
Carbonic Anhydrase Activators for Neurodegeneration: An Overview
by
 , , and
, , and
Valeria Poggetti
, 
Silvia Salerno
*,
Emma Baglini
,
Elisabetta Barresi
,
Federico Da Settimo
and
Sabrina Taliani
Department of Pharmacy, University of Pisa, via Bonanno 6, 56126 Pisa, Italy
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(8), 2544; https://doi.org/10.3390/molecules27082544
Submission received: 15 March 2022
/
Revised: 8 April 2022
/
Accepted: 12 April 2022
/
Published: 14 April 2022
(This article belongs to the Special Issue Carbonic Anhydrases-Chemistry and Biomedical Applications)
Abstract
:Carbonic anhydrases (CAs) are a family of ubiquitous metal enzymes catalyzing the reversible conversion of CO2 and H2O to HCO3− with the release of a proton. They play an important role in pH regulation and in the balance of body fluids and are involved in several functions such as homeostasis regulation and cellular respiration. For these reasons, they have been studied as targets for the development of agents for treating several pathologies. CA inhibitors have been used in therapy for a long time, especially as diuretics and for the treatment of glaucoma, and are being investigated for application in other pathologies including obesity, cancer, and epilepsy. On the contrary, CAs activators are still poorly studied. They are proposed to act as additional (other than histidine) proton shuttles in the rate-limiting step of the CA catalytic cycle, which is the generation of the active hydroxylated enzyme. Recent studies highlight the involvement of CAs activation in brain processes essential for the transmission of neuronal signals, suggesting CAs activation might represent a potential therapeutic approach for the treatment of Alzheimer’s disease and other conditions characterized by memory impairment and cognitive problems. Actually, some compounds able to activate CAs have been identified and proposed to potentially resolve problems related to neurodegeneration. This review reports on the primary literature regarding the potential of CA activators for treating neurodegeneration-related diseases.
1. Introduction
Carbonic anhydrases (CAs) are a superfamily of metalloenzymes that mainly catalyze the interconversion between CO2 and bicarbonate (CO2 + H2O ⇋ HCO3− + H+) by using a metal hydroxide nucleophilic mechanism [1].
These enzymes are ubiquitous and exist both in eukaryotic and prokaryotic organisms. Coding genetic families have a different evolution, and they are: α-CA (vertebrates, bacteria, algae, cytoplasm of green plants), β-CA (bacteria, algae, chloroplasts of dicotyledons and monocotyledons), γ-CA (archaea and other bacteria), δ-CA (some marine diatoms), ζ-CA (some marine diatoms), η-CA (Plasmodium spp.), θ-CA (marine diatom Phaeodactylum tricornutum), and ι-CA (diatom Thalassiosira pseudonana, algae, bacteria, and archaea) [2,3,4,5].
In vertebrates we can find α-CAs and, to date, in human beings, 15 isoforms have been identified with different catalytic activities, subcellular localizations, and tissue distributions: hCA I, II, III, VII, and XIII are cytosolic; hCA IV, IX, XII, and XIV are membrane-bound; hCA VA, and VB are mitochondrial; hCA VI is secreted in saliva and tears; and hCA IX, XII, and XIV are transmembranal. hCA VIII, X, and XI are acatalytic and preponderantly expressed within the brain [3].
The main differences among these isoforms of α-CA are associated with secondary and tertiary organization of the apoprotein, which delineate physical/chemical characteristics, but the active site remains almost unchanged [4,6].
Since CAs catalyze the hydration reaction of CO2, they are involved in several physiological processes related to cellular respiration and to the transfer of CO2 with HCO3− through tissues of metabolization and lungs. They also regulate pH levels; CO2 homeostasis; the secretion of electrolytes in several tissues and organs; a number of biosynthesis processes such as gluconeogenesis, lipogenesis, and ureagenesis; bone resorption; and calcification [3].
In addition to physiological functions, CAs are also involved in pathogenetic processes such as carcinogenesis, obesity, and epilepsy; for this reason, they are considered valid targets to develop potential drugs for the treatment or prevention of these pathologies [2,7].
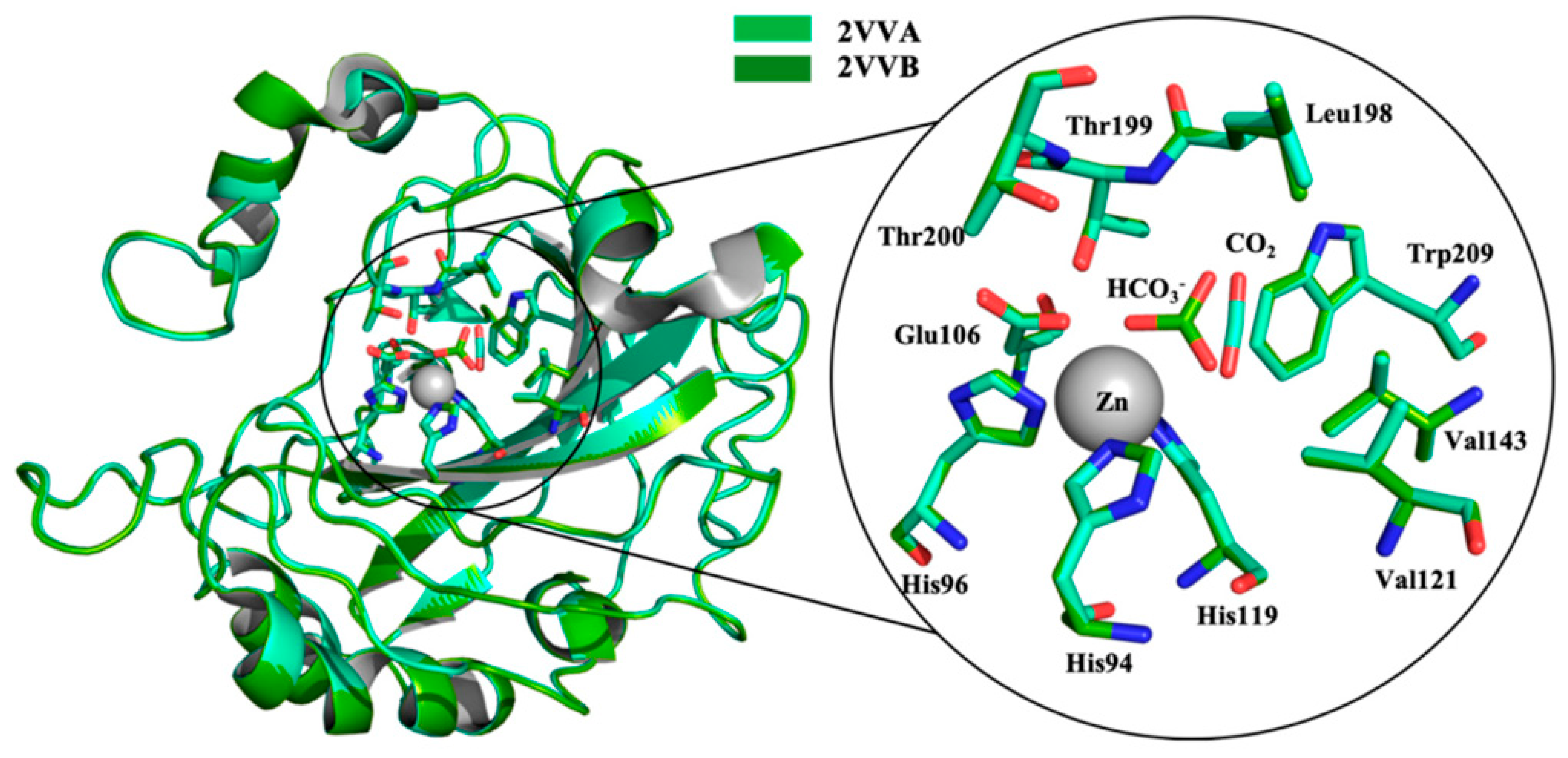
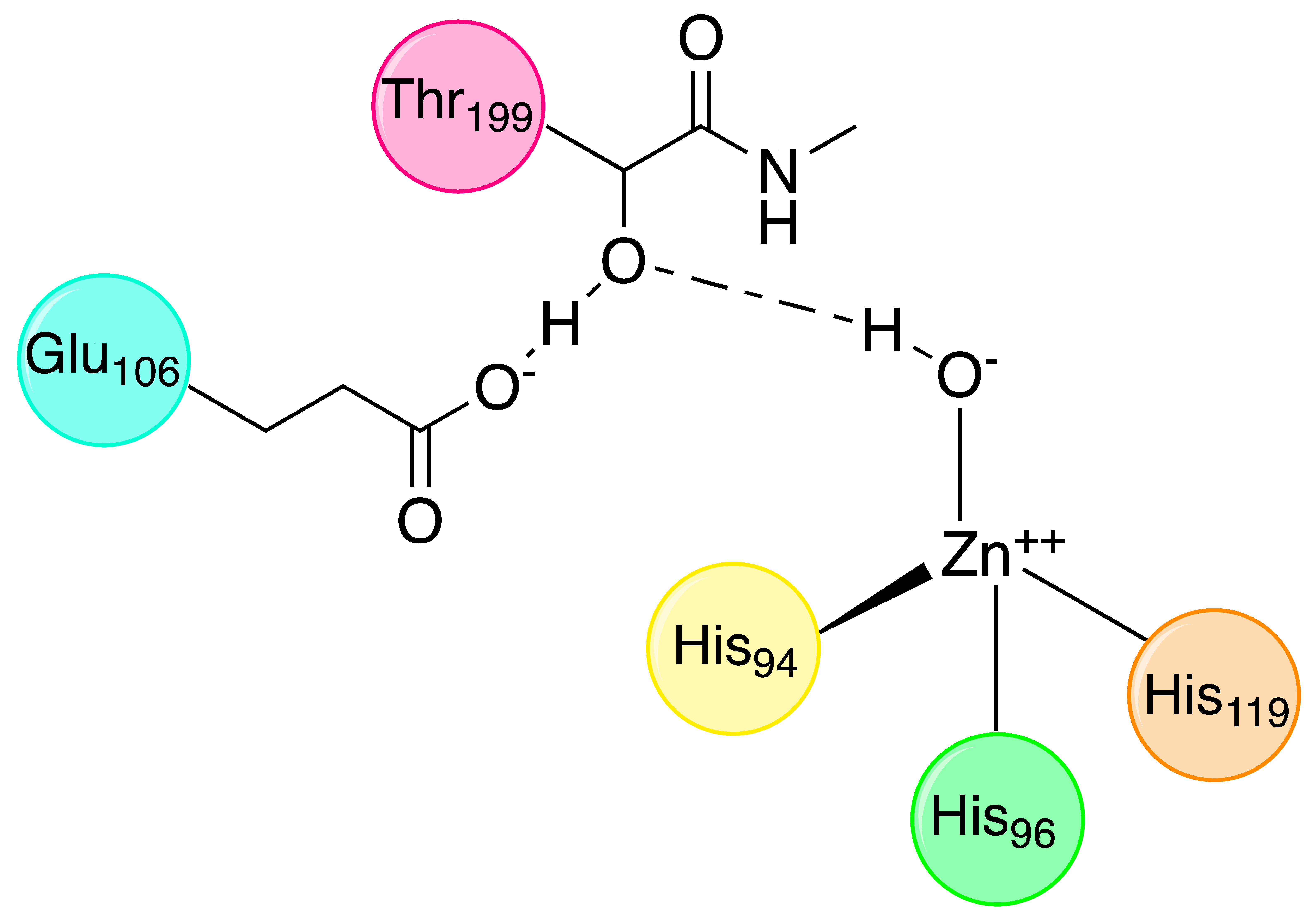
The active site of α-CAs is a hydrophobic pocket 15 Å in depth, constituted by three main components (Figure 1): the metallic (II) ion arranged in a tetrahedral geometry, which is represented by the Zn2+ ion situated at the bottom of the cavity; three histidine residues, His94, His96, and His119, that bind the metal ion thanks to their imidazole moiety; and a water molecule in the inactive state of the enzyme, which loses a proton to produce the hydroxylated state of the enzyme, which represents the active form [8].
The water molecule bound to Zn(II) is also involved in a hydrogen bond interaction with the oxygen of the hydroxyl group of Thr199, which is in turn engaged in another hydrogen bond interaction with the oxygen of the carboxylate group of Glu106 (Figure 2).
These interactions direct the substrate, represented by CO2, in a favourable position for the nucleophilic attack by the zinc-bound hydroxide, deriving from water, which loses a proton and increases its nucleophilicity [10].
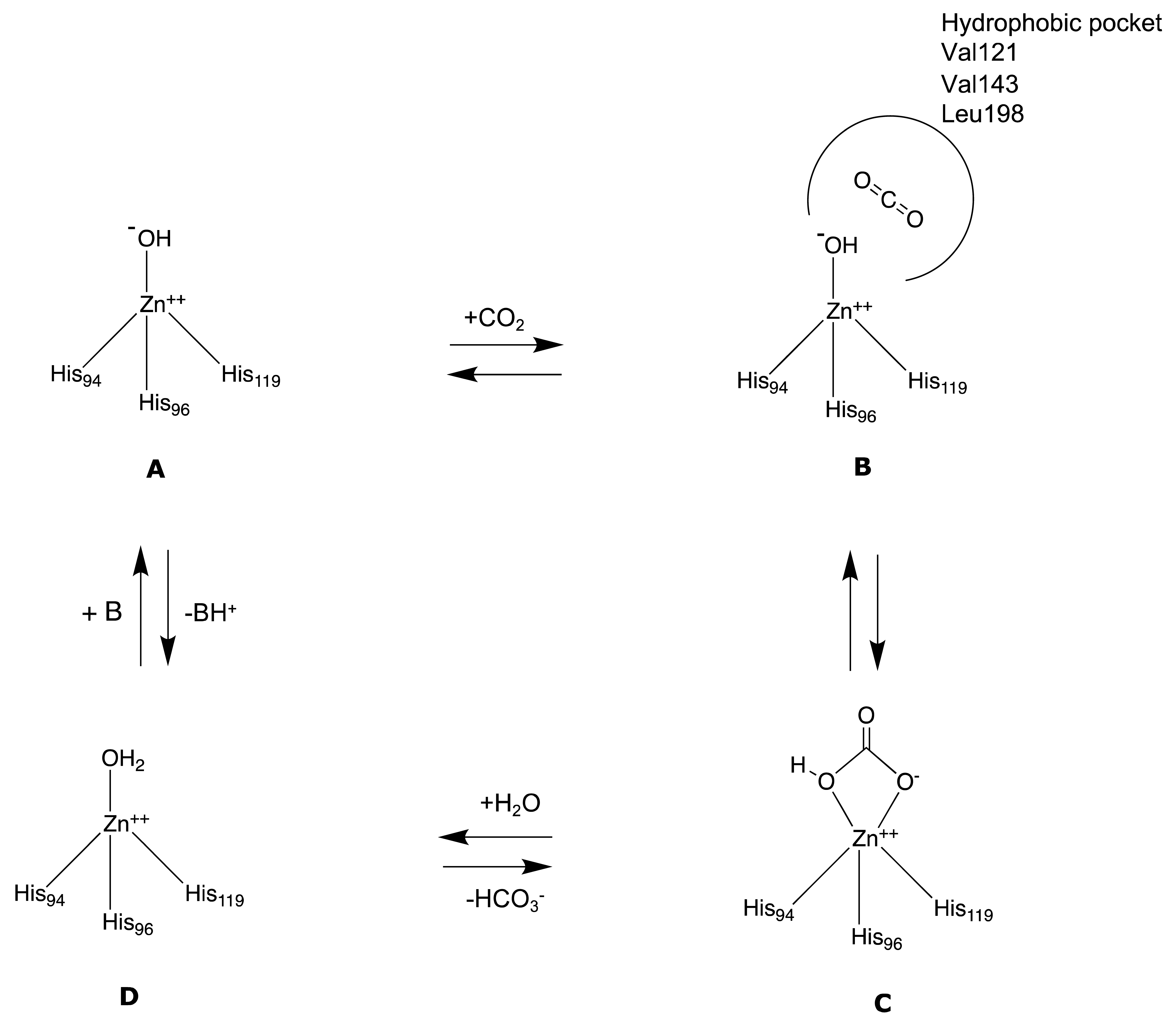
The inactive state of the enzyme is constituted by a water molecule bound to the Zn (II) (Figure 3D). In the first step, the water molecule loses a proton and transforms itself into a hydroxylated species (Figure 3A), which is the active one. The hydroxyl ion acts as a strong nucleophile on the CO2 molecule (Figure 3B), which is situated in a nearby hydrophobic pocket, leading to the formation of a species in which the bicarbonate ion is coordinated to the metal ion (Figure 3C). Subsequently, the bicarbonate ion is displaced from the Zn (II) and released in solution by an incoming water molecule, which rebuilds the inactive and acid form of the enzyme (Figure 3D), and the catalytic cycle can restart [11]. The rate-limiting step in the activation process is the proton transfer from the water molecule bound to the Zn (II) to the surrounding environment, namely the proton shuttling. In the native state of the enzyme, this reaction takes place thanks to the imidazole and basic moiety of His64 (mainly in hCA I, II, IV, V, VII, and IX), which behaves like a proton shuttle because it has a pKa of around 7 [11].
