
Inhibition of Filamentous Thermosensitive Mutant-Z Protein in Bacillus subtilis by Cyanobacterial Bioactive Compounds

 , ,
, ,  , , , , ,
, , , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
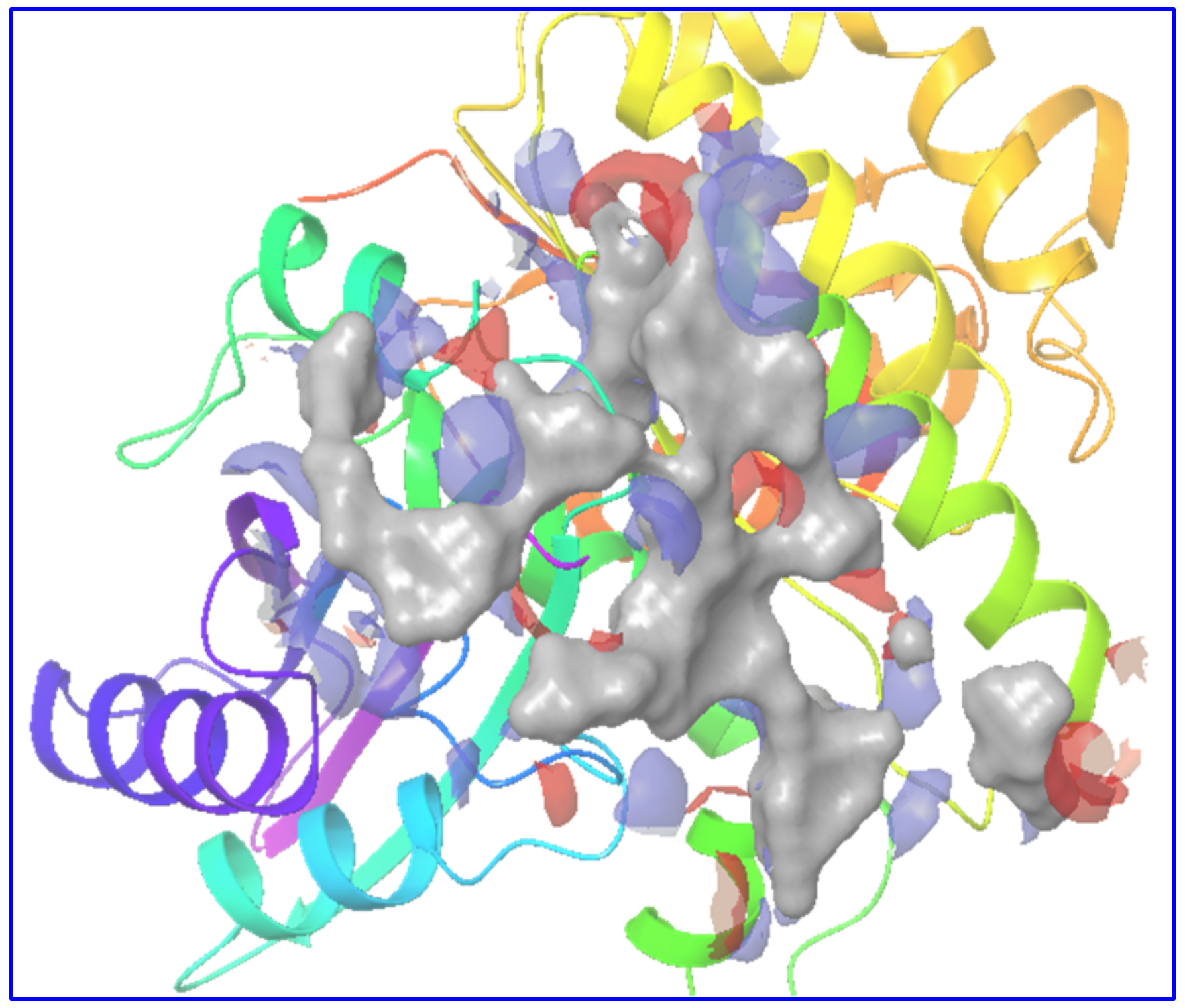
2.1. Preparation of Receptor and Its Binding Site
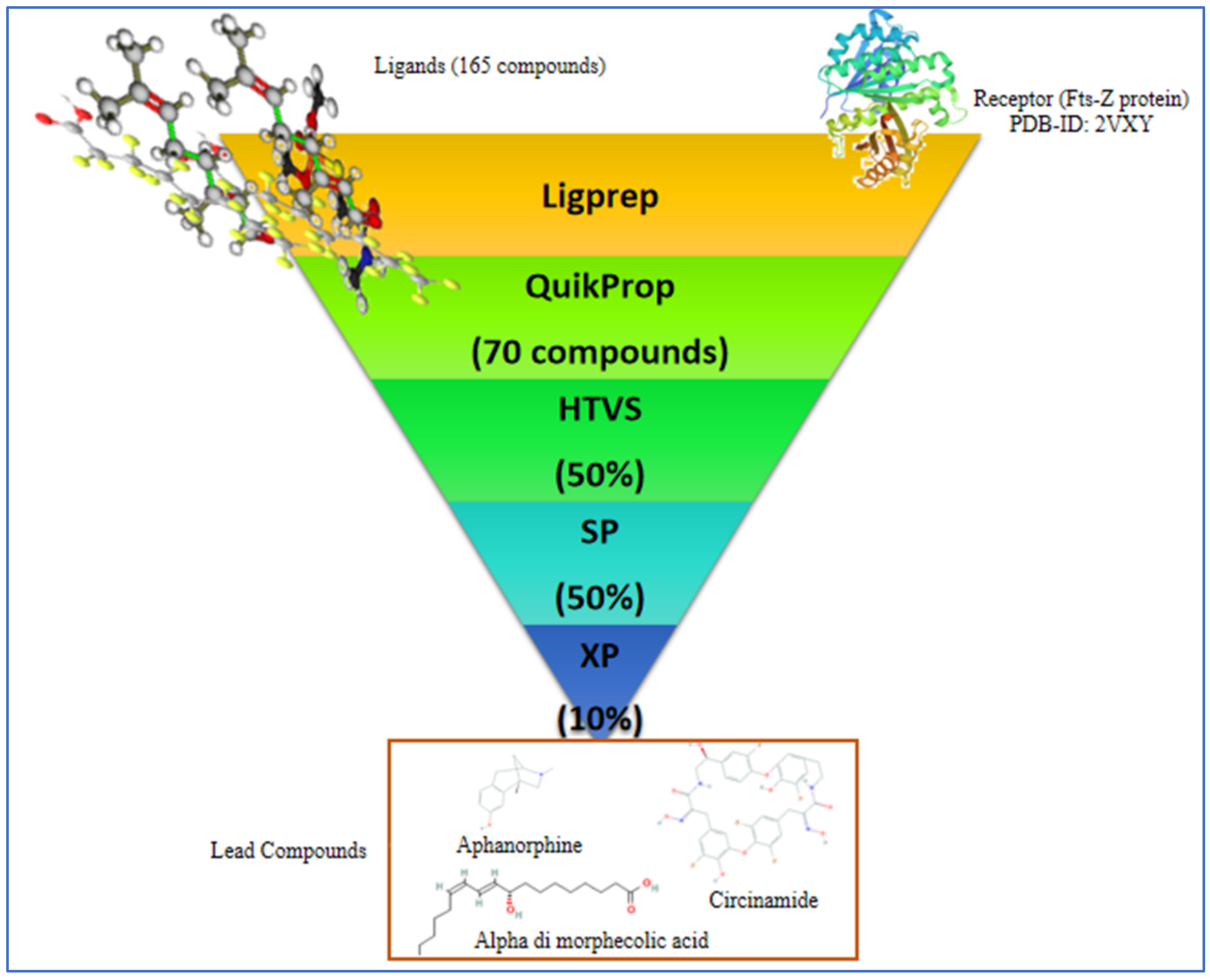
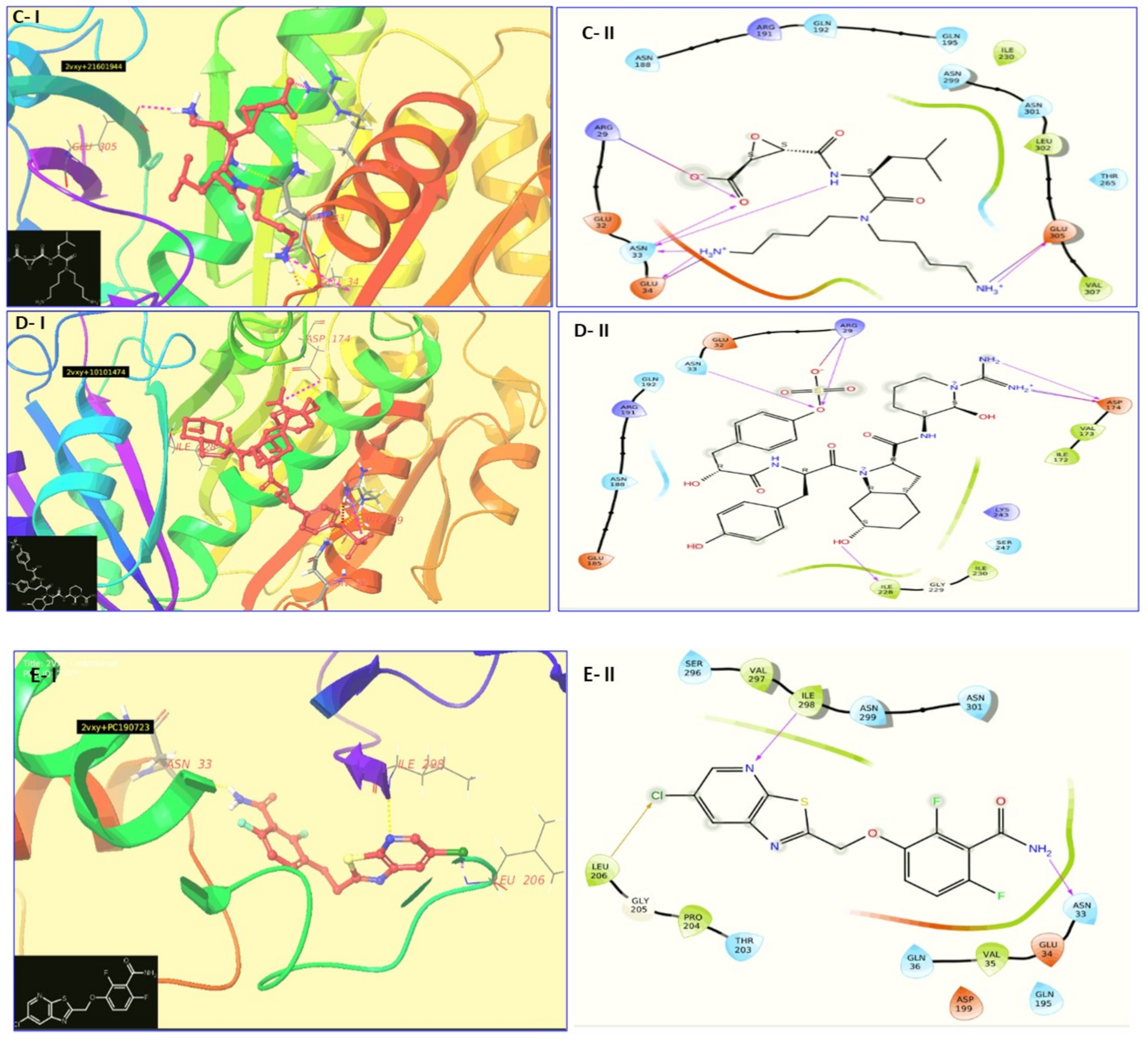
2.2. Virtual Screening and Molecular Docking
2.3. ADME Analysis Using Qik Prop Module
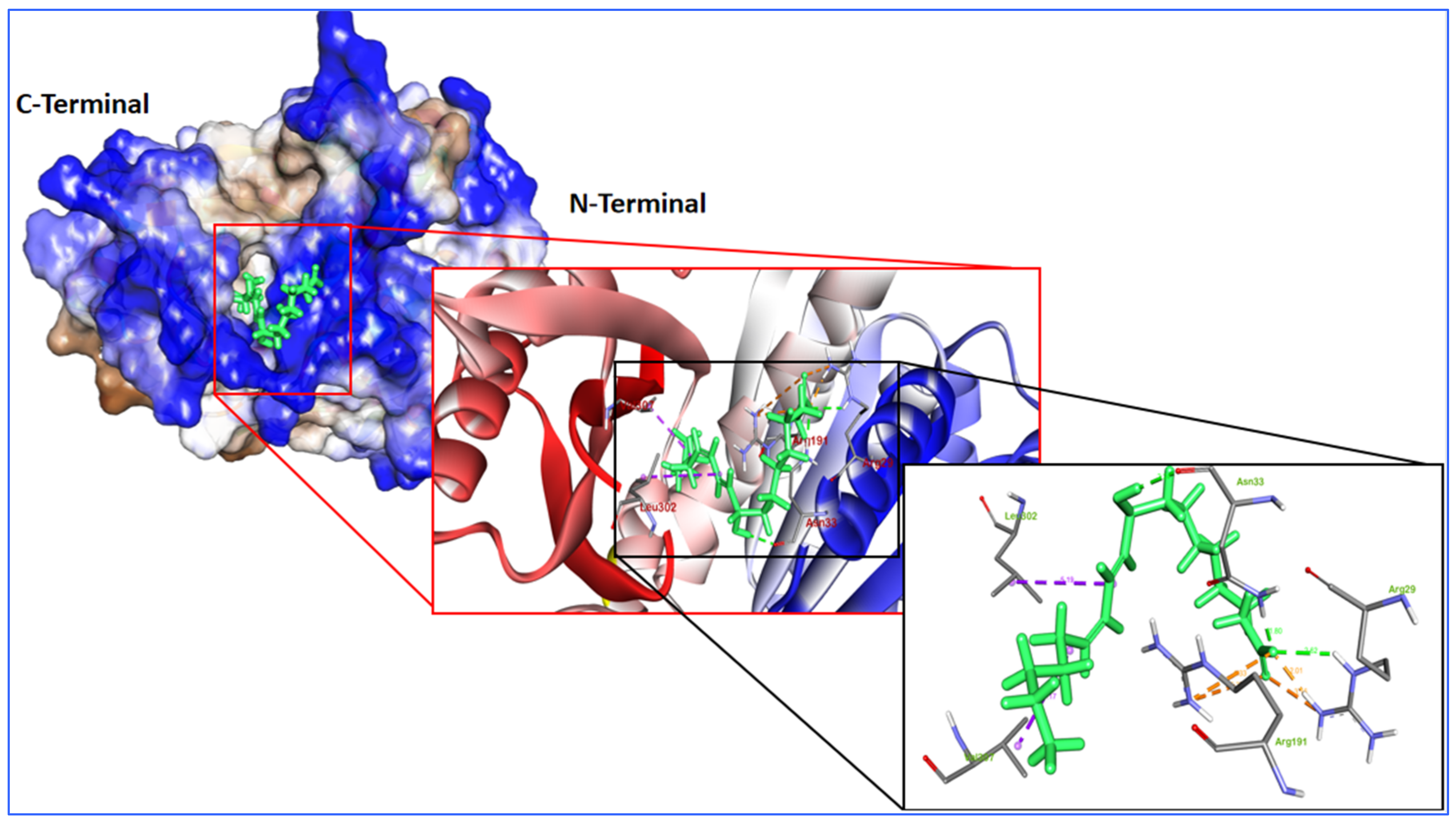
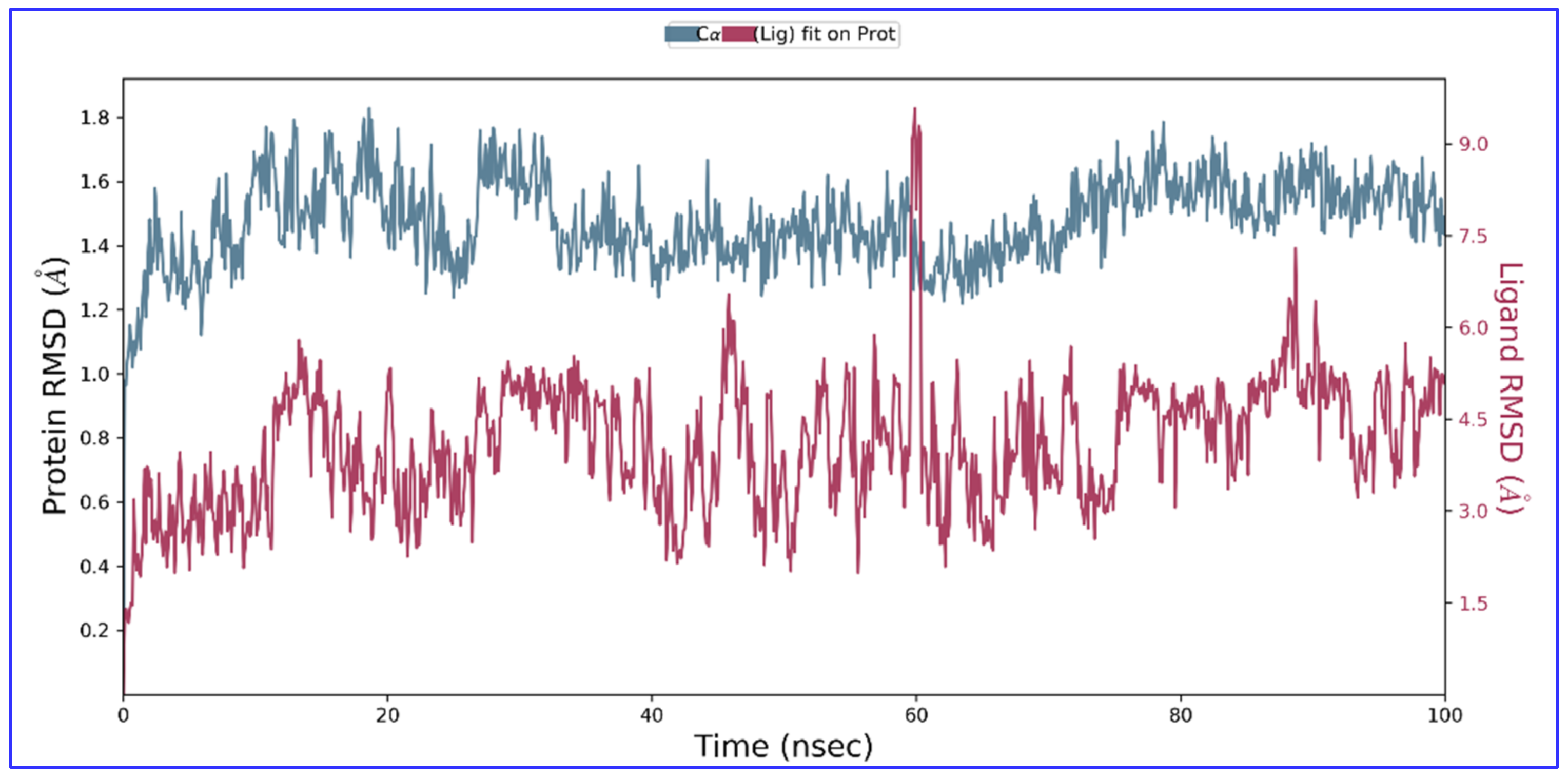
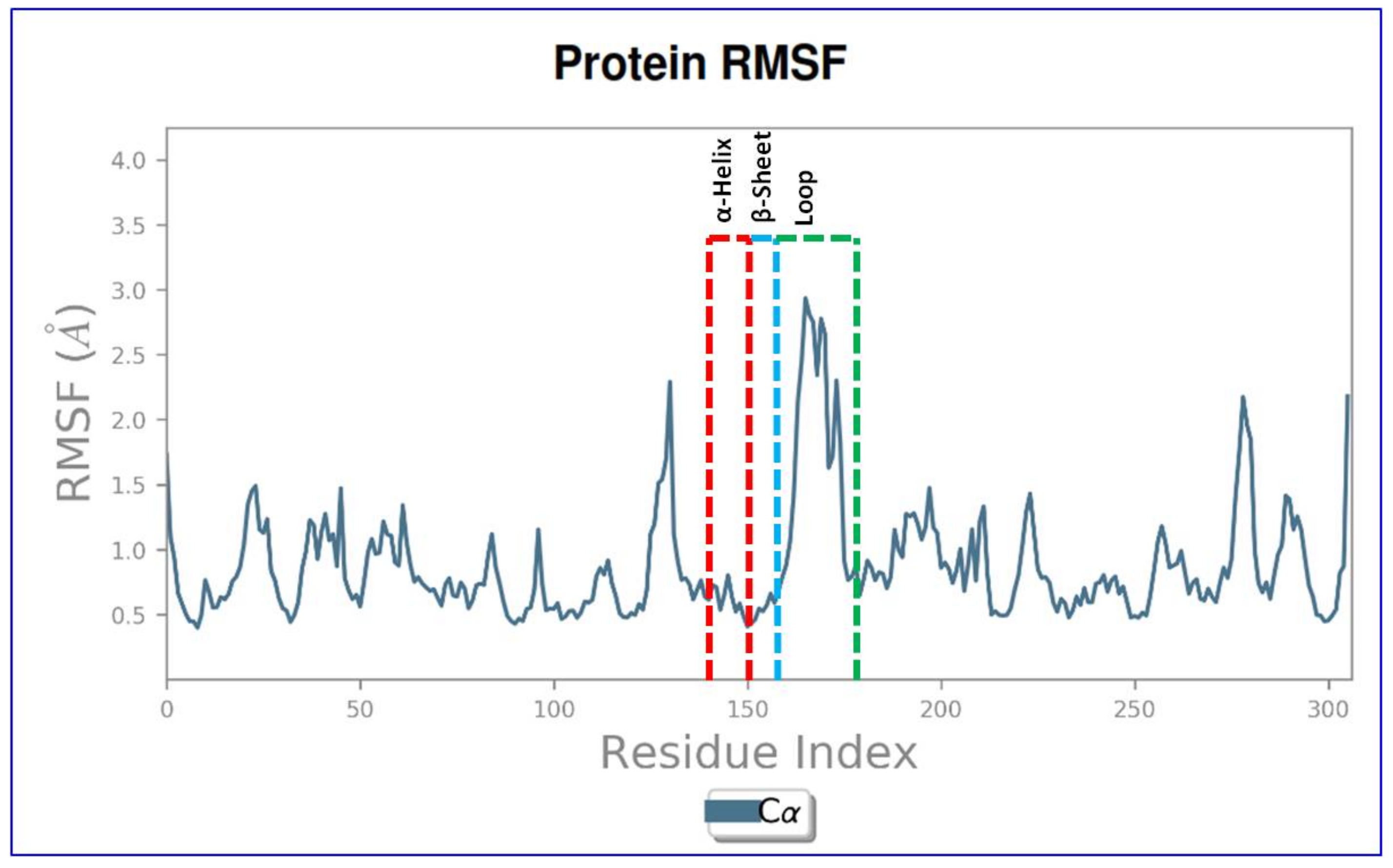
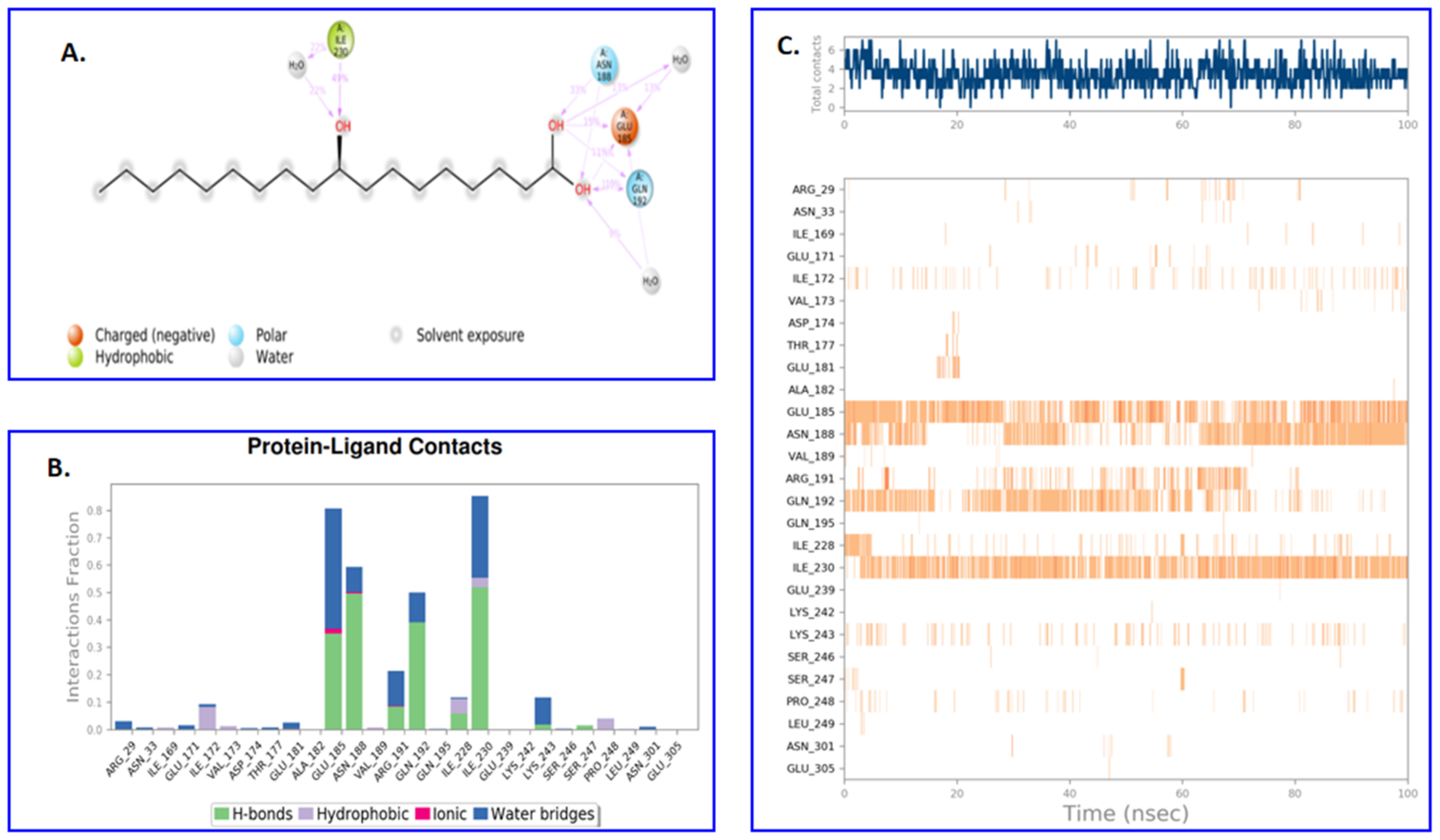
2.4. Molecular Dynamics and Simulation
3. Discussion
4. Materials and Methods
4.1. Target and Ligands Retrieval
4.2. Preparation of Ligands and ADME Analysis
