduced NF-Bp65, DNMT1, HDAC1, and Cyclin D1 reverse expression. Increased caspase-3 and p16INK4 expression. Butyrate + quercetin acted as HDAC inhibitors through epigenetic-NF-κB cascade signaling. | [223] |
| Butyrate (Purchased) | CRL-2577, HT-29, and HCT-116 cells | HDAC activity assessment CpG methylation ChIP assay | Inhibited survivin, DNMT1, and HDAC1. Affected chromatin structure and global DNA methylation. | [206] |
| Butyric acid (Purchased) | Female mice skin | Bisulfite modification of DNA RT-PCR assay | Decreased miR-203 expression. Upregulated the methylation of DNMT, HDAC, and miR-203. | [226] |
| Eicosapentaenoic acid (EPA) (Purchased) | McRH-7777 cells | Flow cytometry qRT-PCR Immunoblot analysis ChIP method | Reduced HDAC1 and DNMT activity and expression. Promoted tumor suppressor gene expression. Activated PPAR by inhibiting HDAC1 expression in hepatocarcinoma cells. Re-expressed the tumor suppressor gene Hic-1 in response to EPA-bound PPAR. | [243] |
| Molecules (Origins) | Used Models | Methods | Key Finding | Ref. |
|---|---|---|---|---|
| Isothiocyanate | ||||
| Phenethyl isothiocyanate (PEITC) (Not reported) | HCT116, SW480, and SW620 cells | Cell viability Western blot analysis Quantitative RT-PCR | Induced stable changes in the expression profile of epigenetic erasers/writers. Induced hypomethylation of PcG target genes, which are generally hypermethylated in cancer. | [244] |
| Phenethyl isothiocyanate (PEITC) (Purchased) | HT29 (Colon cells) | Determination of HDAC1 levels | PEITC + LA reduced the amount of aberrant crypt foci, as well as DNMT1 and HDAC1 levels (in vivo). | [245] |
| Sulforaphane (SFN) (Not reported) | MCF-7 and MDA-MB-231 | RT-PCR Western blot analysis HDAC and HAT activity Bisulfite sequencing analysis ChIP method | Inhibited hTERT in both cells. Reduced the trimethyl-H3K9 and trimethyl-H3K27 in active chromatin indicators in a dose-dependent manner. | [189] |
| Sulforaphane (SFN) (Purchased) | HeLa cells | DNMT and HDAC activity assessment Molecular docking study Bisulfite Modification and MSP RT-PCR | Inhibited the activity of HDAC. Lowered the HDAC1 expression. Reactivated RAR, CDH1, DAPK1, and GSTP1 genes or elevated their expression. | [207] |
| Sulforaphane (SFN) (Purchased) | A549 and H1299 (Lungs cells) | HDAC activity assay qRT-PCR Tumor xenografts | Inhibited the activity of HDAC in lung cancer cells. Increased histone (H4 and H3) acetylation. Suppressed the activity of HDAC in vivo. | [249] |
| Sulforaphane (SFN) (Purchased) | MCF-7 and MDA-MB-231 | HDACs activity assay | SFN + WA decreased HDAC expression. | [250] |
| Sulforaphane (SFN) (Purchased) | A549, HEK293, HT29 cells | Bisulfite genome sequencing RT-PCR Western blot analysis ChIP method HDAC and DNMT activity assessment | Reduced CpG methylation in the miR-9-3 promoter region. Increased H3K4me1 enrichment at the miR-9-3 promoter. Induced the expression of miR-9-3. Reduced the epigenetic modifying enzymes’ expression and activity. | [251] |
| Sulforaphane (SFN) (Purchased) | Breast cancer cells | RT-PCR Western blot analysis HDACs activity Global histone H3 acetylation quantification RNA sequencing analysis | Increased gene transcription in tumor suppressor genes. Decreased the expression of tumor promoting genes. Decreased HDAC1 gene expression and enzymatic activity. Increased levels of histone acetylation. | [252] |
| Sulforaphane (SFN) (Purchased) | HCT116 and RKO (Colon cells) | RT-PCR Western blot analysis HDAC activity | Reduced the oncogenic hTERT mRNA, HDAC, miR-21, protein, and enzyme levels. | [253] |
| Sulforaphane (SFN) (Purchased) | MCF-7 and MDA-MB-231 | RT-PCR Western blot analysis HDAC activity | SFN + GEN suppressed HDAC and HMT. SFN + GEN inhibited the expression of hTERT. | [214] |