The catalytic cycle of the enzyme can be represented by these reactions [2]:
EZn2+—OH− + CO2 ⇆ EZn2+—HCO3− ⇆ EZn2+— OH2 + HCO3−
EZn2+—OH2 ⇆ EZn2+—OH− + H+
Carbonic anhydrases activators (CAAs) are compounds able to behave as additional shuttles towards the His residue, transferring the proton and activating CAs even more.
1.1. Carbonic Anhydrases Activation
CAs activation is a little-explored field compared to CAs inhibition. While CAs inhibitors (CAIs) have been in clinical use for many decades and are already used in therapy for the treatment of some pathologies such as glaucoma [12], epilepsy [13], obesity [14], CAAs, to date, have not shown relevant pharmacological applications and are still under study.
The general mechanism that leads to CAs activation was suggested in 1990 and it can be schematized by the following reactions [3]:
EZn2+—OH2 + A ⇆ [EZn2+—OH2—A]
[EZn2+—OH2—A] ⇆ [EZn2+—HO−—AH+]
[EZn2+—HO−—AH+] ⇆ EZn2+—HO− + AH+
The activator binds to the enzyme in the active site and then, together, they constitute the enzyme–activator complex. Kinetic analysis demonstrated that the activator, once bound to the enzyme, does not compromise the affinity for the substrate (KM), having an impact only on Kcat. Studies on Co (II)-substituted CAs complexed with the activator molecule established that there are no changes in the electronic spectra of complexes, similarly to the pure enzyme, highlighting that the activator does not bind to the metal ion, but to a different site. X-ray crystallographic data evidenced His64, fundamental for its proton-shuttling role, in two different orientations in the hCA II active site, far away from the Zn ion. The first is called “in” conformation in which His64 points toward the metal ion, while the second is named “out” conformation in which His64 points toward the exit of the active site. X-ray crystallographic data, related to the CAA histamine, also demonstrated that, while CAIs bind to the Zn (II), CAAs bind at the entrance of the cavity of the active site, far away from the metal ion. Moreover, activator’s binding site does not overlap with the region in which His64 is located [3].
The CAA is anchored to Gln92 thanks to H-bonds on the side chains of Asn62, Asn67, and Gln92, and to the water molecule, and this leads to a rearrangement of the H-bond network in the active site. In this way, the activator is ready to participate in the rate-limiting step of the catalytic cycle, working as an additional proton shuttle towards His64. In other words, together the activator molecule and His64 lead to the formation of the nucleophilic and active species of the enzyme in which a hydroxyl ion is coordinated to the metal ion instead of the water molecule [8].
1.2. Potential Therapeutic Applications of CAAs
Recent findings reported an involvement of CAs in cognitive and memory disorders, suggesting that the activation of these enzymes may represent a potential effective strategy for the strengthening of synaptic efficacy [15,16] and the development of agents useful for the treatment of neurodegenerative pathologies such as Alzheimer’s disease.
In particular, cerebral isoforms of CAs (I, II, IV, VA, VII, IX, XII, and XIV) may represent a target for an unexplored field to develop new drugs in psychiatric or neurodegenerative disorders.
This hypothesis arises from the observation that CAs activation increases spatial memory in rats [15]. The administration of acetazolamide (a well-known non-selective CAI) to CD1 mice reduces CAs activity in the brain and causes amnesia in recognizing objects (OR), while the treatment with D-phenylalanine (CAA) that unselectively increases CAs activity, strengthens OR memory by activating extracellular signal-regulated kinase (ERK) [17].
Sun and Alkon reported that following the administration of phenylalanine to experimental animals, a remarkable pharmacological improvement of synaptic efficacy, spatial learning, and memory, was produced, demonstrating that CAAs could be used for managing particular conditions in which learning and memory are compromised [15,18].
In addition, it has been proven that the levels of different cerebral isoforms of α-CA (mainly CA I and II) are significantly reduced in patients with Alzheimer’s disease.
Other studies revealed that CA I and II resulted in being oxidized and having a reduced catalytic activity in the frontal cortex and hippocampus of patients with Alzheimer’s disease. Consequently, the CA I and II catalytic activity dysfunction leads to imbalances of intracellular and extracellular pH levels, triggering protein aggregation and contributing to the progression of the disease [19,20]. Very recently, also the idea of potentially repurposing CAIs for the prevention of cerebrovascular and neurovascular pathology in AD and stroke was proposed [21].
Agents able to repristinate the catalytic activity of these isoforms or to increase other cerebral isoforms (such as CA VA, CA VB, and CA VII) could represent innovative approaches for the treatment of Alzheimer’s disease.
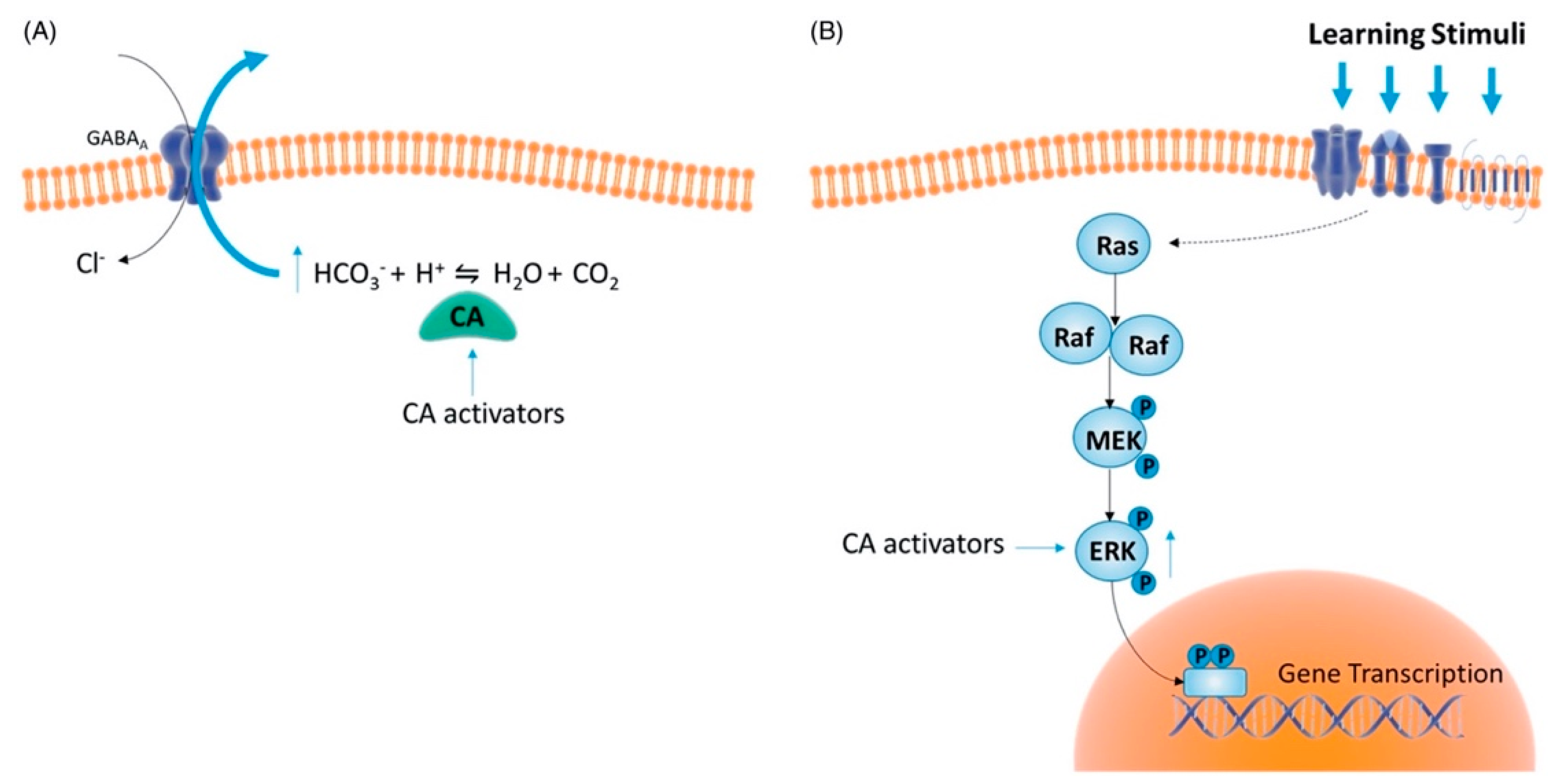
As discussed above, CAs play an important role in several physiological functions, including the regulation of pH levels in the neurons and in the extracellular space through the regulation of ionic gradients of bicarbonate ion. Specifically, CAs contribute to the availability of protons and bicarbonate ions, which is necessary for the transmission of neuronal signalling. This in turn influences the purpose of proton-sensitive membrane proteins, regulating the kinetics and the concentration of pH transition into intra- and extracellular compartments [22,23]. These proteins are gamma aminobutyric acid agonist receptors (GABAARs) [24], N-methyl-D-aspartate receptors (NDMA), and ionic channels [25].
It has been demonstrated that into rats’ hippocampus, the excitation mediated by GABAAR depends on HCO3− concentration, which is regulated by the cytosolic activity of CAs and suppressed by inhibitors [26].
Information processing and memory storage need a synchronized neuronal activity, commonly known as hippocampal theta rhythm, which was demonstrated to be associated to the GABAergic postsynaptic depolarization into pyramidal cells of adult rats’ hippocampus, with an inverted potential from Cl− to HCO3−, and this process is regulated by CAs. Moreover, it has been shown that theta rhythm is inhibited also by CAs inhibitors and that CAs inhibition compromises rats’ spatial learning, but it does not influence other sensory or locomotor behaviours. This evidence implicates that CAs activity has effects both on theta rhythm and on memory consolidation through the signalling transmission mediated by HCO3− [26].
Moreover, there are two putative mechanisms underlying CAs actions on cognition. The first regards GABA and the second regards ERK pathways.
CAAs are reported to increase the efficacy of temporal activity of cholinergic and GABAergic inputs, transforming the hyperpolarizing GABAergic postsynaptic potential (PSP) from inhibitory (IPSP) to excitatory (EPSP), because they reduce intracellular concentrations of HCO3−, favouring its outflow through the channel receptor GABAA (Figure 4A). Thus, the regulation of ionic gradients has several effects on post-synaptic depolarization with benefits of increasing memory and learning [27]. Becoming excitatory, the GABAergic postsynaptic potential potentiates the signal transfer thanks to the activity of CAs, which act as a gate.
On the other hand, a study conducted on CD1 mice demonstrated that D-phenylalanine increases learning thanks to the activation of extracellular pathways of extracellular-signal-regulated kinase (ERK) into the cortex and hippocampus. This kinase is involved in the building of memory, in both the cortex and hippocampus, which are two very important cerebral areas for memory elaboration, especially for long-term memory (Figure 4B). In fact, the activation of ERK pathways in these two cerebral districts leads to an increased genomic response and memory encoding due to structural synaptic variations [17].
In a recent study, P. Blandina et al. (2020) [28] demonstrated, following the treatment with D-phenylalanine, an increased phosphorylation of ERK (1 and 2) in hippocampal and cortical cell homogenates, and these results are in agreement with previous studies showing that ERK phosphorylation in the amygdala was also inhibited by the treatment with acetazolamide [29].
After phosphorylation, ERK pathway induces a genomic response, which is very important for the consolidation of long-term memory. Indeed, this is related to the transformation of short-term learning into long-lasting memory in which CAs play a pivotal role. This study also demonstrated that the administration of acetazolamide causes amnesia during a non-spatial memory test, confirming that CAs are modulators of learning and memory [17]. Based on these findings, a number of CAAs are under study as potential candidates for the development of new drugs for the treatment of neurodegenerative diseases, in particular those related to memory disorders [17].
Actually, even if the hydration reaction of CO2 can occur without a catalyst, anyhow it is too slow at physiological levels of pH and it cannot satisfy metabolic demand, making the catalytic activity of CAs indispensable. The catalytic cycle mentioned above demonstrates that the active state of the enzyme is the basic and hydroxylated one, and it derives from the inactive and acid form. The transfer of the proton from the water molecule to the surrounding environment is the rate-limiting step of the entire process of activation and it takes place thanks to a basic group (imidazole moiety of His64). From this evidence, it was demonstrated that a molecule able to participate in the proton transfer, acting as an additional proton shuttle towards His64, may contribute to activate the enzyme. In this respect, CAAs must feature two main structural requirements: be a small molecule suitable for the active site, and possess at least a protonatable basic moiety with pKa values in the range of 6.5–8.0 for participating in the proton transfer [4].
In addition, also tissue engineering is a field in which CAs activation takes place. As already known, carbonate deposition in animals such as mollusk shells depends on bicarbonate formation catalyzed by CAs [30,31].