4.3. Preparation of Receptor and Its Binding Site
4.4. Virtual Screening and Molecular Docking
4.5. Binding Free Energy Calculation
4.6. Molecular Dynamics and Simulation
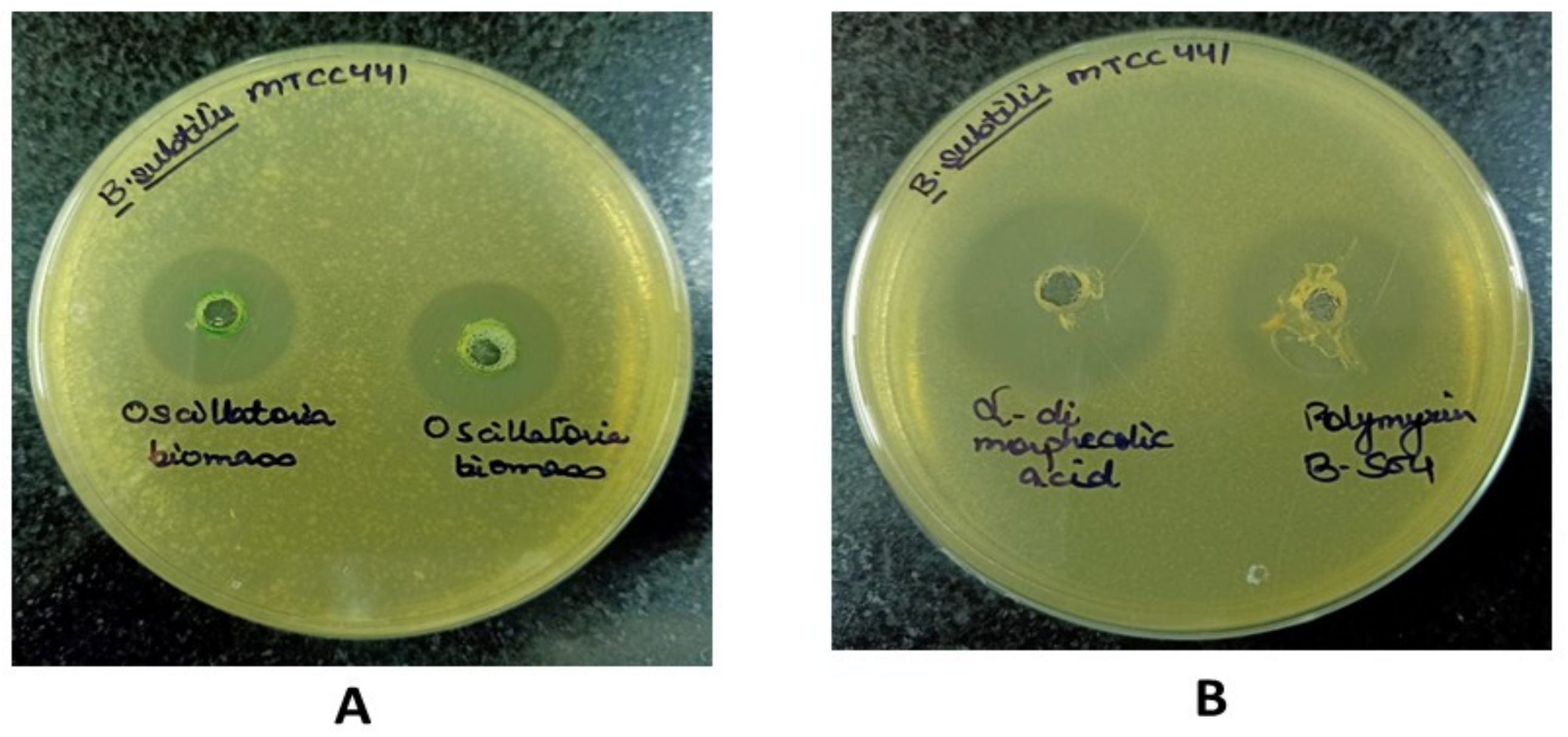
4.7. In Vitro Validation of Lead Compound
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Huang, I.S.; Zimba, P.V. Erratum: Cyanobacterial bioactive metabolites-A review of their chemistry and biology (Harmful algae (2019) 83 (42–94) PII: S1568-9883(19)30058-7). Harmful Algae 2019, 86, 139–209. [Google Scholar] [CrossRef]
- Dixit, R.B.; Suseela, M.R. Cyanobacteria: Potential candidates for drug discovery. Antonie Leeuwenhoek 2013, 103, 947–961. [Google Scholar] [CrossRef]