| Sulforaphane (SFN) (Not reported) | HepG2 and GAS cells | RNA-Seq analysis | Inhibited HDAC activity Downregulated chromatin profile regulating enzymes. | [254] |
| Sulforaphane (SFN) (Purchased) | HepG2 cells | RNA extraction RNA-Seq analysis | Inhibited HDAC activity. Methylation of oncogenic TF binding site motifs affected its activity. | [249] |
| Quinones | ||||
| Laccaic acid (LA) (Purchased) | HT29 cells | HDAC activity assay | LA + PEITC reduced HDAC1 expression levels (in vivo). | [255] |
| Naphthazarin (Naph) (Purchased) | MCF-7 cells | RT-PCR Western blot analysis ChIP method | Activated the p21 promoter by Naph + IR, by inhibiting the binding of multi-domain proteins. | [256] |
| Thymoquinone (Purchased) | JK cell line and MDA-MB-468 cells | Gene analysis RT-PCR analysis | Downregulated numerous major epigenetic factors. | [257] |
| Carotenoid | ||||
| Astaxanthin (Purchased) | LNCaP cells | Cell viability test HDAC activity assay | Inhibited HDAC activity in vitro. | [258] |
| Stilbenes | ||||
| Resveratrol (RSV) (Purchased) | MDA MB 231 and MCF7 | Bisulfite conversion RT-PCR Western blot analysis | Decreased HDAC1 protein levels. | |
| Resveratrol (RSV) (Purchased) | HeLa, SiHa, and Caski cervical cells | Promoter methylation analysis Transient silencing of UHRF1 | Induced PAX1 reactivation via its effect on HDAC, which is mediated through the downregulation of UHRF1, which may affect both histone acetylation and DNA methylation. | [259] |
| Resveratrol (RSV) (Not reported) | B16F10 and Tumor-bearing mouse model | Western blot analysis Luciferase reporter assays Bisulfite sequencing PCR ChIP assay | HDAC1 was recruited to the focal adhesion kinase (FAK) promoter region. | [260] |
| Resveratrol (RSV) (Purchased) | MDA-MB-231 and MCF-7 cells | MTT assay Clonogenic method HDAC activity | Reduced HDAC activity. | [261] |
| Curcumin (Cur) (Purchased) | Hep3B cells | Histone purification Histone acetylation assay Western blot analysis HAT assay HDAC assay RT-PCR | Inhibited histone acetylation in the presence or absence of TSA. Inhibited the activity of HAT both in vitro and in vivo. Induced histone hypoacetylation in vivo. | [262] |
| Curcumin (Cur) (Purchased) | Raji cells | Immunocytochemistry analysis Western blot analysis | Reduced HDAC1, HDAC3, and HDAC8 protein expression levels. Increased Ac-histone H4 protein expression. | [263] |
| Curcumin (Cur) (Purchased) | Raji cells | MTT assay Western blot analysis RT-PCR | Decreased the amounts of HDAC3, HDAC1, and p300. Reversed the protection degradation of p300 and HDAC1 by MG-132. | [264] |
| Curcumin (Cur) (Purchased) | Hela cells | HDAC activity assessment | Inhibited the HDAC activity (IC50 = 115 μM). | [265] |
| Curcumin (Cur) (Purchased) | DAOY, D283 Med, and D341 Med (medulloblastoma) | HDAC activity In vivo medulloblastoma xenografts | Reduced the HDAC4 activity and expression. Increased tubulin acetylation. | [266] |
| Curcumin (Cur) (Purchased) | LNCaP cells | Western blot analysis DNA extraction and bisulfite genomic sequencing PCR array RNA isolation and RT-PCR ChIP assay Histone H3 methylated lys27 ELISA HDAC activity assay | Altered HDAC expression. Increased the expression of HDAC1, 4, 5, and 8, but decreased the HDAC3. Decreased HDAC activity. Decreased H3K27me3 enrichment at the Neurog1 promoter region and at the global level. | [267] |
| Curcumin (Cur) (Purchased) | MCF7 and MDA MB 231 | Bisulfite conversion and MSP RT-PCR Western blot analysis | Decreased protein levels of HDAC1. | |
| Curcumin (Cur) (Purchased) | HeLa, SiHa, and Caski cells | MTT assay Quantitative expression of PAX1, UHRF1, and p21 PAX1 promoter methylation analysis Transient silencing of UHRF1 | Induced reactivation of PAX1, due to its effect on HDAC. | [259] |
| Calebin-A (Not reported) | MPNST cells | Cell proliferation Western blot analysis Flow cytometry ChIP assay | Reduced the expression of acetyl H3 proteins. | [268] |