In fact, Muller et al., studying human osteogenic SaOS-2 cells, demonstrated that following their exposition to calcium bicarbonate in vitro, there was an increased Ca-deposit formation and also an amplified upregulation of hCA II gene expression [31].
Calcium carbonate formation is an important step in the biomineralization and bone formation processes since it behaves as a bioseed for the precipitation of calcium phosphate, and it has been shown that in sponges the presence of CAAs leads to an increased formation of the first one [30]. These findings paved the way to an additional application of CAA that resides in the bone formation process.
2. Carbonic Anhydrase Activators (CAAs)
In this chapter, the activators of carbonic anhydrase (CAAs) reported so far in the literature are described and classified, focusing the attention on their chemical structures, updating previously published reviews [3,4].
Of note, CAAs biological profiles on the various CA isoforms are discussed with particular attention to those compounds able to behave as effective CAAs in vitro and that therefore could be interesting for animal studies regarding their involvement in cognitive processes. When possible, structure–activity relationships (SARs) are also discussed.
2.1. Amino Acids and Amines
Since an important structural requirement to activate CAs is a basic protonatable moiety as a mimic of the proton shuttling residue, the first molecules studied as CAAs were amino acids and their related amines.
As already discussed, the first X-ray crystallographic data on the complex between hCA II and the activator histamine [32] demonstrated and confirmed the theory that the activator-binding site is situated far away from the Zn(II) ion, reaching the edge of the cavity of the active site in the same region occupied by the two conformations of His64 [3].
In this regard, other compounds such as D- and L-phenylalanine [33], D- and L-histidine [34], D-tryptophan [35], and L-adrenaline [36] have been studied as activators, and they all bound to hCA II in a region called activator-binding site A [3], which is located on the opposite part of the region occupied by His64.
There is only one exception that regards D-Trp, which, in addition to activate binding site A, also binds to a region called activator-binding site B, which is further out with respect to A [35].
Subsequently, several amino acids and also some related natural and synthetic amines have been studied [37] as CA activators, and the most interesting data were collected and reviewed by Supuran in 2018 [3] and by Angeli et al., in 2021 [4]. In particular, amino acid compounds 1–6 and also some amines 7–14 (Table 1) have been tested on 13 catalytically active human CAs isoforms: hCA I–VII, IX, and XII–XV, and their activation data were expressed as activation constants (KA).
In Table 1, the most interesting results are reported.
From a general point of view, all compounds 1–14 activate all tested hCA isoforms with diverse profiles showing KA values ranging from the low nanomolar (9.0 nM of 12 for hCA IX) to the high micromolar (86 μM of D-Phe 1 for hCA I) range.
Some isoforms are better activated by amino acids, rather than amines. For example, hCA I is better activated by D-Phe 1, D-His and L-His 2 with KA 30–90 nM, compared to histamine 7, which activates hCA I with KA 2.1 μM [3].
Moreover, some amino acids are characterized by a high eudysmic ratio, for example L-Phe 1 is more active than D-Phe 1 on hCA I with KA values of 70 nM and 86 μM, respectively. Conversely, D-Phe 1 is more active on hCA VB (KA 70 nM) and on hCA XIII (KA 50 nM) with respect to L-Phe 1 (KA values of 10.45 μM and 1.02 μM, respectively).
hCA XIII isoform is also well activated by D-Trp 4 with KA 0.81 μM and by D-DOPA 3 with KA 0.81 μM, while L-DOPA 3 is endowed with a great affinity for hCA VA and VB (KA values of 36 nM and 63 nM, respectively) [4].
This reveals that also small structural differences comprising the enantiomeric form can lead to a more or less strong activation. In fact, after binding with the enzyme, the activator interacts in several ways and with different amino acids residues and water molecules of the binding site. These interactions can be both favorable and unfavorable depending on the residues involved and this leads to a better or a worse activation of the different isoforms [3].
Interestingly, D- and L-His 2 are the only amino acids that strongly activate hCA VII (KA values of 0.71 μM and 0.92 μM, respectively), the predominantly cerebral isoform that is activated also by D- and L-DOPA 3 and neurotransmitters such as dopamine 8 and serotonin 9. These neurotransmitters, as well as histamine 7, showed also to potently activate the mitochondrial isoforms, hCA VA, which are also present in the nervous tissues [4].
Some biogenic amines maintain the same activating features of their amino acidic precursors (serotonine—Trp), but some others change them (histamine—His). This happens because for some amino acids like His, the carboxylic group is necessary for strengthening the ability to activate CAs [4].
From the analysis of the data reported so far for compounds 1–14 [3,4], it is clear that none of the amino acids 1–6 or amines 7–14 are isoform-selective CAA.
Anyhow, a certain selectivity is observed for example for histamine 7, which shows a KA value of 10 nM towards the mitochondrial isoform hCA VA and the transmembrane one hCA XIV, while it results as a moderate activator (KA values in the micromolar range) for all the other CA isoforms.
The most interesting compound from the point of view of neurodegeneration, however, is histidine 2, which activates the isoforms most expressed in the brain, namely hCA I, VA, and VII.
2.2. Histamine-Based Compounds
Histamine 7 was one of the first CAAs that was studied, and it was used as a lead compound for the development of novel and more potent CAAs.
Over the years, several modifications have been made to the structure of histamine for the development of CAAs.
These changes can be schematically classified as follows: replacement of the imidazole ring with other heterocycles, derivatization of the primary amino group, insertion of halogens, and insertion of another imidazole ring. Subsequently, several classes of histamine-inspired compounds were developed.
2.2.1. Replacement of the Imidazole Ring
In one of the first studies conducted by Supuran et al. in 1996, the imidazole ring of histamine was replaced with several heterocycles like 2,4,6-trisubstitued pyridinium (15), 1,3,4-thiazole (16), or both (17) (Table 2) [38].
The compounds of series 15–17 showed to be efficient hCA II activators at 10 μM concentration, displaying activation rates between 147 and 184%.
2.2.2. Derivatization of the Primary Amino Group
In the following years, some X-ray crystallographic studies demonstrated that the amino group of histamine does not interact with the enzyme, so this moiety was substituted with different groups in order to obtain compounds such as carboxamides 18, ureas or thioureas 19 [39], sulfonamides 20, arylsulfonylureido derivatives 21 [40], and histamine dimers comprising polyaminoacyl moieties like EDTA 22 [39] and aminoacyl histamines 23 [41] (Table 2). All these compounds showed to efficiently activate hCA I, II (h = human), and bCA IV (b = bovine), with KA values in the low nanomolar-low micromolar range, as reported in Table 2 [4].
In particular, for compounds 18, 19, and 22, an efficient activation was observed towards all three isozymes, but especially for hCA I and bCA IV, with the best performing compounds displaying KA values in the nanomolar range. Anyhow, some of them also showed to activate hCA II with KA values around 10–25 nM [39]. The same trend was also observed for aminoacyl histamines 23 [41].
Sulfonamides 20 activated all three isozymes. The hCA I showed to be particularly sensitive (KA 6.0 nM–0.28 μM). The arylsulfonylureido derivatives 21 were the best CA activators of these series, with KA values in the 3–6 nM range for hCA I, 8–150 nM for hCA II, and 10–30 nM for bCA IV, thus being 1500 times stronger hCA II activators compared to histamine, although they are non-selective [40].
These series of CAAs might lead to the development of drugs/diagnostic agents for genetic disease of bone, kidneys, and above all, brain, since they mainly activate the CA isoforms most expressed in these compartments (namely hCA I, hCA II, and hCA VII) [39,40,41].
In these studies, it was shown that the derivatization of the aminoethyl moiety that does not interact with the enzyme is useful to make it point outwards to the active site or to increase the stabilization of the activator–enzyme complex. Based on the earlier study on the ureido histamine derivatives [39], Licsandru et al. [11] decorated the aminoethyl chain of histamine and synthetized ureido 19 (X = O) and bis-ureido 24 (Table 2) derivatives featuring alkylof different length, cycloalkyl, and aryl moieties at R, R′.
Both new ureido- 19 (X = O) and diureido-derivative 24 do not activate hCA II as their KA values are > 200 μM, but they moderately activate hCA I with KA values ranging between 0.73 and 3.4 μM. In particular, the best hCA I activator 24a (Table 2) (KA 0.73 μM, 2.74 times more active than histamine) belongs to diureido derivatives of series 24 [11].
In general, ureido derivatives are worse hCA I activators than bisureido derivatives, and this is probably due to the long alkyl chain that is unfavorable for the binding of these compounds at the entrance of the active site due to its hydrophilic nature [1].
The data reported by Licsandru et al. [11] show a different ability of the compounds investigated to activate two distinct isoforms, hCA I and II. This undoubtedly depends on the nature of the entrance of the active site cavity: The one of hCA II is more hydrophilic than the hCA I one, which is characterized by the presence of six histidine residues. In this view, the lipophilic nature of compounds 19 (X = O) and 24 prevents the binding to the active site, and thus, the activation of the hCA II isoform while favouring that of hCA I.
In 2011, Dave et al. derivatized histamine with pyridinium salt to obtain compound 25 (Table 2), which was crystallized in complex with hCA II, showing remarkable binding (hCA II 156% activation rate at 20 μM) [42].
On the basis of this interesting study, compound 25 was further functionalized and new pyridinium histamine derivatives 26 (Table 2) were developed [43].
Compounds of general formula 26 were investigated as activators of three cytosolic isoforms, hCA I, II, and VII, which are widely expressed in the brain, and they displayed activities ranging from the subnanomolar to the micromolar range.
While histamine 7 acts towards hCA I as a low micromolar activator (KA 2 μM), its substituted pyridinium derivatives 26 show a range of activities, with KA values of 0.5–93 μM.
The physiologically dominant cytosolic isoform hCA II is weakly activated by histamine 7 (KA 125 μM), whereas all compounds of series 26 showed to be better CAAs with KA values in the range of 9–78 μM. Finally, hCA VII is activated by histamine (KA 37.5 μM) and is also activated by all pyridinium salts 26, but with a variable profile. Based on the substitution pattern at the pyridinium ring, it was possible to identify in the compounds 26 three groups of hCA VII activators. The 4-phenyl-2,6-disubstituted salts incorporating bulkier alkyl moieties and trialkyl-substituted derivatives were shown to be weak hCA VII activators (KA 19–81 μM). The second group, with heterogeneous substitution patterns, such as the disubstituted one, and the trisubstituted ones with only alkyl moieties or incorporating the styryl moiety, showed better affinity for hCA VII (KA 2.15–12.5 μM). The diethylpyridinium derivative, the trisubstituted ones (with only alkyl moieties at the pyridinium ring), and the tetrasubstituted pyridinium salts showed to be excellent hCA VII activators (KA 0.8 nM–1.16 μM) [43].
In a more recent study, (hetero)aryl substituted thiazol-2,4-yl derivatives of general formula 27 (Table 2) were developed [44].
In particular, compound 28 was shown to be a good activator mainly towards isoforms hCA I, II, and VII, activating I and II isoforms with KA values an order of magnitude greater (KA 63.4 μM and 68.1 μM, respectively) than isoform VII (KA 7.5 μM) [44].
2.2.3. Insertion of Halogens
Two different studies [45,46] were conducted on histamine analogues in which halogens were inserted on the imidazole ring. The presence of halogens on the histamine structure leads to changes in its physico-chemical features, because of the withdrawing properties of halogens themselves.
Both mono- and di-halogenated derivatives (29 and 30, respectively, Table 3) were investigated, featuring chlorine, bromine, or iodine. Moreover, also N-tert-butyloxycarbonyl derivatives 31 and 32 (Table 3) were synthetized and tested as CAAs on isoforms hCA I and II. Boc-protected compounds showed to have a lower capacity to activate CAs than the deprotected ones. In fact, the first have KA values in the range of 5.4–29.3 μM towards hCA I and of13.6–50.2 μM towards hCA II, and the second have KA values in the range of0.7–21 nM for hCA I and of 1.0–115 nM for hCA II. The obtained data revealed also that the mono-halogenated derivatives are more active than the di-halogenated ones, and the activation of the enzyme increases with the growth of the molecular weight of the compounds [45].
2.2.4. Insertion of Another Imidazole Ring
With respect to the insertion of another imidazole ring, in 2014, Draghici et al. developed compounds of general structure 33 (Table 3), in which two imidazole rings were linked by means of an ethyl chain and featured substituents at position 2, characterized by increasing steric hindrance (H, Me, Et, i-Pr, and Ph) [47].
More specifically, one of the two imidazoles behaves as an additional proton shuttle towards His64, and the other acts as a binding point to the CA-active site edge [51]. The presence of small groups such as methyl or hydrogen groups at position 2 of the imidazole result in very strong (nanomolar KA values) and selective activators of the hCA VA (KA 9.0–131 nM) and VII (KA 15–89 nM) isoforms present in the human brain [47].
2.2.5. Histamine-Inspired Compounds
Synthetic compounds 34–36 have been studied as activators too and included into the “histamine-inspired compounds” group, as well as natural compounds such as L-(+)-ergothioneine (37), melatonin (38), and spinacine (39) (Table 3) [48].
Both synthetic (34–36) and natural (37–39) derivatives were assayed as activators of four human isoforms of CA, namely hCA I, II, IV, and VII. Most of compounds 34–39 activated hCA I and VII in the micromolar range, with KA values ranging between 0.12 µM and 34.7 µM, whereas they were shown to be weak activators of hCA IV (KA 53.4–80.5 μM) and not active towards hCA II (KA values > 100 µM). In this series, two natural compounds, L-(+)-ergothioneine (37) and melatonin (38), are noteworthy and deserve to be further developed as they were shown to be selective and quite potent hCA VII activators (KA values of 820 nM and 120 nM respectively). Instead, spinacine (39) was shown to be the best hCA I activator with a KA value of 7.21 μM that, unfortunately, is higher than the histamine one [48].
Subsequently, Akocak et al. synthetized spinaceamine-substituted compounds 40 (Table 3). In particular, 14 spinaceamine derivatives from this class were obtained and evaluated for their ability to activate hCA I, II, IV, and VII isoforms [49].
Spinaceamine is a bicyclic alkaloid present in the skin of Australian amphibians and structurally is considered a product that originates from the cyclization of histamine. It has got two units of protonatable nitrogen in its structure that in turn can participate in the rate-limiting step of the activation process [52].
Isoform hCA I is moderately activated by all the tested compounds 40 with KA values in the range of 2.52–21.5 μM. These compounds also activate hCA II with KA values in the range of 0.60–17.2 μM. As regards the hCA IV isoform, it is little activated by all compounds 40 (KA 0.52–63.8 μM)
Finally, the cerebral isoform hCA VII is potently and quite selectively activated by these derivatives; in fact, apart from a few compounds that display KA greater than 1 μM, all the other tested compounds potently activate this isoform with nanomolar KAs (82–840 nM) [49].