- Singh, R.; Parihar, P.; Singh, M.; Bajguz, A.; Kumar, J.; Singh, S.; Singh, V.P.; Prasad, S.M. Uncovering potential applications of cyanobacteria and algal metabolites in biology, agriculture, and medicine: Current status and future prospects. Front. Microbiol. 2017, 8, 515. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Kate, B.N.; Banecjee, U.C. Bioactive compounds from cyanobacteria and microalgae: An overview. Crit. Rev. Biotechnol. 2005, 25, 73–95. [Google Scholar] [CrossRef]
- Swain, S.S.; Padhy, R.N.; Singh, P.K. Anticancer Compounds from Cyanobacterium Lyngbya Species: A Review; Springer International Publishing: New York, NY, USA, 2015; Volume 108, ISBN 1048201504872. [Google Scholar]
- Swain, S.S.; Paidesetty, S.K.; Padhy, R.N.; Singh, P.K. Computational approach for locating effective cyanobacterial compounds against Mycobacterium tuberculosis. Indian J. Pharm. Educ. Res. 2017, 51, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Uzair, B.; Tabassum, S.; Rasheed, M.; Rehman, S.F. Exploring marine cyanobacteria for lead compounds of pharmaceutical importance. Sci. World J. 2012, 2012, 179782. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, S. Cyanobacterial Diversity: A Potential Source of Bioactive Compounds. Adv. Biotechnol. Microbiol. 2017, 4, 19–21. [Google Scholar] [CrossRef]
- Ducat, D.C.; Way, J.C.; Silver, P.A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011, 29, 95–103. [Google Scholar] [CrossRef]
- Ramsamy, Y.; Hardcastle, T.C.; Muckart, D.J.J. Surviving Sepsis in the Intensive Care Unit: The Challenge of Antimicrobial Resistance and the Trauma Patient. World J. Surg. 2017, 41, 1165–1169. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33. [Google Scholar] [CrossRef]
- Su, M.; Zhao, C.; Li, D.; Cao, J.; Ju, Z.; Kim, E.L.; Jung, Y.S.; Jung, J.H. Viriditoxin stabilizes microtubule polymers in SK-OV-3 cells and exhibits antimitotic and antimetastatic potential. Mar. Drugs 2020, 18, 445. [Google Scholar] [CrossRef]
- Friedrich, A.W. Control of hospital acquired infections and antimicrobial resistance in Europe: The way to go. Wien. Med. Wochenschr. 2019, 169, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, P. Antibacterial discovery and development—The failure of success? Nat. Biotechnol. 2006, 24, 1497–1503. [Google Scholar] [CrossRef]
- WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. Saudi Med. J. 2017, 38, 444.