| Pterostilbene (PTS) (Purchased) | MDA-MB-157, HCC1806, and MCF10A | RT-PCR Western blot analysis SIRT activity assay DNMT activity | RSV + PTS downregulated the SIRT1 (a type III HDAC). | [269] |
| Steroids | ||||
| Guggulsterone (Purchased) | Breast cancer cells | Bisulfite conversion and MSP RT-PCR Western blot analysis | Decreased protein levels of HDAC1. | |
| Withaferin A (WA) (Purchased) | Breast cancer cells | Bisulfite conversion and MSP RT-PCR Western blot analysis | Decreased HDAC1 and MeCP2 protein levels. | |
| Withaferin A (WA) (Purchased) | Breast cancer cells | qRT-PCR Western blot analysis HDAC activity | WA + SFN decreased the HDAC activity and expression. | [250] |
| Phenolic acids | ||||
| Chlorogenic acid (Purchased) | Breast cancer cells | In vitro HDAC inhibition | Inhibited the HDAC activity (IC50 = 375 μM). | [265] |
| Caffeic acid (Purchased) | Breast cancer cells | In vitro HDAC inhibition | Moderately inhibited the HDAC activity (IC50 = 2.54 mM). | [265] |
| Secoiridoids | ||||
| Oleacein (Olea europaea L.) | U266, NCI-H929, RPMI-8226, MM1s, and JJN3 cells | Western blot analysis qRT-PCR HDAC activity | Accumulated α-tubulin and acetylated histones. Downregulated multiple Class I/II HDACs. Inhibited HDACs with downregulation of Sp1. | [270] |
| Tannins | ||||
| Ellagic acid (EA) (Purchased) | hASCs cells | qPCR Western blot analysis HDAC activity Immunocytochemistry of H3R17me2 and HDAC9 | Inhibited HDAC 9 downregulation. Reduced the histone acetylation levels. | [271] |
| Tanshinone | ||||
| Tanshinone IIA (Purchased) | HepG2-C8, JB6 P+, JB6-shNrf2 cells | Luciferase reporter activity assay RNA extraction and qRT-PCR Western blot analysis HDAC activity Bisulfite sequencing ChIP method | Inhibited HDAC activity. | [272] |
| Others natural compounds | ||||
| Arsenic trioxide (AS2O3) (Purchased) | HeLa or HEK293T cells | Luciferase reporter assay RT-PCR ChIP method Flow cytometry | Reduced global histone through direct binding. Increased HDAC4 expression. Inhibited the hMOF activity. | [273] |
| Calcium glucarate (CAG) (Purchased) | Female mice cells | RT-PCR Bisulfite sequencing assay Western blot analysis | Downregulated the levels of miR-203 Upregulated DNMT, HDAC, and promoter methylation of miR-203. | [226] |
| Proanthocyanidins (GSPs) | Breast cancer cells | Western blot analysis HDAC activity | Reduced HDAC activity. | [261] |
| Curcumol (Not reported) | CSLCs cells | qRT-PCR Western blot analysis Global DNA methylation HDAC activity Xenograft tumorigenicity assay Immunohistochemistry | Blocked DNMT/HDAC activity and CSLC self-renewal in vivo and in vitro. Affected CLSCs by HDAC regulation. | [274] |
| Selenium (Se) (Not reported) | LNCaP cells | HDAC activity | Reduced HDAC activity. No alteration of mRNA and protein levels of HDACs. Reduced methylated histone H3 on lysine 9 (H3-K9) levels. Increased acetylated H3-K9 levels. | [275] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouyahya, A.; El Hachlafi, N.; Aanniz, T.; Bourais, I.; Mechchate, H.; Benali, T.; Shariati, M.A.; Burkov, P.; Lorenzo, J.M.; Wilairatana, P.; et al. Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates. Molecules 2022, 27, 2568. https://doi.org/10.3390/molecules27082568
Bouyahya A, El Hachlafi N, Aanniz T, Bourais I, Mechchate H, Benali T, Shariati MA, Burkov P, Lorenzo JM, Wilairatana P, et al. Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates. Molecules. 2022; 27(8):2568. https://doi.org/10.3390/molecules27082568
Chicago/Turabian StyleBouyahya, Abdelhakim, Naoufal El Hachlafi, Tarik Aanniz, Ilhame Bourais, Hamza Mechchate, Taoufiq Benali, Mohammad Ali Shariati, Pavel Burkov, José M. Lorenzo, Polrat Wilairatana, and et al. 2022. "Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates" Molecules 27, no. 8: 2568. https://doi.org/10.3390/molecules27082568