Additionally, Akocak et al. worked on the modification of histamine by inserting a second imidazole ring and thus developed bis-spinaceamine-substituted derivatives (41) and bis-histamine Schiff bases (42) (Table 3) [50].
Three bis-spinaceamines 41a,b,d and four Schiff bases 42a–d were synthetized and tested as activators on hCA I, II, IV, and VII isoforms, as reported in Table 3.
The data obtained evinced that hCA VII is the most sensitive isoform to be activated by all the tested compounds. In particular, the three bis-spinaceamines 41a,b,d are the most interesting compounds since they can be considered as quite potent and selective activators (KA values in the range of 32–39 nM), regardless of the spacer (X) that featured. Instead, bis-histamine Schiff bases 42a–d display moderate (KA values ranging from 3.28 to 42.1 μM) activity on all four isoforms with compound 42d (furyl substituted), which selectively activates the hCA VII isoform (KA 85 nM) [50].
2.3. Histidine- and Carnosine-Based Derivatives
As well as histamine 7, histidine 2 (Table 1) was also used as lead compound for the design of CAAs and one of the first changes was done on the primary amino moiety. In fact, in 2001, Scozzafava et al. modified histidine and its dipeptide with β-alanine, carnosine 43 (Table 4), and some structurally related dipeptides bearing basic amino acids such as arginine or lysine (R), thus obtaining arylsulfonylureido tri-/tetra-peptide derivatives of general formula 44 (Table 4) [53].
Many of the compounds 44 displayed KA values in the 1–20 nM range for hCA I and bCA IV, and in the range of 10–40 nM for hCA II. Interestingly, ex vivo experiments revealed that some of them were able to strongly enhance cytosolic red cells CA activity in human erythrocytes, thus being able to behave as effective in vivo CAAs, and might thus constitute interesting candidates for animal studies regarding their involvement in cognitive processes [53].
In another study in 2009, Abdo et al. developed arylsulfonylhydrazido-L-histidine derivatives bearing a 4-substitued aryl moiety; these compounds were shown to be less active than histidine itself, except for 45 (Table 4), which activates hCA II with a KA 0.21 μM [54].
Like histamine, also histidine methyl ester (46) and carnosine methyl ester (47) were derivatized, introducing electron-withdrawing halogens, and obtaining mono (X2 = H)- and di-halogenated (X2 = Cl, Br, I) analogues (Table 4) [46].
The obtained compounds of series 46 and 47 resulted in activating in a diverse manner the hCA I, II VII, XII, and XIV isoforms tested. The most interesting results are summarized in Table 4.
The differences in the activation properties of the synthetized compounds can be correlated both to the size of the halogens inserted into the molecular structure and their electron-withdrawing properties, as they modify the distribution of electrons into the ring and, in turn, the nucleophilicity and the basicity of the entire molecule that could interfere with the ability to activate the enzyme.
From a general point of view, mono- and di-halogenated L-His-OMe derivatives 46 are more effective hCA I and II activators compared to the L-Car-OMe 47 counterparts, but are less effective than the corresponding histamine derivatives (series 29–30, Table 3).
The best hCA I and II activator is compound 46a (KA 0.9 nM and 12 nM, respectively), which is a mono-halogenated Boc-substituted derivative. Its analogue with the deprotected amine (46b, KA 1.2 nM and 0.32 μM, respectively) or the corresponding di-halogen derivative (46c, KA 1.5 nM and 13 nM, respectively) are instead less effective activators of hCA I and II. This general trend is observed for all the compounds of the series.
The corresponding derivative L-Car-OMe of compound 46a, namely 47a, weakly activated hCAI and II (KA 4.3 μM and 5.2 μM, respectively), while it showed to be an effective activator of isoform VII (KA 0.81 μM), which is more effective than the corresponding L-His-OMe derivative 46d (KA 9.7 μM). In fact, in general, L-Car-OMe derivatives 47 proved to be effective activators of the cerebral isoform hCA VII, which is weakly activated by L-His-OMe derivatives 46 and also by the corresponding histamine derivatives (series 29–30, Table 3).
The best-performing hCA VII activator is compound 47b (KA 8.1 nM), which is a di-halogenated derivative; the relative Boc- analogue 47c is instead a less effective hCA VII activator (KA 4.0 μM). Thus, series 47 displayed an opposite trend with respect to series 46 for what concerns the activation of hCA I and II isoforms.
hCA XII and XIV are both little activated by the two series 46 and 47, with values of KA, aside for very few exceptions, in the range of 0.83–64.7 μM for hCA XII and 0.92–40.8 μM for hCA XIV [46].
In 2020, Vistoli et al. [55] biologically evaluated in hCA I, II, VA, and IX, some dipeptides that contain carnosine 43 (Table 4) and histidine 2 (Table 1) of general formula 48 (Table 4). In fact, L-carnosine is found in excitable tissues together with other histidine-containing peptides such as carnicine and L-carnosinamide (derivatized on -COOH), L-homocarnosine and Gly-L-His (different lengths of amino terminal residues), and the N-acetyl-carnosine derivative [56].
In general, compounds 48 activate the four human isoforms hCA I, II, VA, and IX in the low to high micromolar range. In particular, concerning hCA I, they showed KA values in the range of 16.6–80.4 μM, with D-carnosinamide (48a, Table 4) and carnicine (48b, Table 4) being the most active (KA 16.6 μM) compounds for this isoform. Isoform hCA II was poorly activated (KA > 76.6 μM) by all the compounds tested. Also, hCA VA was shown to be weakly activated (KA 6.4–52.8 μM), and the most active compound for this isoform was shown to once again be carnicine 48b (KA 6.4 μM).
2.4. Gold Nanoparticles of Histamine, Histidine- and Carnosine Derivatives
Gold nanoparticles coated with bioactive substances demonstrated to be very fascinating for innovative biomedical applications because they seem to improve the site-specific drug delivery.
In this view, histamine, L-carnosine-methyl ester, and L-His-methyl ester were conjugated with lipoic acid to obtain compounds 49, 50, and 51 respectively, (Table 5) [57].
Compounds 49–51 were biologically evaluated on isoforms hCA I, II, IV, VA, VII, and XIV in comparison with their starting compounds, namely, histamine 7 (Table 1), L-His 2 (Table 1), L-His methyl ester, L-carnosine 43 (Table 4), and L-carnosine methyl ester, and the obtained data are reported in Table 6.
Compounds 49–51 showed to be very potent CAAs both in vitro (KA values ranging from 1.0 to 9.0 nM) and ex-vivo (in whole blood experiments, with an increase of 200–280% of the CA activity) of all the CA isoforms tested.
This is the first example of enzyme activation with nanoparticles and may lead to interesting biomedical applications [57].
2.5. Sulfur, Selenium and Tellurium Containing Amines
In 2019, Tanini et al. synthetized three different series of β-aminochalcogenides containing sulfur 52, selenium 53, and tellurium 54 (Table 5) and investigated the CA-activating properties of the derivatives obtained for hCA I, II, VA, and VII isoforms [58].
The amphetamine represents the lead compound for the development of these new chalcogen-containing compounds, based on the results of a previous study conducted in 2017 in which psychoactive compounds demonstrated to be strong CAAs [59].
All the synthesized compounds of series 52–54 showed to be moderate activators of hCA I (KA 7.7–43.7 μM), hCA VA (KA 5.2–23.3 μM), and the brain-associated cytosolic CA isoform, hCA VII (KA 78.9–44.9 μM), without distinction about the presence of S (52), Se (53), or Te (54); hCA II was not activated by these compounds (almost all KA values > 100 μM) (Table 5).
In addition, the presence of Se and Te makes derivatives of series 53 and 54 antioxidant compounds able to inhibit the formation of ROS metabolites, thus preventing cellular stress and damage, which are characteristics of neurodegenerative diseases. More specifically, they were tested as mimics of glutathione peroxidase and the best compounds were the aminotellurides (54). For this reason, these compounds can be considered able to contrast the progression of neurodegenerative processes due to their dual action: inhibition of oxidative stress (thanks to tellurium and selenium properties) and strengthening of synapses and neuronal activity (thanks to CAs activation) [58].
2.6. Drug Repurposing
Several drugs already used in therapy for other aims and carrying proton-shuttling groups in their structure such as timolol [60] and propranolol (β-blockers), fluoxetine, sertraline and citalopram [61] (serotonin reuptake inhibitors), sildenafil [62] (phosphodiesterase IV inhibitor), and amphetamine/methamphetamine derivates [59] (psychoactive substances), were studied as CAAs.
These compounds feature a central heterocyclic scaffold, which is not directly correlated to histamine or histidine, functionalized with an amino group linked to an alkyl spacer.
Timolol 55 (Table 7) is an anti-hypertensive drug, belonging to the class of β-blocker featuring of an aromatic and heterocyclic moiety and a secondary and protonatable amino group. Following enzymatic studies, it has been proven that timolol activates hCA I and II (KA values of 12 μM and 9.3 μM, respectively), establishing a ternary complex with the enzyme and the substrate. It binds at the entrance of the active site, where His64 is located, and in a different position compared to the substrate, as timolol binding does not impact on the affinity of the substrate to its target, only increasing vmax in enzymatic kinetics [60].
More cutting-edge research is represented by the SSRIs: fluoxetine 56, sertraline 57, and citalopram 58 (Table 7) are drugs used in therapy for the treatment of depression, and they bear one protonatable group in their molecular structure that makes them potential CAAs.
All three SSRIs 56–58 demonstrated to be able to dose-dependently activate hCAI and II, with similar activation rates as those of histamine 7 and phenylalanine 1 (Table 1). In particular, hCA I is better activated by histamine 7 (170% at 1.0 μM) and fluoxetine 56 (175% at 1.0 μM), instead of hCA II, which is better activated by citalopram 58 (170% at 1.0 μM), and phenylalanine 1 (175% at 1.0 μM). Sertraline is the weaker activator of both hCA I and hCA II (145% and 140%, respectively, at 1.0 μM). These data are important because they pave the way to a new therapeutic approach that is particularly direct against Alzheimer’s disease associated with major depression [61].
Sildenafil 59 (Table 7) is a phosphodiesterase-5 (PDE5) inhibitor used in therapy for the treatment of erectile dysfunction. Its structure is very bulky, but it bears a piperazine moiety similar to that of some CAAs, giving the hope that also sildenafil can activate CAs [62]. From this view, in 2009, Sildenafil 59 was tested on hCA I-XIV isoforms [62]. From the obtained data (some interesting ones reported in Table 7), sildenafil 59 mostly activates hCA I, VB and VI with KA values of 1.08 μM, 6.54 μM, and 2.37 μM, respectively, while isoforms hCA III, IV, and VA are less activated with KA values in the range of 13.4–16.8 μM. Isoforms hCA II, IX, XIII, and XIV are little activated by sildenafil with KA in the range of 27.5–34.0 μM. In the end, hCA VII and XII are the least activated isoform by sildenafil with KA about 73 μM [62].
Sildenafil 59 has been also tested in vivo on rats. Unfortunately, CAs activity was shown to be decreased compared to baseline levels and this could be related both to the production of the metabolite “desmethylsildenafil” and to the production of NO/nitrate following PDE5 inhibition. Indeed, the first could bind with its NH group to the zinc-bound water molecule in the same way as phenols, a class of CAIs [63], while the second behaves as potent inhibitor [64].
Many psychoactive substances possess the phenethylamine scaffold and the general formula Ar–CH2CH(R)NHR’, and, as reported, this type of amine could effectively activate CAs [32,35].
In this contest, in 2017, five amines structurally related to amphetamine, namely amphetamine 60, methamphetamine 61, phentermine 62, mephentermine 63, and chlorphenteramine 64 (Table 7) were the first psychoactive substances to be investigated as CAAs and tested on 11 hCA isoforms (hCA I, II, IV, VA, VB, VI, VII, IX, XII, XIII, and XIV) [59].
Compounds 60–64 are used in therapy for treating attention deficit hyperactivity disorder, narcolepsy, obesity, and nasal congestion, but unfortunately, they have a lot of side effects including psychosis [65].
The data obtained (some interesting ones reported in Table 7) evinced that compounds 60–64 cannot be considered CAAs of the hCA I and II isoforms (KA > 150 μM), while they moderately activate hCA XIII and XIV (KA ranges 24.1–79.5 μM and 6.81–18.1 μM, respectively). Isoform hCA IX resulted not significantly activated by the amines investigated in this study, while the other tumor-associated isoform hCA XII demonstrated to be significantly activated by amphetamine 60, methamphetamine 61, and chlorphenteramine 64 with KA values in the range of 0.64–0.94 μM, but less activated by the other amines (KA range 3.24–6.12 μM). The most activated isoform is hCA IV (KA values in the range of 51 nM–1.03 μM). In particular, the best hCA IV activators are methamphetamine 61 and chlorphenteramine 64 with KA values of 51 nM and 55 nM, respectively, followed by phentermine 62 and amphetamine 60 with KA values of 74 nM and 94 nM, respectively. Isoforms hCA VA and VB are effectively activated by compounds 60–64 with KA values in the range of 0.24–2.56 μM and, particularly, the best activator of hCA VA showed to be chlorphenteramine 64 with a KA 310 nM; the best activator of hCA VB demonstrated to be mephentermine 63 with a KA 240 nM.
Of note, the cerebral isoform hCA VII was successfully activated by all five compounds with KA values in the range of 98–0.93 μM, with chlorphenteramine 64 being the best hCA VII activator of this series (KA 98 nM).
Since some CAs (hCA VII, VA, VB, and XII) are abundant in the brain, some of the cognitive effects of these psychoactive compounds might be related to the activation of these enzymes, bringing new light to the intricate relationship between CA activation by these substances and their multitude of pharmacologic activities [59].
Very recently, the CAs activating effects of a series of histamine receptor (H1, H2, H3, and H4) agonists/antagonists towards four hCA isoforms expressed in the human brain, namely hCA I, II, IV, and VII, were investigated [66].
In particular, 30 compounds (including histamine itself 7, Table 1, and many well-known drugs, for example pyrilamine and loratadine among the H1 antagonists, and cimetidine and ranitidine among the H2 antagonists) were tested, and they were all shown to be moderate activators of hCA I (KA range from 52 nM to >100 μM), hCA II (KA range from 82 nM to >100 μM), hCA IV (KA range from 1.02 μM to >100 μM), and hCA VII (KA range from 110 nM to >100 μM, with hCA IV being the worst activated isoform. The most interesting results are reported in Table 8. In general, the best activators of hCA I isoform were the methyl histamine/histidines 65, 66, 67 (KA range from 52 μM to 0.36 μM), the thiazole derivative 68 (KA 0.87 μM), burimamide 69 (KA 0.88 μM), metiamide 70 (KA 0.98 μM), and impromidine 71 (KA 0.72 μM), which are structurally related to histamine as they all bear the imidazole ring in their structure, except for compound 68, which has a thiazole ring. In particular, 1-methyl histidine 67 has an additional proton transfer group represented by the carboxyl moiety, thus resulting in being much more potent (KA 52 nM).