- Hofer, U. The CO. Nat. Rev. Microbiol. 2019, 17, 1–14. [Google Scholar]
- Moo, C.-L.; Yang, S.-K.; Yusoff, K.; Ajat, M.; Thomas, W.; Abushelaibi, A.; Lim, S.-H.-E.; Lai, K.-S. Mechanisms of Antimicrobial Resistance (AMR) and Alternative Approaches to Overcome AMR. Curr. Drug Discov. Technol. 2019, 17, 430–447. [Google Scholar] [CrossRef]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Battaje, R.R.; Panda, D. Lessons from bacterial homolog of tubulin, FtsZ for microtubule dynamics. Endocr. Relat. Cancer 2017, 24, T1–T21. [Google Scholar] [CrossRef] [Green Version]
- Štumpf, S.; Hostnik, G.; Primožič, M.; Leitgeb, M.; Bren, U. Generation times of e. Coli prolong with increasing tannin concentration while the lag phase extends exponentially. Plants 2020, 9, 1680. [Google Scholar] [CrossRef]
- Štumpf, S.; Hostnik, G.; Primožič, M.; Leitgeb, M.; Salminen, J.P.; Bren, U. The effect of growth medium strength on minimum inhibitory concentrations of tannins and tannin extracts against E. coli. Molecules 2020, 25, 2947. [Google Scholar] [CrossRef]
- Walsh, C.T. Enzymes in the D-alanine branch of bacterial cell wall peptidoglycan assembly. J. Biol. Chem. 1989, 264, 2393–2396. [Google Scholar] [CrossRef]
- Šarac, Z.; Matejić, J.S.; Stojanović-Radić, Z.Z.; Veselinović, J.B.; Džamić, A.M.; Bojović, S.; Marin, P.D. Biological activity of Pinus nigra terpenes-Evaluation of FtsZ inhibition by selected compounds as contribution to their antimicrobial activity. Comput. Biol. Med. 2014, 54, 72–78. [Google Scholar] [CrossRef]
- Marston, H.D.; Dixon, D.M.; Knisely, J.M.; Palmore, T.N.; Fauci, A.S. Antimicrobial resistance. JAMA J. Am. Med. Assoc. 2016, 316, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Haranahalli, K.; Tong, S.; Ojima, I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ. Bioorg. Med. Chem. 2016, 24, 6354–6369. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, S.; Sahu, S.K. FtsZ inhibitors as a new genera of antibacterial agents. Bioorg. Chem. 2019, 91, 103169. [Google Scholar] [CrossRef]
- Dhaked, H.P.S.; Bhattacharya, A.; Yadav, S.; Dantu, S.C.; Kumar, A.; Panda, D. Mutation of Arg191 in FtsZ Impairs Cytokinetic Abscission of Bacillus subtilis Cells. Biochemistry 2016, 55, 5754–5763. [Google Scholar] [CrossRef]
- Wang, J.; Galgoci, A.; Kodali, S.; Herath, K.B.; Jayasuriya, H.; Dorso, K.; Vicente, F.; González, A.; Cully, D.; Bramhill, D.; et al. Discovery of a Small Molecule that Inhibits Cell Division by Blocking FtsZ, a Novel Therapeutic Target of Antibiotics. J. Biol. Chem. 2003, 278, 44424–44428. [Google Scholar] [CrossRef] [Green Version]
- Léonard, C.; Alsteens, D.; Dumitru, A.C.; Mingeot-Leclercq, M.-P.; Tyteca, D. Lipid Domains and Membrane (Re)Shaping: From Biophysics to Biology. In The Biophysics of Cell Membranes; Springer Series in Biophysics; Springer: Singapore, 2017; Volume 19. [Google Scholar] [CrossRef]
- Vollmer, W.; Massidda, O.; Tomasz, A. The Cell Wall of Streptococcus pneumoniae. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Booth, S.; Lewis, R.J. Structural basis for the coordination of cell division with the synthesis of the bacterial cell envelope. Protein Sci. 2019, 28, 2042–2054. [Google Scholar] [CrossRef]
- Jensen, S.O.; Thompson, L.S.; Harry, E.J. Cell division in Bacillus subtilis: FtsZ and FtsA association is Z-ring independent, and FtsA is required for efficient midcell Z-ring assembly. J. Bacteriol. 2005, 187, 6536–6544. [Google Scholar] [CrossRef] [Green Version]
- Squyres, G.R.; Holmes, M.J.; Barger, S.R.; Pennycook, B.R.; Ryan, J.; Yan, V.T.; Garner, E.C. Single-molecule imaging reveals that Z-ring condensation is essential for cell division in Bacillus subtilis. Nat. Microbiol. 2020, 6, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.W.; Errington, J. Bacterial cell division: Assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 2009, 7, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Zhang, Y.; Parhi, A.K.; Lavoie, E.J.; Tuske, S.; Arnold, E.; Kerrigan, J.E.; Pilch, D.S. Enterococcal and streptococcal resistance to PC190723 and related compounds: Molecular insights from a FtsZ mutational analysis. Biochimie 2013, 95, 1880–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, H.; Lai, P.; Gao, Y. Chemical composition, antibacterial activity, and synergistic effects with conventional antibiotics and nitric oxide production inhibitory activity of essential oil from Geophila repens (L.) I.M. Johnst. Molecules 2017, 22, 1561. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Han, H.; Wang, Z.; Li, T.; Teng, D.; Mao, R.; Hao, Y.; Yang, N.; Wang, X.; Wang, J. Recent progress of bacterial FtsZ inhibitors with a focus on peptides. FEBS J. 2021, 288, 1091–1106. [Google Scholar] [CrossRef]
- Li, X.; Ma, S. Advances in the discovery of novel antimicrobials targeting the assembly of bacterial cell division protein FtsZ. Eur. J. Med. Chem. 2015, 95, 1–15. [Google Scholar] [CrossRef]
- Ito, H.; Ura, A.; Oyamada, Y.; Tanitame, A.; Yoshida, H.; Yamada, S.; Wachi, M.; Yamagishi, J.I. A 4-aminofurazan derivative—A189—Inhibits assembly of bacterial cell division protein FtsZ In-vitro and in vivo. Microbiol. Immunol. 2006, 50, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Margalit, D.N.; Romberg, L.; Mets, R.B.; Hebert, A.M.; Mitchison, T.J.; Kirschner, M.W.; RayChaudhuri, D. Targeting cell division: Small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 11821–11826. [Google Scholar] [CrossRef] [Green Version]
- Beuria, T.K.; Santra, M.K.; Panda, D. Sanguinarine blocks cytokinesis in bacteria by inhibiting FtsZ assembly a bundling. Biochemistry 2005, 44, 16584–16593. [Google Scholar] [CrossRef]
- Domadia, P.N.; Bhunia, A.; Sivaraman, J.; Swarup, S.; Dasgupta, D. Berberine targets assembly of Escherichia coli cell division protein FtsZ. Biochemistry 2008, 47, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Xu, Z.R.; Li, W.F.; Shuai, J.B.; Lu, P.; Hu, C.X. Protein secretion pathways in Bacillus subtilis: Implication for optimization of heterologous protein secretion. Biotechnol. Adv. 2007, 25, 1–12. [Google Scholar] [CrossRef]
- Su, Y.; Liu, C.; Fang, H.; Zhang, D. Bacillus subtilis: A universal cell factory for industry, agriculture, biomaterials and medicine. Microb. Cell Fact. 2020, 19, 173. [Google Scholar] [CrossRef]
- Kaushik, P.; Chauhan, A.; Chauhan, G.; Goyal, P. Antibacterial Potential and UV-HPLC Analysis of Laboratory- Grown Culture of Anabaena variabilis. Internet J. Food Saf. 2009, 11, 11–18. [Google Scholar]
- Crombie, L.; Holloway, S.J. The biosynthesis of calendic acid, octadeca-(8E,10E, 12Z)-trienoic acid, by developing marigold seeds: Origins of (E,E,Z) and (Z,E,Z) conjugated triene acids in higher plants. J. Chem. Soc. Perkin Trans. 1 1985, 2425–2434. [Google Scholar] [CrossRef]
- Ragasa, C.Y.; Guardamano, J.D.; Tan, M.C.S.; Ulep, R.A.; van Altena, I.A. Isolation and structure elucidation of 9-hydroxy-10E, 12Z-octadecadienoic acid from Dioscorea luzonensis. Res. J. Pharm. Biol. Chem. Sci. 2016, 7, 1233–1236. [Google Scholar]
- Liu, H.; Joshi-Barve, S.; Barve, S.; McClain, C.; Ramsden, C.; Kirpich, I. Effects of Ethanol and Oxidized Metabolites of Linoleic Acid on Caco-2 cell Model of Intestinal Epithelial Barrier. FASEB J. 2013, 27, 890.20. [Google Scholar] [CrossRef]
- Naidu, S.V.; Gupta, P.; Kumar, P. Enantioselective syntheses of (-)-pinellic acid, α- and β-dimorphecolic acid. Tetrahedron 2007, 63, 7624–7633. [Google Scholar] [CrossRef]
- Watson, J.D.; Laskowski, R.A.; Thornton, J.M. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 2005, 15, 275–284. [Google Scholar] [CrossRef]
- Fujita, J.; Harada, R.; Maeda, Y.; Saito, Y.; Mizohata, E.; Inoue, T.; Shigeta, Y.; Matsumura, H. Identification of the key interactions in structural transition pathway of FtsZ from Staphylococcus aureus. J. Struct. Biol. 2017, 198, 65–73. [Google Scholar] [CrossRef]
- Ranjan, A.; Chauhan, A.; Jindal, T. In-silico and In-vitro evaluation of human acetylcholinesterase inhibition by organophosphates. Environ. Toxicol. Pharmacol. 2018, 57, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M. Radius of gyration as an indicator of protein structure compactness Aggregation of ribosomal protein S1 View project amyloids View project. Artic. Mol. Biol. 2008, 42, 701–706. [Google Scholar] [CrossRef]
- Bakr, R.O.; Zaghloul, S.S.; Hassan, R.A.; Sonousi, A.; Wasfi, R.; Fayed, M.A.A. Antimicrobial activity of Vitex agnus-castus Essential Oil and Molecular Docking Study of Its Major Constituents. J. Essent. Oil-Bear. Plants 2020, 23, 184–193. [Google Scholar] [CrossRef]
- Furlan, V.; Bren, U.; Micaroni, M.; Oddi, S. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules 2021, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Modi, N.H.; Panda, D.; Roy, N. Probing the binding site of curcumin in Escherichia coli and Bacillus subtilis FtsZ—A structural insight to unveil antibacterial activity of curcumin. Eur. J. Med. Chem. 2010, 45, 4209–4214. [Google Scholar] [CrossRef]
- Xie, J.; Wu, Y.Y.; Zhang, T.Y.; Zhang, M.Y.; Peng, F.; Lin, B.; Zhang, Y.X. New antimicrobial compounds produced by endophytic Penicillium janthinellum isolated from Panax notoginseng as potential inhibitors of FtsZ. Fitoterapia 2018, 131, 35–43. [Google Scholar] [CrossRef]
- Ballu, S.; Itteboina, R.; Sivan, S.K.; Manga, V. Structural insights of Staphylococcus aureus FtsZ inhibitors through molecular docking, 3D-QSAR and molecular dynamics simulations. J. Recept. Signal Transduct. 2018, 38, 61–70. [Google Scholar] [CrossRef]
- Chan, F.Y.; Sun, N.; Neves, M.A.C.; Lam, P.C.H.; Chung, W.H.; Wong, L.K.; Chow, H.Y.; Ma, D.L.; Chan, P.H.; Leung, Y.C.; et al. Identification of a new class of FtsZ inhibitors by structure-based design and In-vitro screening. J. Chem. Inf. Model. 2013, 53, 2131–2140. [Google Scholar] [CrossRef]
- Mundt, S.; Kreitlow, S.; Jansen, R. Fatty acids with antibacterial activity from the cyanobacterium Oscillatoria redekei HUB 051. J. Appl. Phycol. 2003, 15, 263–267. [Google Scholar] [CrossRef]