Moreover, some compounds such as α-methyl histamine 65 and 1-methyl histidine 67 were shown to strongly activate the hCA II isoform with KA values of 82 nM and 0.57 μM, respectively.
Interestingly, hCA VII showed to be the most activated isoform, and impromidine 71 is the most active derivative (KA 0.10 μM), followed by other compounds ranging between 0.11 and 7.05 μM. In particular, some interesting SARs were observed for this isoform, resulting from the presence of more lipophilic groups (as in 68, 72, 73 or 74) able to promote a greater and more selective hCA VII activation [66].
2.7. Miscellaneous
2.7.1. Ureas and di-Ureas Incorporating 1,2,4-triazole Derivatives
In 2017, Le Duc et al. substituted the imidazole moiety of a histamine with 1,2,4-triazole, developing two different series represented by mono-urea 75 and di-urea 76 compounds (Table 9) [67].
Fourteen derivatives were synthetized, in which R and R′ can both be alkyl and aryl groups, introduced for modulating the linker-length between the two triazole moieties (in series 76) or for modifying lipophilicity and, in turn, the activity of the enzyme (in series 75).
Both series bear an ureido moiety, as it has been proven that this group gives flexibility to the various compounds to better accommodate at the entrance of the CA-active site cavity [69].
Mono-ureas 75 and di-ureas 76 were biologically evaluated on hCA I and II isoforms and they demonstrated to strongly activate hCA I with KA values in the range of 0.81–993 nM. The best effective hCA I activators belong to series 76 and are compounds 76a (KA 0.81 nM) and 76b (KA 0.94 nM) (Table 9). Whereas the best hCA I activator of series 75 is compound 75a (KA 6.1 nM). Moreover, for series 75, it was proven that the activity for hCA I diminishes with the increasing of the chain-length; SARs are instead more complex for series 76. In general, regarding hCA I activation properties, KA values are influenced by the length of the chain for series 75 and by the linker between the two rings for series 76 [67].
Even the hCA II isoform results in being well activated by the compounds of the two series 75 and 76, with KA values in the range of 0.05–6.7 μM. In particular, in series 75, the best hCA II activator is compound 75a (KA = 1.7 nM), as well as for hCA I (KA 6.1 nM), whereas compounds that activate the most hCA II belong to series 76. The best hCA II activator showed to be 76b (KA 0.05 nM), which also strongly activates hCA I (KA 0.94 nM), resulting in a very potent dual hCA I/II activator. In this series, also compound 76c, with the 1,4-phenylene linker and the amino group, well activated the hCA II isoform (KA 0.12 nM).
Interestingly, compound 76b is the most effective hCA II activator (KA 0.05 nM) ever reported, and has shown to be a lead compound for the development of clinical candidates in cognitive impairment or other diseases characterized by a deficit of hCA I and hCA II isoforms, such as Alzheimer’s disease or aging [67].
2.7.2. Amino Alcohol Oxime Ethers
In 2021, Nocentini et al. [68] based their research on a previous study, in which it was demonstrated that the β-blocker timolol 55 (Table 7) was able to activate CAs. More specifically, this drug proved to bind at the entrance of the active site cavity and to activate hCA I and II isoforms by establishing a ternary complex with the enzyme and CO2 [60]. Thus, timolol 55 was identified as a lead compound for the design of a series of new activators.
In this context, Nocentini et al. studied a series of amino alcohol oxime ether derivatives with a general structure 77 (Table 9), already known as β-blockers, analgesics, and antiarrhythmics, and tested their activation ability towards hCA I, II, IV, and VII [68].
All amino alcohol derivatives of series 77 showed to be good activators of the tested CAs, with KA values spanning from low micromolar to nanomolar range. Specifically, hCA II and VII are most potently activated by these derivatives (KA range of 79–420 nM) with respect to hCA I and IV (KA range from 10- to 100-fold higher), with hCA IV being the less-activated isoform (KA 1.01–12.9 μM).
In particular, compounds belonging to the tert-butylamino series turned out to better activate isoforms hCA I and II compared to those belonging to the iso-propylamino one. In fact, tert-butylamine 77a (Table 9) is the most potent and selective hCA II activator (KA 79 nM) of this study, while the iso-propylamines 77b and 77c (Table 9) showed potent hCA VII activation profiles (KA 82 nM and 91 nM, respectively) and 10- to 100-fold selectivity with respect to the other CA isoforms tested [68].
2.7.3. Imidazoline and Other Related Five-Membered N-heterocycle Derivatives
Imidazoline ring is considered a bioisoster of the imidazole moiety and it is present in the antihypertensive agent clonidine 78 (Table 10). In 2020, Chiaramonte et al. developed a series of 2-aminoimidazolines 79 (Table 10), structurally related to clonidine 78, which showed to be able to activate several hCA isoforms (hCA I, IV, VA, VII, IX, XII, and XIII) with potency in the micromolar range, while it was inactive on hCA II. In particular, clonidine showed to best activate hCA VII and XIII isoforms with KA values of 8.4 μM and 7.8 μM, respectively [70].
The 2-aminoimidazoline derivatives 79 were tested on five hCA isoforms (I, II, VA, VII, and XIII) and, as clonidine, they showed to be inactive on hCA II, while resulting good activators of hCA I, VA, VII and XIII isoforms (Table 10), with KA values in the micromolar range.
In general, from the data obtained, it is evinced that most of the derivatives of series 79 were better activators than clonidine on all hCA isoforms tested. Moreover, some structure–activity relationships (SAR) were derived for these compounds that can be applied all over the hCA isoforms: the substitution with methyl at N-1 (R1 = CH3) improves the performance of compounds with respect to the unsubstituted ones (R1 = H), as well as the alkyl aryl chain compared to the alkyl one (R2). Finally, the amino or methylamino group is preferable to S as a linker (X) between the heterocycle and the alkyl-aryl chain. In addition, some other SAR were obtained and showed to be different on the various isoforms, suggesting that, in series 79, it could be possible to develop new compounds more selective for a particular CA isoform. In particular, the most interesting compounds are 79a and 79b, showing to be potent (KA 0.9 μM) and quite selective activators of hCA V and VII, respectively.
In an effort to improve both potency and selectivity, in 2022, Chiaramonte et al. developed histamine-related compounds by replacing the imidazole ring with several five-membered heterocycles (pyrrole, N1-substitued imidazole, pyrazole, etc..), and changing the length of the aliphatic chain that leads to the primary amino group [71].
According to these modifications, 23 compounds were synthetized and biologically tested on hCA I, II, VA, VII, and XIII isoforms, but only a few of them demonstrated to activate these isoforms better than the lead compound histamine 7 (Table 1). In general, none of the new compounds displayed activity on hCA II, and the most activated isoform was hCA I with KA values in the low micromolar range (0.9–93.5 μM), while the other isoforms were activated with higher KA values (11.2–78.5 μM for hCA V, 9.7–74.6 μM for hCA VII, and from 28.2 to >100 μM for hCA XIII).
Specifically, above all derivatives synthesized, compounds 80, 81, and 82 showed an interesting selectivity for activating hCA I over hCA II, VA, VII, and XIII (Table 10). Compounds 80–82 could represent lead molecules to obtain new potent and selective hCA I activators [71].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 10.
Clonidine (78), imidazoline (79), and the other related five-membered N-heterocyclic derivatives 80–82 as CAAs.
Table 10.
Clonidine (78), imidazoline (79), and the other related five-membered N-heterocyclic derivatives 80–82 as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 78 Clonidine |  | hCA I: KA = 76.3 μM hCA VA: KA = 42.6 μM hCA VII: KA = 8.4 μM hCA XIII: KA = 7.8 μM | [70] |
| 79 |  R1 = H, CH3; R2 = alkyl, alkylaryl X = NH, NCH3, S | hCA I: KA = 4.18- >100 μM hCA VA: KA = 0.9–52.7 μM hCA VII: KA = 0.9–46.7 μM hCA XIII: KA = 6.5- >100 μM | [70] |
| 79a |  | hCA I: KA = 30.2 μM hCA VA: KA = 0.9 μM hCA VII: KA = 6.5 μM hCA XIII: KA = 17.4 μM | [70] |
| 79b |  | hCA I: KA = 16.9 μM hCA VA: KA = 3.7 μM hCA VII: KA = 0.9 μM hCA XIII: KA = 19.1 μM | [70] |
| 80 |  | hCA I: KA = 2.16 μM hCA VA: KA = 29.8 μM hCA VII: KA = 44.6 μM hCA XIII: KA > 100 μM | [71] |
| 81 |  | hCA I: KA = 2.19 μM hCA VA: KA = 78.5 μM hCA VII: KA > 100 μM hCA XIII: KA > 100 μM | [71] |
| 82 |  | hCA I: KA = 0.9 μM hCA VA: KA = 11.2 μM hCA VII: KA = 13.2 μM hCA XIII: KA > 100 μM | [71] |
2.7.4. Indazole, Pyrazole and Oxazole Derivatives Carrying Amino Acidic Tails
In 2016, Maccallini et al. [72] attempted to join the favorable characteristics for activating CAs of heterocyclic compounds such as indazole, pyrazole, and oxazole rings with amino acids characterized by the ability to inhibit neuronal nitric oxide synthase (nNOS), which is an important activity for the treatment of neurodegenerative diseases that are characterized by an abnormal nitrergic signal joined with a low activity and expression of CAs. Thus, in this context, five different series of compounds 83–87 (Table 11) that were potentially able to inhibit nNOS and activate CAs were developed. In these series 83–87 (Table 11) a heterocyclic moiety (indazole, pyrazole, and oxazole) coupled with an amino acid residue (Ala, Tyr, and Glu) was present.
Among all derivatives of series 83–87, five substituted indazoles 85 turned out to inhibit nNOS with high potency and high selectivity with respect to iNOS and eNOS.
Moreover, compounds 83–87 were evaluated in vitro on hCA I, II, IV, and VII isozymes, and the obtained results revealed a slight activation of isoforms II and IV in the micromolar range (KA 4.0–43.2 μM and 7.1–48.6 μM, respectively) for all tested compounds. Some derivatives, 84b (R = p-OH-C6H4), 85a (R = CH3), and 85c (R = CH2CH2COOH) activated the hCA VII isoform with KA in the low micromolar range (KA values of 0.69 μM, 0.59 μM, and 0.51 μM, respectively), but without a clear correlation between enzyme activation and chemical structure. The most activated isoform was hCA I with KA values ranging from 9.0 to 6.39 μM for all compounds 83–87: of note, the 1H-indazole series 84 (KA values ranging from 9.0 nM to 1.25 μM), with compound 84b (R = p-OH-C6H4) being the best hCA I activator (KA 9 nM).
Anyhow, the most encouraging compound is the indazole derivative 85b (KA 15 nM towards hCA I), which also displayed the best percentage of nNOS inhibition, thus showing to be a dual agent able to perform a selective nNOS inhibition and hCA I activation. Derivative 85b is therefore a promising lead compound to develop new drug candidates for the therapy of neurodegeneration [72].
2.7.5. Indole-Based Derivatives
In 2021, Barresi et al. [73] developed three series (88–90, Table 12) of compounds with an indole central scaffold, which is known to be a “privileged scaffold” for drug discovery, due to its versatility and usefulness [74].
The indole-based derivatives 88–90 were decorated at N1 with a benzyl group in all series, while different substitution patterns at 3- and 5-positions were exploited. Specifically, in series 88 and 89, 3-position features a glyoxylamide (88) or carboxamide (89) moiety in which the N-atom is substituted (R1) with polar (hydroxypropyl or hydroxyethyl), protonatable groups (dimethylaminopropyl or diethylaminoethyl) or with a benzyl group; conversely, in series 90 an ethyl ester is present at 3-position of the scaffold. The indole 5-position is substituted with hydroxyethyl or with protonatable groups (dimethylaminopropyl or dimethylaminoethyl or diethylaminoethyl) [73].
Nine compounds were synthetized and tested on the main isoforms expressed in human brain, namely hCA I, II, VA, and VII. Isoforms hCA I and II showed not to be significantly activated by all compounds, with KA values ranging between 69.1 μM and values > 100 μM. Isoform hCA VA demonstrated to be quite activated by all the tested compounds with KA values in the range of 24.4–59.8 μM. The cerebral isoform hCA VII proved to be the most sensitive to the activation by all compounds with KA values ranging between 7.2 and 10.8 μM, independently from the position of the basic group, leading to speculation that what matters is its presence, rather than its position at the scaffold. The best hCA VII activators belong to series 89 and 90, namely compounds 89a, 90a, and 90b, with KA values of 7.5, 7.2, and 8.2 μM, respectively [73]. Due to these results being extremely encouraging, the three compounds were furtherly biologically characterized.
In particular, compounds 89a, 90a, and 90b were first tested for their cytotoxicity on microglial cells by treating human C20 cells with micromolar concentrations of the target compounds and, then, cell viability was evaluated by MTS assay. The data obtained revealed that, while compounds 90a and 90b caused a slight reduction in cell viability, compound 89a did not induce cytotoxicity in the human microglial cell line C20 at any tested concentration.
Given these results, compound 89a was selected for further determination of its capability to produce BDNF (brain-derived neurotrophic factor) on the same cell line and, interestingly, it showed to induce the production of BDNF as good as microglia [73,75].
Going deeper, there is a link between the process of activating CAs and the process which leads to the release of BDNF, and this link is the correct acidification process. Indeed, it is important for activating CAs because an activator behaves as an additional proton shuttle towards His64 and so it must have a protonatable moiety in its molecular structure; in addition, it is also important for releasing BDNF from secretory granules, since correct acidification is a fundamental step during the release of BDNF [76].
Furthermore, 89a was found to possess the physicochemical parameters suitable for reaching the CNS.
3. Conclusions
Carbonic anhydrases activators (CAAs) are still poorly studied compared to carbonic anhydrases inhibitors (CAIs). Indeed, while CAIs are already used in therapy for the treatment of a number of pathologies such as glaucoma, epilepsy, and obesity, CAAs to date, have not shown relevant pharmacological applications and are still under study.
CAAs are proposed to participate in the proton transfer, acting as additional proton shuttles towards His64 in the rate-limiting step of CA catalytic cycle, that is, the generation of the active hydroxylated state of the enzyme from the inactive acid form. In this respect, CAAs must feature two main structural requirements: to be a small molecule suitable for the active site and to possess at least a protonatable basic moiety with pKa values in the range of 6.5–8.0 for participating in the proton transfer.
Interest in CAAs has increased thanks to recent findings reporting an involvement of CAs in cognitive and memory disorders, suggesting that the activation of this enzyme may represent a potential effective strategy for the strengthening of synaptic efficacy. In particular, cerebral isoforms of CAs (mainly hCA I, II, VA, and VII) could be a valid target for the development of new agents useful for the treatment of neurodegenerative pathologies such as Alzheimer’s disease in which these isoforms showed to be less expressed.
Indeed, it was demonstrated that the administration of an activator to experimental animals determined the enhancement of synaptic efficiency and the consolidation of cognition, spatial memory, and learning.
Based on these findings, a number of CAAs are under study as potential candidates for the development of new drugs for the treatment of disorders in which memory and cognition are impaired. In this context, this paper reviewed the current state of the art concerning CAAs, focusing attention on those compounds that potently and selectively activate CAs, with particular regard to cerebral isoforms, and for which preliminary biological evaluations were carried out in order to evaluate their potential application in therapy.
Amino acids and amine represent the most investigated compounds in the field of CAs activation. In fact, many of the compounds here described were obtained using histamine or histidine as lead compounds, and involved, thanks to previous SAR studies, the modification of either the heterocyclic ring, the amino portion, or the introduction of portions that could improve the activity and selectivity for the CA isoforms of interest. Some of them showed to strongly activate hCAI, hCA II, and/or hCA VII isoform with activation constant (KA) values in the very low nanomolar range.
As a summary, the hCA activation properties of the most interesting compounds reviewed are highlighted in Table 13.
In conclusion, in the present review we highlighted the peculiar decorations of CAAs, describing their involvement in the proton shuttling in the rate-limiting step of the CA catalytic cycle. Furthermore, this report provides an exciting outlook on existing data that might help the medicinal chemist in the design and development of more efficient and selective new leads for future in vitro and in vivo studies, so that novel drug candidates can be unveiled and introduced onto the market and into the clinical pipeline.
Author Contributions
Conceptualization, F.D.S., and S.T.; methodology, V.P., S.S. and S.T.; writing and draft preparation, V.P., S.S., E.B. (Emma Baglini), and E.B. (Elisabetta Barresi). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Italian MIUR under grant PRIN 2017, 2017XYBP2R.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Supuran, C.T. Structure and Function of Carbonic Anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A. Carbonic-Anhydrase Inhibitors and Their Therapeutic Potential. Expert. Opin. Ther. Pat. 2000, 10, 575–600. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrase Activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Berrino, E.; Carradori, S.; Supuran, C.T.; Cirri, M.; Carta, F.; Costantino, G. Amine-and Amino Acid-Based Compounds as Carbonic Anhydrase Activators. Molecules 2021, 26, 7331. [Google Scholar] [CrossRef]
- D’Ambrosio, K.; Di Fiore, A.; Buonanno, M.; Monti, S.M.; De Simone, G. η- and θ-Carbonic Anhydrases. In Carbonic Anhydrases: Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target; Academic Press: Cambridge, MA, USA, 2019; pp. 139–148. [Google Scholar]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, A.; Zoccola, D.; Tambutté, S.; Vullo, D.; Supuran, C.T. Carbonic Anhydrase Activators. The First Activation Study of a Coral Secretory Isoform with Amino Acids and Amines. Bioorg. Med. Chem. 2010, 18, 2300–2303. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic Anhydrase Activators. Activation of Isozymes I, II, IV, VA, VII, and XIV with L- and D-Histidine and Crystallographic Analysis of Their Adducts with Isoform II: Engineering Proton-Transfer Processes within the Active Site of an Enzyme. Chem. A Eur. J. 2006, 12, 7057–7066. [Google Scholar] [CrossRef]
- Ciccone, L.; Cerri, C.; Nencetti, S.; Orlandini, E. Carbonic Anhydrase Inhibitors and Epilepsy: State of the Art and Future Perspectives. Molecules 2021, 26, 6380. [Google Scholar] [CrossRef]
- Nishimori, I.; Onishi, S.; Vullo, D.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activators: The First Activation Study of the Human Secretory Isoform VI with Amino Acids and Amines. Bioorg. Med. Chem. 2007, 15, 5351–5357. [Google Scholar] [CrossRef]
- Licsandru, E.; Tanc, M.; Kocsis, I.; Barboiu, M.; Supuran, C.T. A Class of Carbonic Anhydrase I–Selective Activators. J. Enzym. Inhib. Med. Chem. 2017, 32, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Masini, E.; Carta, F.; Scozzafava, A.; Supuran, C.T. Antiglaucoma Carbonic Anhydrase Inhibitors: A Patent Review. Expert. Opin. Ther. Pat. 2013, 23, 705–716. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure-Based Drug Discovery of Carbonic Anhydrase Inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T.; Carta, F. Antiobesity Carbonic Anhydrase Inhibitors: A Literature and Patent Review. Expert. Opin. Ther. Pat. 2013, 23, 725–735. [Google Scholar] [CrossRef]
- Miao-Kun, S.; Alkon, D.L. Pharmacological Enhancement of Synaptic Efficacy, Spatial Learning, and Memory through Carbonic Anhydrase Activation in Rats. J. Pharmacol. Exp. Ther. 2001, 297, 961–967. [Google Scholar]
- Schmidt, S.D.; Nachtigall, E.G.; Marcondes, L.A.; Zanluchi, A.; Furini, C.R.G.; Passani, M.B.; Supuran, C.T.; Blandina, P.; Izquierdo, I.; Provensi, G.; et al. Modulation of Carbonic Anhydrases Activity in the Hippocampus or Prefrontal Cortex Differentially Affects Social Recognition Memory in Rats. Neuroscience 2022, in press. [Google Scholar] [CrossRef]
- Canto de Souza, L.; Provensi, G.; Vullo, D.; Carta, F.; Scozzafava, A.; Costa, A.; Schmidt, S.D.; Passani, M.B.; Supuran, C.T.; Blandina, P. Carbonic Anhydrase Activation Enhances Object Recognition Memory in Mice through Phosphorylation of the Extracellular Signal-Regulated Kinase in the Cortex and the Hippocampus. Neuropharmacology 2017, 118, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.K.; Alkon, D.L. Carbonic Anhydrase Gating of Attention: Memory Therapy and Enhancement. Trends Pharmacol. Sci. 2002, 23, 83–89. [Google Scholar] [CrossRef]
- Meier-Ruge, W.; Iwangoff, P.; Reichlmeier, K. Neurochemical Enzyme Changes in Alzheimer’s and Pick’s Disease. Arch. Gerontol. Geriatr. 1984, 3, 161–165. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Identification of the Oxidative Stress Proteome in the Brain. Free Radic. Biol. Med. 2011, 50, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Lemon, N.; Canepa, E.; Ilies, M.A.; Fossati, S. Carbonic Anhydrases as Potential Targets Against Neurovascular Unit Dysfunction in Alzheimer’s Disease and Stroke. Front. Aging Neurosci. 2021, 13, 785. [Google Scholar] [CrossRef]
- Chesler, M. Regulation and Modulation of PH in the Brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Kaila, K. Carbonic Anhydrases and Brain PH in the Control of Neuronal Excitability. Subcell. Biochem. 2014, 75, 271–290. [Google Scholar] [PubMed]
- Pasternack, M.; Voipio, J.; Kaila, K. Intracellular Carbonic Anhydrase Activity and Its Role in GABA-Induced Acidosis in Isolated Rat Hippocampal Pyramidal Neurones. Acta Physiol. Scand. 1993, 148, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Makani, S.; Chen, H.Y.; Esquenazi, S.; Shah, G.N.; Waheed, A.; Sly, W.S.; Chesler, M. NMDA Receptor-Dependent Afterdepolarizations Are Curtailed by Carbonic Anhydrase 14: Regulation of a Short-Term Postsynaptic Potentiation. J. Neurosci. 2012, 32, 16754–16762. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.; Mondal, U.K.; Supuran, C.T.; Ilies, M.A.; McKenna, R. Crystal Structure of Carbonic Anhydrase II in Complex with an Activating Ligand: Implications in Neuronal Function. Mol. Neurobiol. 2018, 55, 7431–7437. [Google Scholar] [CrossRef]
- Sun, M.K.; Zhao, W.Q.; Nelson, T.J.; Alkon, D.L. Theta Rhythm of Hippocampal CA1 Neuron Activity: Gating by GABAergic Synaptic Depolarization. J. Neurophysiol. 2001, 85, 269–279. [Google Scholar] [CrossRef]
- Blandina, P.; Provensi, G.; Passsani, M.B.; Capasso, C.; Supuran, C.T. Carbonic Anhydrase Modulation of Emotional Memory. Implications for the Treatment of Cognitive Disorders. J. Enzym. Inhib. Med. Chem. 2020, 35, 1206–1214. [Google Scholar] [CrossRef]
- Sun, Y.J.; Chen, Y.; Pang, C.; Wu, N.; Li, J. Acetazolamide Attenuates Chemical-Stimulated but Not Thermal-Stimulated Acute Pain in Mice. Acta Pharmacol. Sin. 2013, 35, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Schröder, H.C.; Schlossmacher, U.; Neufurth, M.; Feng, Q.; Diehl-Seifert, B.; Müller, W.E.G. Modulation of the Initial Mineralization Process of SaOS-2 Cells by Carbonic Anhydrase Activators and Polyphosphate. Calcif. Tissue Int. 2013, 94, 495–509. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Schröder, H.C.; Schlossmacher, U.; Grebenjuk, V.A.; Ushijima, H.; Wang, X. Induction of Carbonic Anhydrase in SaOS-2 Cells, Exposed to Bicarbonate and Consequences for Calcium Phosphate Crystal Formation. Biomaterials 2013, 34, 8671–8680. [Google Scholar] [CrossRef]
- Briganti, F.; Mangani, S.; Orioli, P.; Scozzafava, A.; Vernaglione, G.; Supuran, C.T. Carbonic Anhydrase Activators: X-Ray Crystallographic and Spectroscopic Investigations for the Interaction of Isozymes I and II with Histamine. Biochemistry 1997, 36, 10384–10392. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Vullo, D.; Supuran, C.T. Carbonic Anhydrase Activators. Activation of Isoforms I, II, IV, VA, VII, and XIV with L- and D-Phenylalanine and Crystallographic Analysis of Their Adducts with Isozyme II: Stereospecific Recognition within the Active Site of an Enzyme and Its Consequenc. J. Med. Chem. 2006, 49, 3019–3027. [Google Scholar] [CrossRef]
- Temperini, C.; Scozzafava, A.; Puccetti, L.; Supuran, C.T. Carbonic Anhydrase Activators: X-ray Crystal Structure of the Adduct of Human Isozyme II with L-Histidine as a Platform for the Design of Stronger Activators. Bioorg. Med. Chem. Lett. 2005, 15, 5136–5141. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activators: Kinetic and X-Ray Crystallographic Study for the Interaction of D- and L-Tryptophan with the Mammalian Isoforms I-XIV. Bioorg. Med. Chem. 2008, 16, 8373–8378. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic Anhydrase Activators: L-Adrenaline Plugs the Active Site Entrance of Isozyme II, Activating Better Isoforms I, IV, VA, VII, and XIV. Bioorg. Med. Chem. Lett. 2007, 17, 628–635. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. I. Stop-Flow Kinetic Studies on the Native Human Isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Supuran, C.T.; Barboiu, M.; Luca, C.; Pop, E.; Brewster, M.E.; Dinculescu, A. Carbonic Anhydrase Activators. Part 14. Syntheses of Mono and Bis Pyridinium Salt Derivatives of 2-Amino-5-(2-Aminoethyl)- and 2-Amino-5-(3-Aminopropyl)-1,3,4-Thiadiazole and Their Interaction with Isozyme II. Eur. J. Med. Chem. 1996, 31, 597–606. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activators—Part 21. Novel Activators of Isozymes I, II and IV Incorporating Carboxamido and Ureido Histamine Moieties. Eur. J. Med. Chem. 2000, 35, 31–39. [Google Scholar] [CrossRef]
- Briganti, F.; Scozzafava, A.; Supuran, C.T. Novel Carbonic Anhydrase Isozymes I, II and IV Activators Incorporating Sulfonyl-Histamino Moieties. Bioorg. Med. Chem. Lett. 1999, 9, 2043–2048. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A. Carbonic Anhydrase Activators: Amino Acyl/Dipeptidyl Histamine Derivatives Bind with High Affinity to Isozymes I, II and IV and Act as Efficient Activators. Bioorg. Med. Chem. 1999, 7, 2915–2923. [Google Scholar] [CrossRef]
- Dave, K.; Ilies, M.A.; Scozzafava, A.; Temperini, C.; Vullo, D.; Supuran, C.T. An Inhibitor-like Binding Mode of a Carbonic Anhydrase Activator within the Active Site of Isoform II. Bioorg. Med. Chem. Lett. 2011, 21, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Dave, K.; Scozzafava, A.; Vullo, D.; Supuran, C.T.; Ilies, M.A. Pyridinium Derivatives of Histamine Are Potent Activators of Cytosolic Carbonic Anhydrase Isoforms I, II and VII. Org. Biomol. Chem. 2011, 9, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Rami, M.; Winum, J.Y.; Supuran, C.T.; Melnyk, P.; Yous, S. (Hetero)Aryl Substituted Thiazol-2,4-Yl Scaffold as Human Carbonic Anhydrase I, II, VII and XIV Activators. J. Enzym. Inhib. Med. Chem. 2019, 34, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saada, M.C.; Vullo, D.; Montero, J.L.; Scozzafava, A.; Winum, J.Y.; Supuran, C.T. Carbonic Anhydrase I and II Activation with Mono- and Dihalogenated Histamine Derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 4884–4887. [Google Scholar] [CrossRef]
- Saada, M.C.; Vullo, D.; Montero, J.L.; Scozzafava, A.; Supuran, C.T.; Winum, J.Y. Mono- and Di-Halogenated Histamine, Histidine and Carnosine Derivatives Are Potent Carbonic Anhydrase I, II, VII, XII and XIV Activators. Bioorg. Med. Chem. 2014, 22, 4752–4758. [Google Scholar] [CrossRef]