- Naz, F.; Mashkoor, M.; Sharma, P.; Haque, M.A.; Kapil, A.; Kumar, M.; Kaur, P.; Abdul Samath, E. Drug repurposing approach to target FtsZ cell division protein from Salmonella Typhi. Int. J. Biol. Macromol. 2020, 159, 1073–1083. [Google Scholar] [CrossRef]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Chauhan, P.K.; Collins, I.; Davies, D.T.; Gavade, M.; Kumar, D.; Lancett, P.; Macdonald, R.; et al. Design, synthesis and structure–activity relationships of substituted oxazole–benzamide antibacterial inhibitors of FtsZ. Bioorg. Med. Chem. Lett. 2014, 24, 353–359. [Google Scholar] [CrossRef]
- Handler, A.A.; Lim, J.E.; Losick, R. Peptide inhibitor of cytokinesis during sporulation in Bacillus subtilis. Mol. Microbiol. 2008, 68, 588–599. [Google Scholar] [CrossRef] [Green Version]
- Chandra, N.D.; Suresh, C.S.; Kumar, G.A.; Sandeep, P.M.; Sabu, A.; Haridas, M. Implication of biotransformation of berberine and its derivatives on FtsZ protein: An In-silico study. Int. J. Comput. Biol. Drug Des. 2017, 10, 1–11. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Vijayalakshmi, P.; Nisha, J.; Rajalakshmi, M. Virtual screening of potential inhibitor against FtsZ protein from Staphylococcus aureus. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 331–339. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Greenwood, J.; Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 2, 6474–6487. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yamaguchi, Y.; Namai, T.; Hirukawa, T.; Yamaguchi, Y.; Namai, T. Oxygenated Fatty Acids with Anti-rice Blast Fungus Activity in Rice Plants. Biosci. Biotechnol. Biochem. 1993, 57, 283–287. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioactive Molecules | Docking Score (Kcalmol−1) | MMGBSA-dG Binding (Kcalmol−1) | Interactions (Hydrogen Bonds) | ∆G (Kcalmol−1) [std] | Lig. Efficiency (Kcalmol−1) | Compounds Reported in |
|---|---|---|---|---|---|---|
| Alpha di-morphecolic acid (5312830) | −6.574 | −44.12 | Arg 29, Asn 33, Asn 299 | −8.52 [−0.35] | −0.41 | Oscillatoria redekei |
| Aphanorphine (189594) | −6.010 | − | Asn 33, Ile 298 | −5.45 [−0.35] | −0.36 | Aphanizomenon flos aquae |
| Circinamide (21601944) | −7.366 | − | Arg 29, Asn 33, Glu 34, Glu 305 | −7.00 [−0.36] | −0.26 | Anabeana circinalis |
| Aeruginosin 102 A (10101474) | −8.666 | − | Arg 29, Asn 33, Asp 174, Ile 228 | −8.23 [−0.51] | −0.16 | Microcyctis viridis |
| PC 190723 (2501641) | −4.135 | −53.36 | Asp 33, Leu 206, Ile 298 | −7.68 [−0.52] | −0.33 | Known Inhibitor |
| Top Hit | Acceptable Range | Compound ID | ||
|---|---|---|---|---|
| 5,312,830 | 189,594 | 10,101,474 | ||
| mol MW (gMol−1) | <500 | 296.4 | 203.28 | 386.5 |
| Donor HB | 0.0–5.0 | 1 | 0 | 1 |
| Accpt HB | 2.0–20.0 | 2.75 | 3.50 | 5.75 |
| QPlog Poct | 8.0–35 | 10.267 | 12.999 | 18.416 |
| QPlog Pw | 4.0–45.0 | 5.804 | 9.579 | 5.53 |
| QPlog Po/w | −2.0–6.5 | 1.972 | 3.654 | 2.881 |
| QPlog S | −6.5–0.5 | −1.751 | −5.559 | −3.646 |
| QPlog HERG | below −5 | −4.01 | −5.967 | −5.298 |
| QPP Caco | <25 poor and >500 great | 729.418 | 1059 | 1180 |
| QP LogBB | −3.0–1.2 | 0.465 | −0.83 | −0.61 |
| QPP MDCK | <25 poor and >500 great | 389.156 | 526 | 1275 |
| QPLog Khsa | −1.5–1.5 | 0.079 | 0.449 | –6.288 |
| QPLog Kp | −8.0–1.0 | −4.142 | −2.147 | −1.761 |
| PHOA | >80% is strong and <25% is weak | 89.734 | 100 | 100 |
| Ro5 | Max., 4.0 | 0 | 0 | 0 |
| Test Organism | Assays | Lead Compound | Known Inhibitor | Oscillatoria Biomass | |
|---|---|---|---|---|---|
| Alpha di Morphecolic Acid | Aphanorphine | Polymyxin B-Sulphate | Methanol Extracts | ||
| B. subtilis (MTCC-441) | Agar Well diffusion assay (Zone of Inhibition in mm) | 23.12 ± 0.1 | 0.0 Nil | 21.33 ± 0.47 | 16.14 ± 0.6 |
| MIC (µgmL−1) | 512 | NC | 256 | 1024 | |
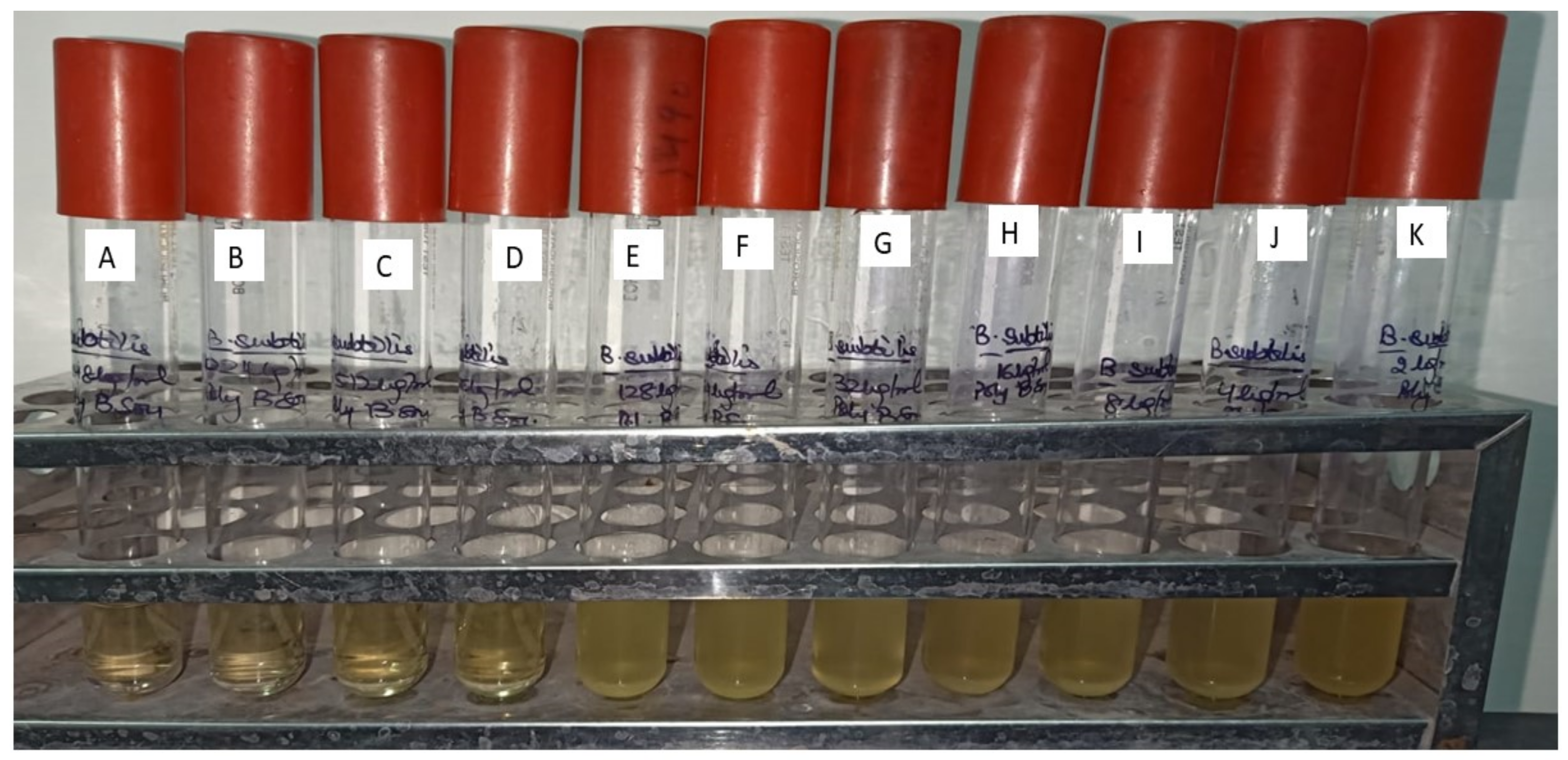
| Concentration in (µgmL−1) | MIC (µgmL−1) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1024 | 512 | 256 | 128 | 64 | 32 | 16 | 8 | 4 | 2 | 1 | ||
| Methanolic Extract | − | + | + | + | + | + | + | + | + | + | + | 1024 |
| Alpha Di Morphecolic Acid | − | − | + | + | + | + | + | + | + | + | + | 512 |
| Polymyxin B Sulfate | − | − | − | + | + | + | + | + | + | + | + | 256 |
| Aphanorphine | + | + | + | + | + | + | + | + | + | + | + | >1024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurnani, M.; Rath, P.; Chauhan, A.; Ranjan, A.; Ghosh, A.; Lal, R.; Mukerjee, N.; Aljarba, N.H.; Alkahtani, S.; Rajput, V.D.; et al. Inhibition of Filamentous Thermosensitive Mutant-Z Protein in Bacillus subtilis by Cyanobacterial Bioactive Compounds. Molecules 2022, 27, 1907. https://doi.org/10.3390/molecules27061907
Gurnani M, Rath P, Chauhan A, Ranjan A, Ghosh A, Lal R, Mukerjee N, Aljarba NH, Alkahtani S, Rajput VD, et al. Inhibition of Filamentous Thermosensitive Mutant-Z Protein in Bacillus subtilis by Cyanobacterial Bioactive Compounds. Molecules. 2022; 27(6):1907. https://doi.org/10.3390/molecules27061907
Chicago/Turabian StyleGurnani, Manisha, Prangya Rath, Abhishek Chauhan, Anuj Ranjan, Arabinda Ghosh, Rup Lal, Nobendu Mukerjee, Nada H. Aljarba, Saad Alkahtani, Vishnu D. Rajput, and et al. 2022. "Inhibition of Filamentous Thermosensitive Mutant-Z Protein in Bacillus subtilis by Cyanobacterial Bioactive Compounds" Molecules 27, no. 6: 1907. https://doi.org/10.3390/molecules27061907