- Draghici, B.; Vullo, D.; Akocak, S.; Walker, E.A.; Supuran, C.T.; Ilies, M.A. Ethylene Bis-Imidazoles Are Highly Potent and Selective Activators for Isozymes VA and VII of Carbonic Anhydrase, with a Potential Nootropic Effect. Chem. Commun. 2014, 50, 5980–5983. [Google Scholar] [CrossRef] [Green Version]
- Mollica, A.; Macedonio, G.; Stefanucci, A.; Carradori, S.; Akdemir, A.; Angeli, A.; Supuran, C.T. Five- and Six-Membered Nitrogen-Containing Compounds as Selective Carbonic Anhydrase Activators. Molecules 2017, 22, 2178. [Google Scholar] [CrossRef] [Green Version]
- Akocak, S.; Lolak, N.; Bua, S.; Nocentini, A.; Karakoc, G.; Supuran, C.T. α-Carbonic Anhydrases Are Strongly Activated by Spinaceamine Derivatives. Bioorg. Med. Chem. 2019, 27, 800–804. [Google Scholar] [CrossRef]
- Akocak, S.; Lolak, N.; Bua, S.; Nocentini, A.; Supuran, C.T. Activation of Human α-Carbonic Anhydrase Isoforms I, II, IV and VII with Bis-Histamine Schiff Bases and Bis-Spinaceamine Substituted Derivatives. J. Enzym. Inhib. Med. Chem. 2019, 34, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; Kondeti, B.; Tu, C.; Maupin, C.M.; Silverman, D.N.; McKenna, R. Structural Insight into Activity Enhancement and Inhibition of H64A Carbonic Anhydrase II by Imidazoles. IUCrJ 2014, 1, 129–135. [Google Scholar] [CrossRef]
- Akocak, S.; Lolak, N.; Vullo, D.; Durgun, M.; Supuran, C.T. Synthesis and Biological Evaluation of Histamine Schiff Bases as Carbonic Anhydrase I, II, IV, VII, and IX Activators. J. Enzym. Inhib. Med. Chem. 2017, 32, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activators: High Affinity Isozymes I, II, and IV Activators, Incorporating a β-Alanyl-Histidine Scaffold. J. Med. Chem. 2001, 45, 284–291. [Google Scholar] [CrossRef]
- Abdo, M.R.; Vullo, D.; Saada, M.C.; Montero, J.L.; Scozzafava, A.; Winum, J.Y.; Supuran, C.T. Carbonic Anhydrase Activators: Activation of Human Isozymes I, II and IX with Phenylsulfonylhydrazido l-Histidine Derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 2440–2443. [Google Scholar] [CrossRef]
- Vistoli, G.; Aldini, G.; Fumagalli, L.; Dallanoce, C.; Angeli, A.; Supuran, C.T. Activation Effects of Carnosine- and Histidine-Containing Dipeptides on Human Carbonic Anhydrases: A Comprehensive Study. Int. J. Mol. Sci. 2020, 21, 1761. [Google Scholar] [CrossRef] [Green Version]
- Cararo, J.H.; Streck, E.L.; Schuck, P.F.; Ferreira, G.d.C. Carnosine and Related Peptides: Therapeutic Potential in Age-Related Disorders. Aging Dis. 2015, 6, 369–379. [Google Scholar]
- Saada, M.C.; Montero, J.L.; Vullo, D.; Scozzafava, A.; Winum, J.Y.; Supuran, C.T. Carbonic Anhydrase Activators: Gold Nanoparticles Coated with Derivatized Histamine, Histidine, and Carnosine Show Enhanced Activatory Effects on Several Mammalian Isoforms. J. Med. Chem. 2011, 54, 1170–1177. [Google Scholar] [CrossRef] [Green Version]
- Tanini, D.; Capperucci, A.; Supuran, C.T.; Angeli, A. Sulfur, Selenium and Tellurium Containing Amines Act as Effective Carbonic Anhydrase Activators. Bioorg. Chem. 2019, 87, 516–522. [Google Scholar] [CrossRef]
- Angeli, A.; Vaiano, F.; Mari, F.; Bertol, E.; Supuran, C.T. Psychoactive Substances Belonging to the Amphetamine Class Potently Activate Brain Carbonic Anhydrase Isoforms VA, VB, VII, and XII. J. Enzym. Inhib. Med. Chem. 2017, 32, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, A.; Ikeda, H.; Tsukamoto, H.; Kihira, K.; Ishioka, M.; Hirose, J.; Hata, T.; Fujioka, H.; Ono, Y. Timolol Activates the Enzyme Activities of Human Carbonic Anhydrase I and II. Biol. Pharm. Bull. 2010, 33, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Caccia, S.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Activators. The Selective Serotonin Reuptake Inhibitors Fluoxetine, Sertraline and Citalopram Are Strong Activators of Isozymes I and II. Bioorg. Med. Chem. Lett. 2003, 13, 2765–2768. [Google Scholar] [CrossRef]
- Abdülkadir Coban, T.; Beydemir, Ş.; Gücin, I.; Ekinci, D.; Innocenti, A.; Vullo, D.; Supuran, C.T. Sildenafil Is a Strong Activator of Mammalian Carbonic Anhydrase Isoforms I–XIV. Bioorg. Med. Chem. 2009, 17, 5791–5795. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Ludwig, P.A.; Christianson, D.W. Two-Site Binding of Phenol in the Active Site of Human Carbonic Anhydrase II: Structural Implications for Substrate Association. J. Am. Chem. Soc. 2002, 116, 3659–3660. [Google Scholar] [CrossRef]
- Mincione, F.; Scozzafava, A.; Supuran, C.T. Antiglaucoma Carbonic Anhydrase Inhibitors as Ophthalomologic Drugs. In Drug Design of Zinc-Enzyme Inhibitors: Functional, Structural, and Disease Applications; Wiley: Hoboken, NJ, USA, 2009; pp. 139–153. [Google Scholar]
- Forray, A.; Sofuoglu, M. Future Pharmacological Treatments for Substance Use Disorders. Br. J. Clin. Pharmacol. 2014, 77, 382–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provensi, G.; Nocentini, A.; Passani, M.B.; Blandina, P.; Supuran, C.T. Activation of Carbonic Anhydrase Isoforms Involved in Modulation of Emotional Memory and Cognitive Disorders with Histamine Agonists, Antagonists and Derivatives. J. Enzym. Inhib. Med. Chem. 2021, 36, 719–726. [Google Scholar] [CrossRef]
- Le Duc, Y.; Licsandru, E.; Vullo, D.; Barboiu, M.; Supuran, C.T. Carbonic Anhydrases Activation with 3-Amino-1H-1,2,4-Triazole-1-Carboxamides: Discovery of Subnanomolar Isoform II Activators. Bioorg. Med. Chem. 2017, 25, 1681–1686. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, A.; Cuffaro, D.; Ciccone, L.; Orlandini, E.; Nencetti, S.; Nuti, E.; Rossello, A.; Supuran, C.T. Activation of Carbonic Anhydrases from Human Brain by Amino Alcohol Oxime Ethers: Towards Human Carbonic Anhydrase VII Selective Activators. J. Enzym. Inhib. Med. Chem. 2021, 36, 48–57. [Google Scholar] [CrossRef]
- Ilies, M.; Supuran, C.T.; Scozzafava, A. Carbonic Anhydrase Activators. In Carbonic Anhydrase—Its Inhibitors and Activators; CRC Press: Boca Raton, FL, USA, 2004; pp. 317–352. [Google Scholar]
- Chiaramonte, N.; Maach, S.; Biliotti, C.; Angeli, A.; Bartolucci, G.; Braconi, L.; Dei, S.; Teodori, E.; Supuran, C.T.; Romanelli, M.N. Synthesis and Carbonic Anhydrase Activating Properties of a Series of 2-Amino-Imidazolines Structurally Related to Clonidine1. J. Enzym. Inhib. Med. Chem. 2020, 35, 1003–1010. [Google Scholar] [CrossRef]
- Chiaramonte, N.; Gabellini, A.; Angeli, A.; Bartolucci, G.; Braconi, L.; Dei, S.; Teodori, E.; Supuran, C.T.; Romanelli, M.N. New Histamine-Related Five-Membered N-Heterocycle Derivatives as Carbonic Anhydrase I Activators. Molecules 2022, 27, 545. [Google Scholar] [CrossRef]
- Maccallini, C.; Di Matteo, M.; Vullo, D.; Ammazzalorso, A.; Carradori, S.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Pandolfi, A.; Supuran, C.T.; et al. Indazole, Pyrazole, and Oxazole Derivatives Targeting Nitric Oxide Synthases and Carbonic Anhydrases. ChemMedChem 2016, 11, 1695–1699. [Google Scholar] [CrossRef]
- Barresi, E.; Ravichandran, R.; Germelli, L.; Angeli, A.; Baglini, E.; Salerno, S.; Marini, A.M.; Costa, B.; Da Pozzo, E.; Martini, C.; et al. Carbonic Anhydrase Activation Profile of Indole-Based Derivatives. J. Enzym. Inhib. Med. Chem. 2021, 36, 1783–1797. [Google Scholar] [CrossRef]
- Taliani, S.; Da Settimo, F.; Martini, C.; Laneri, S.; Novellino, E.; Greco, G. Exploiting the Indole Scaffold to Design Compounds Binding to Different Pharmacological Targets. Molecules 2020, 25, 2331. [Google Scholar] [CrossRef]
- Gomes, C.; Ferreira, R.; George, J.; Sanches, R.; Rodrigues, D.I.; Gonçalves, N.; Cunha, R.A. Activation of Microglial Cells Triggers a Release of Brain-Derived Neurotrophic Factor (BDNF) Inducing Their Proliferation in an Adenosine A2A Receptor-Dependent Manner: A2A Receptor Blockade Prevents BDNF Release and Proliferation of Microglia. J. Neuroinflamm. 2013, 10, 780. [Google Scholar] [CrossRef] [Green Version]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Lume, M.; Matsumoto, T.; Hara, T.; Yamawaki, S.; Takahashi, M.; Shiosaka, S.; Itami, C.; et al. BDNF Pro-Peptide Actions Facilitate Hippocampal LTD and Are Altered by the Common BDNF Polymorphism Val66Met. Proc. Natl. Acad. Sci. USA 2015, 112, E3067–E3074. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
α-CA catalytic site: superposition of CA II crystal structure in complex with CO2 (pdb 2VVA) and HCO3− (pdb 2VVB). In the circle, an enlargement of the catalytic domain [9]. Under CC BY 4.0 license.
Figure 1.
α-CA catalytic site: superposition of CA II crystal structure in complex with CO2 (pdb 2VVA) and HCO3− (pdb 2VVB). In the circle, an enlargement of the catalytic domain [9]. Under CC BY 4.0 license.

Figure 2.
Zinc-bound hydroxylic ion interactions.

Figure 3.
Catalytic cycle of CAs. (A)The water molecule transforms itself into a hydroxylated species. (B) The hydroxyl ion attacks CO2. (C) Formation of the species in which HCO3− is coordinated to the zinc ion. (D) Rebuilding of the inactive state of the enzyme.
Figure 3.
Catalytic cycle of CAs. (A)The water molecule transforms itself into a hydroxylated species. (B) The hydroxyl ion attacks CO2. (C) Formation of the species in which HCO3− is coordinated to the zinc ion. (D) Rebuilding of the inactive state of the enzyme.

Figure 4.
Mechanisms underlying the actions of CAs on cognition [28]: effects on GABAergic post-synaptic potential (A); effects on ERK pathways (B). Under CC BY 4.0 license.
Figure 4.
Mechanisms underlying the actions of CAs on cognition [28]: effects on GABAergic post-synaptic potential (A); effects on ERK pathways (B). Under CC BY 4.0 license.

Table 1.
Amino acids and amines studied as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 1 Phenylalanine |  | hCA I (L-): KA = 70 nM hCA I (D-): KA = 86 μM hCA II (L-): KA = 13 nM hCA II (D-): KA = 35 nM hCA XIV (L-): KA = 0.24 μM hCA XIV (D-): KA = 7.21 μM | [33] |
| 2 Histidine |  | hCA I (L-): KA = 30 nM hCA I (D-): KA = 90 nM hCA VII (L-): KA = 0.92 μΜ hCA VII (D-): KA = 0.71 μΜ hCA VA (D-): KA = 0.12 μΜ | [34] |
| 3 DOPA |  | hCA VA (L-): KA = 36 nM hCA VB (L-): KA = 63 nM hCA XIII (D-): KA = 0.81 μM | [4] |
| 4 Tryptophan |  | hCA XIII (L-): KA = 0.81 μM | [35] |
| 5 Tyrosine |  | [3,4] | |
| 6 4-NH2-L-Phe |  | [3] | |
| 7 Histamine |  | hCA VA: KA = 10 nM hCA XIV: KA = 10 nM | [3,4] |
| 8 Dopamine |  | hCA VA: KA = 0.13 μM hCA VII: KA = 0.89 μM | [3,4] |
| 9 Serotonine |  | [3,4] | |
| 10n = 1 11 n = 2 |  | [3,4] | |
| 12 X = NH 13 X = O |  | hCA (13): KA = 0.13–0.43 μM (except for hCA VB and VII) | [3,4] |
| 14 Adrenaline |  | hCA I: KA = 90 nM hCA XII: KA = 0.41 μM | [36] |
Table 2.
CAAs based on histamine structure.
| Compound n | Structure | CA Activation | Ref. |
|---|---|---|---|
| 15 |  | hCA II (R = CH3; n = 2): 147% activation rate at 10 μM | [38] |
| 16 |  | hCA II (n = 2): 163% activation rate at 10 μM | [38] |
| 17 |  R = alkyl, aryl; n = 2,3 | hCA II (R = CH3; n = 2): 184% activation rate at 10 μM | [38] |
| 18 |  | hCA I: KA = 4.0 nM–0.27 μM hCA II: KA = 0.10–0.86 μM bCA IV: KA = 20 nM–21 μM | [39] |
| 19 |  X = O, S | hCA I: KA = 4.0 nM–36 μM hCA II: KA = 80 nM–16 μM bCA IV: KA = 20 nM–12 μM hCA II: KA > 200 μM | [11,39] |
| 20 |  | hCA I: KA = 6.0 nM–0.28 μM hCA II: KA = 80 nM–34 μM bCA IV: KA = 10 nM–7.0 μM | [40] |
| 21 |  | hCA I: KA = 3.0–6.0 nM hCA II: KA = 80 nM–0.15 μM bCA IV: KA = 10–30 nM | [40] |
| 22 |  | hCA I: KA = 6.0 nM hCA II: KA = 0.12 μM bCA IV: KA = 30 nM | [39] |
| 23 |  | hCA I: KA = 1.0 nM–0.21 μM hCA II: KA = 10 nM–11 μM bCA IV: KA = 3.0 nM–4.6 μM | [41] |
| 24 |  | hCA I: KA = 0.73–3.4 μM hCA II: KA > 200 μM | [11] |
| 24a |  | hCA I: KA = 0.73 μM | [11] |
| 25 |  | hCA II: 156% activation rate at 20 μM | [42] |
| 26 |  | hCA I: KA = 0.5 nM–93 μM hCA II: KA = 9 nM–78 μM hCA VII: KA = 0.8 nM–1.16 μM | [43] |
| 27 |  X, Y = CH, N | [44] | |
| 28 |  | hCA I: KA = 63.4 μM hCA II: KA = 68.1 μM hCA VII: KA = 7.5 μM | [44] |
Table 3.
Histamine based compounds as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 29 |  X = Cl, Br, I | hCA I: KA = 0.7–21 nM hCA II: KA = 1.0–115 nM | [45] |
| 30 |  X = Cl, Br, I | hCA I: KA = 0.7–21 nM hCA II: KA = 1.0–115 nM | [45] |
| 31 |  X = Cl, Br, I | hCA I: KA = 5.4–29.3 μM hCA II: KA = 13.6–50.2 μM | [45] |
| 32 |  X = Cl, Br, I | hCA I: KA = 5.4–29.3 μM hCA II: KA = 13.6–50.2 μM | [45] |
| 33 |  R = H, Me, Et, i-Pr, Ph | hCA VA: KA = 9.0–131 nM hCA VII: KA = 15–89 nM | [47] |
| 34 |  | [48] | |
| 35 |  | [48] | |
| 36 |  | [48] | |
| 37 L-(+)-Ergothioneine |  | [48] | |
| 38 Melatonin |  | [48] | |
| 39 Spinacine |  | [48] | |
| 40 |  R = Aryl, furyl | hCA VII selective: KA = 82–840 nM | [49] |
| 41 |   | hCA VII selective: KA = 32–39 nM | [50] |
| 42 |   | hCA I, II, IV, VII: KA = 3.28–42.1 μM hCA VII (42d selective): KA = 85 nM | [50] |
Table 4.
Histidine- and carnosine-based derivatives 43–48 as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 43 Carnosine |  | [53] | |
| 44 |  X = 4-F, 2-Me R1, R2 = amino acidic residues | hCA I, bCA IV: KA = 1.0–20 nM hCA II: KA = 10–40 nM | [53] |
| 45 |  | hCA II: KA = 0.21 μM | [54] |
| 46 |  X1 = Cl, Br; X2 = H, Cl, Br, I R = H, Butyloxycarbonyl (Boc) | [46] | |
| 47 |  X1 = Cl, Br; X2 = H, Cl, Br, I R = H, Butyloxycarbonyl (Boc) | [46] | |
| 46a |  | hCA I: KA = 0.9 nM hCA II: KA = 12 nM | [46] |
| 46b |  | hCA I: KA = 1.2 nM hCA II: KA = 0.32 μM | [46] |
| 46c |  | hCA I: KA = 1.5 nM hCA II: KA = 13 nM | [46] |
| 46d |  | hCA VII: KA = 9.7 μM | [46] |
| 47a |  | hCA I: KA = 4.3 μM hCA II: KA = 5.2 μM hCA VII: KA = 0.81 μM | [46] |
| 47b |  | hCA VII: KA = 8.1 nM | [46] |
| 47c |  | hCA VII: KA = 4.0 μM | [46] |
| 48 |  X = H, CH3; R1 = H, acetyl, Gly; R = H, COOH, CONH2, CH2OH; n = 1, 2, 3 | [55] | |
| 48a D-Carnosinamide |  | hCA I: KA = 16.6 μM | [55] |
| 48b Carnicine |  | hCA I: KA = 16.6 μM hCA VA: KA = 6.4 μM | [55] |
| 48c L-Anserine |  | hCA IX: KA = 1.14 μM | [55] |
Table 5.
Gold nanoparticles of histamine, histidine, and carnosine derivatives 49–51, and sulfur-, selenium- and tellurium-containing amines 52–54 as CAAs.
Table 5.
Gold nanoparticles of histamine, histidine, and carnosine derivatives 49–51, and sulfur-, selenium- and tellurium-containing amines 52–54 as CAAs.
| Compound n | Structure | CA Activation | Ref. |
|---|---|---|---|
| 49 |  | KA = 1–7 nM (details in Table 6) | [57] |
| 50 |  | KA = 1–8 nM (details in Table 6) | [57] |
| 51 |  | KA = 1–9 nM (details in Table 6) | [57] |
| 52 |  | hCA I: KA = 7.7–13.5 μM hCA VA: KA = 10.2–43.7 μM hCA VII: KA = 11.4–23.4 μM | [58] |
| 53 |  | hCA I: KA = 5.2–22.1 μM hCA VA: KA = 6.6–20.9 μM hCA VII: KA = 10.1–23.3 μM | [58] |
| 54 |  | hCA I: KA = 4.6–13.3 μM hCA VA: KA = 3.3–20.2 μM hCA VII: KA = 8.9–44.9 μM | [58] |
Table 6.
Activation data of human CA isoforms I, II, VA, and VII with compounds 49–51 in comparison with their starting compounds, using a stopped flow CO2 hydrase assay [37].
Table 6.
Activation data of human CA isoforms I, II, VA, and VII with compounds 49–51 in comparison with their starting compounds, using a stopped flow CO2 hydrase assay [37].
| Isoform/ Compound | KA (μM) | |||||
|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA IV | hCA VA | hCA VII | hCA XIV | |
| Histamine (7) | 2.1 | 125 | 25.3 | 0.010 | 37.5 | 0.010 |
| 49 | 0.005 | 0.002 | 0.001 | 0.001 | 0.003 | 0.007 |
| L-His (2) | 0.03 | 10.9 | 7.3 | 1.34 | 0.92 | 0.90 |
| L-His-OMe | 0.02 | 10.4 | 6.8 | 1.86 | 0.88 | 0.93 |
| 50 | 0.002 | 0.008 | 0.001 | 0.002 | 0.003 | 0.001 |
| L-carnosine (43) | 1.1 | 33 | 19 | 1.54 | 0.75 | 0.64 |
| L-carnosine-OMe | 10.9 | 32 | 18 | 1.36 | 0.84 | 0.71 |
| 51 | 0.009 | 0.007 | 0.002 | 0.002 | 0.001 | 0.001 |
Table 7.
Structures of drugs 55–64 as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 55 Timolol |  | hCA I: KA = 12 μM hCA II: KA = 9.3 μM | [60] |
| 56 Fluoxetine |  | hCA I: 175% activation rate at 1 μM hCA II: 165% activation rate at 1 μM | [61] |
| 57 Sertraline |  | hCA I: 145% activation rate at 1 μM hCA II: 140% activation rate at 1 μM | [61] |
| 58 Citalopram |  | hCA I: 134% activation rate at 1 μM hCA II: 170% activation rate at 1 μM | [61] |
| 59 Sildenafil |  | hCA I: KA = 1.08 μM hCA VB: KA = 6.54 μM hCA VI: KA = 2.37 μM | [62] |
| 60 Amphetamine |  | hCA IV: KA = 94 nM hCA VA: KA = 0.81 μM hCA VB: KA = 2.56 μM hCA VII: KA = 0.91 μM | [59] |
| 61 Methamphetamine |  | hCA IV: KA = 51 nM hCA VA: KA = 0.92 μM hCA VB: KA = 0.78 μM hCA VII: KA = 0.93 μM | [59] |
| 62 Phentermine |  | hCA IV: KA = 74 nM hCA VA: KA = 0.53 μM hCA VB: KA = 0.62 μM hCA VII: KA = 0.89 μM | [59] |
| 63 Mephentermine |  | hCA IV: KA = 1.03 μM hCA VA: KA = 0.37 μM hCA VB: KA = 0.24 μM hCA VII: KA = 0.64 μM | [59] |
| 64 Chlorphenteramine |  | hCA IV: KA = 55 nM hCA VA: KA = 0.31 μM hCA VB: KA = 0.75 μM hCA VII: KA = 98 nM | [59] |
Table 8.
Structures of histamine receptors agonists/antagonists 65–74 as CAAs.
| Compound n/Name | Structure | CA Activation | Ref. |
|---|---|---|---|
| 65 α-methyl histamine |  | hCA I: KA = 0.12 μM hCA II: KA = 82 nM hCA VII: KA = 1.25 μM | [66] |
| 66 4-methyl histamine |  | hCA I: KA = 0.36 μM hCA II: KA = 5.4 μM hCA VII: KA = 0.39 μM | [66] |
| 67 1-methyl histamine |  | hCA I: KA = 52 nM hCA II: KA = 0.57 μM hCA VII: KA = 0.19 μM | [66] |
| 68 2-(2-aminoethyl) thiazole |  | hCA I: KA = 0.87 μM hCA II: KA = 7.45 μM hCA VII: KA = 0.7 μM | [66] |
| 69 Burimamide |  | hCA I: KA = 0.88 μM hCA II: KA = 8.39 μM hCA VII: KA = 0.43 μM | [66] |
| 70 Metiamide |  | hCA I: KA = 0.98 μM hCA II: KA = 8.75 μM hCA VII: KA = 1.01 μM | [66] |
| 71 Impromidine |  | hCA I: KA = 0.72 μM hCA II: KA = 2.14 μM hCA VII: KA = 0.10 μM | [66] |
| 72 Methimmepip |  | hCA I: KA = 3.16 μM hCA II: KA = 5.24 μM hCA VII: KA = 0.12 μM | [66] |
| 73 Proxyfan |  | hCA I: KA = 3.15 μM hCA II: KA = 7.66 μM hCA VII: KA = 0.52 μM | [66] |
| 74 Ciproxyfan |  | hCA I: KA = 4.29 μM hCA II: KA = 9.9 μM hCA VII: KA = 0.11 μM | [66] |
Table 9.
Ureas and di-ureas incorporating 1,2,4-triazole derivatives 75–76 and amino alcohol oxime ethers 77 as CAAs.
Table 9.
Ureas and di-ureas incorporating 1,2,4-triazole derivatives 75–76 and amino alcohol oxime ethers 77 as CAAs.
| Compound n | Structure | CA Activation | Ref. |
|---|---|---|---|
| 75 |  R, R′ = alkyl, aryl Y = NH2, H Z = H, NH2, COOH | [67] | |
| 76 |  R, R′ = alkyl, aryl Y = NH2, H Z = H, NH2, COOH | [67] | |
| 75a |  | hCA I: KA = 6.1 nM hCA II: KA = 1.7 nM | [67] |
| 76a |  | hCA I: KA = 0.81 nM hCA II: KA = 14.4 nM | [67] |
| 76b |  | hCA I: KA = 0.94 nM hCA II: KA = 0.05 nM | [67] |
| 76c |  | hCA I: KA = 65 nM hCA II: KA = 0.12 nM | [67] |
| 77 |  R1 = aryl, cicloalkyl R2 = i-propyl, t-butyl | [68] | |
| 77a |  | hCA I: KA = 7.10 μM hCA II: KA = 79 nM hCA IV: KA = 6.01 μM hCA VII: KA = 0.42 μM | [68] |
| 77b |  | hCA I: KA = 12.1 μM hCA II: KA = 2.50 μM hCA IV: KA = 7.73 μM hCA VII: KA = 82 nM | [68] |
| 77c |  | hCA I: KA = 8.15 μM hCA II: KA = 1.94 μM hCA IV: KA = 1.08 μM hCA VII: KA = 91 nM | [68] |
Table 11.
Indazole, pyrazole, and oxazole derivatives 83–87 as CAAs.
| Compound n | Structure | CA Activation | Ref. |
|---|---|---|---|
| 83a–c |  a: R = CH3 (Ala) b: R = p-OH-C6H4 (Tyr) c: R = CH2CH2COOH (Glu) | [72] | |
| 84a–c |  a: R = CH3 (Ala) b: R = p-OH-C6H4 (Tyr) c: R = CH2CH2COOH (Glu) | [72] | |
| 85a–c |  a: R = CH3 (Ala) b: R = p-OH-C6H4 (Tyr) c: R = CH2CH2COOH (Glu) | [72] | |
| 86a–c |  a: R = CH3 (Ala) b: R = p-OH-C6H4 (Tyr) c: R = CH2CH2COOH (Glu) | [72] | |
| 87a–c |  a: R = CH3 (Ala) b: R = p-OH-C6H4 (Tyr) c: R = CH2CH2COOH (Glu) | [72] | |
| 84b |  | hCA I: KA = 9.0 nM hCA VII: KA = 0.69 μM | [72] |
| 85a |  | hCA I: KA = 6.39 nM hCA VII: KA = 0.59 μM | [72] |
| 85b |  | hCA I: KA = 15 nM hCA VII: KA = 10.8 μM | [72] |
| 85c |  | hCA I: KA = 4.12 μM hCA VII: KA = 0.51 μM | [72] |
Table 12.
Indole-based derivatives 88–90 as CAAs.
| Compound n/ | Structure | CA Activation | Ref. |
|---|---|---|---|
| 88 |  R1 = CH2CH2OH, CH2CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2, CH2Ph R2 = CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2 | [73] | |
| 89 |  R1 = CH2CH2OH, CH2CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2, CH2Ph R2 = CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2 | [73] | |
| 90 |  R1 = CH2CH2OH, CH2CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2, CH2Ph R2 = CH2CH2OH, CH2CH2N(CH3)2, CH2CH2N(C2H5)2, CH2CH2CH2N(CH3)2 | [73] | |
| 89a |  | hCA VII: KA = 7.5 μM | [73] |
| 90a |  | hCA VII: KA = 7.2 μM | [73] |
| 90b |  | hCA VII: KA = 8.2 μM | [73] |
Table 13.
Properties of the most interesting compounds reviewed.
| Compound | Class | In Vitro Activity | Other Correlate Activities | Ref. | |
|---|---|---|---|---|---|
| hCA Activation Activity (KA) | Selectivity | ||||
| 21 | Arylsulfonylureido derivatives of histamine | 3–6 nM | hCA I | - | [40] |
| 22 | Histamine dimers | 6 nM | hCA I | - | [39] |
| 29–30 | Histamine-based halogenated compounds | 0.7–21 nM 1.0–115 nM | dual hCA I/ hCA II | - | [45] |
| 41 | Histamine inspired-compounds | 32–39 nM | hCA VII | - | [49] |
| 42d | Histamine inspired-compounds | 85 nM | hCA VII | - | [49] |
| 44 | Carnosine-based derivatives | 1–20 nM 10–40 nM | dual hCA I/ hCA II | Enhancement of red cell CA activity | [53] |
| 46a | Histidine-based derivatives | 0.9 nM | hCA I | - | [46] |
| 47b | Carnosine-based derivatives | 8.1 nM | hCA VII | - | [46] |
| 49–51 | Gold nanoparticles of histamine, histidine, carnosine derivatives | 1–9 nM | no-selectivity (I, II, IV, VA, VII, IVX) | - | [45] |
| 53–54 | Selenium and tellurium containing amines | 3.3–44.9 μM | no-selectivity (I, VA, VII) | ROS inhibition | [58] |
| 76b | Di-ureas incorporating 1,2,4-triazole derivatives | 0.05 nM | hCA II | - | [67] |
| 77a | Amino alcohol oxime ethers | 79 nM | hCA II | - | [68] |
| 77b | Amino alcohol oxime ethers | 82 nM | hCA VII | - | [68] |
| 85b | Indazole derivartives | 15 nM | hCA I | nNOS inhibitor | [72] |
| 89a | Indole-based derivatives | 7.5 μM | hCA VII | BDNF production | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Poggetti, V.; Salerno, S.; Baglini, E.; Barresi, E.; Da Settimo, F.; Taliani, S. Carbonic Anhydrase Activators for Neurodegeneration: An Overview. Molecules 2022, 27, 2544. https://doi.org/10.3390/molecules27082544
AMA Style
Poggetti V, Salerno S, Baglini E, Barresi E, Da Settimo F, Taliani S. Carbonic Anhydrase Activators for Neurodegeneration: An Overview. Molecules. 2022; 27(8):2544. https://doi.org/10.3390/molecules27082544
Chicago/Turabian StylePoggetti, Valeria, Silvia Salerno, Emma Baglini, Elisabetta Barresi, Federico Da Settimo, and Sabrina Taliani. 2022. "Carbonic Anhydrase Activators for Neurodegeneration: An Overview" Molecules 27, no. 8: 2544. https://doi.org/10.3390/molecules27082544