
Therapeutic Potential of Highly Selective A3 Adenosine Receptor Ligands in the Central and Peripheral Nervous System

 ,
,  ,
,  , ,
, , 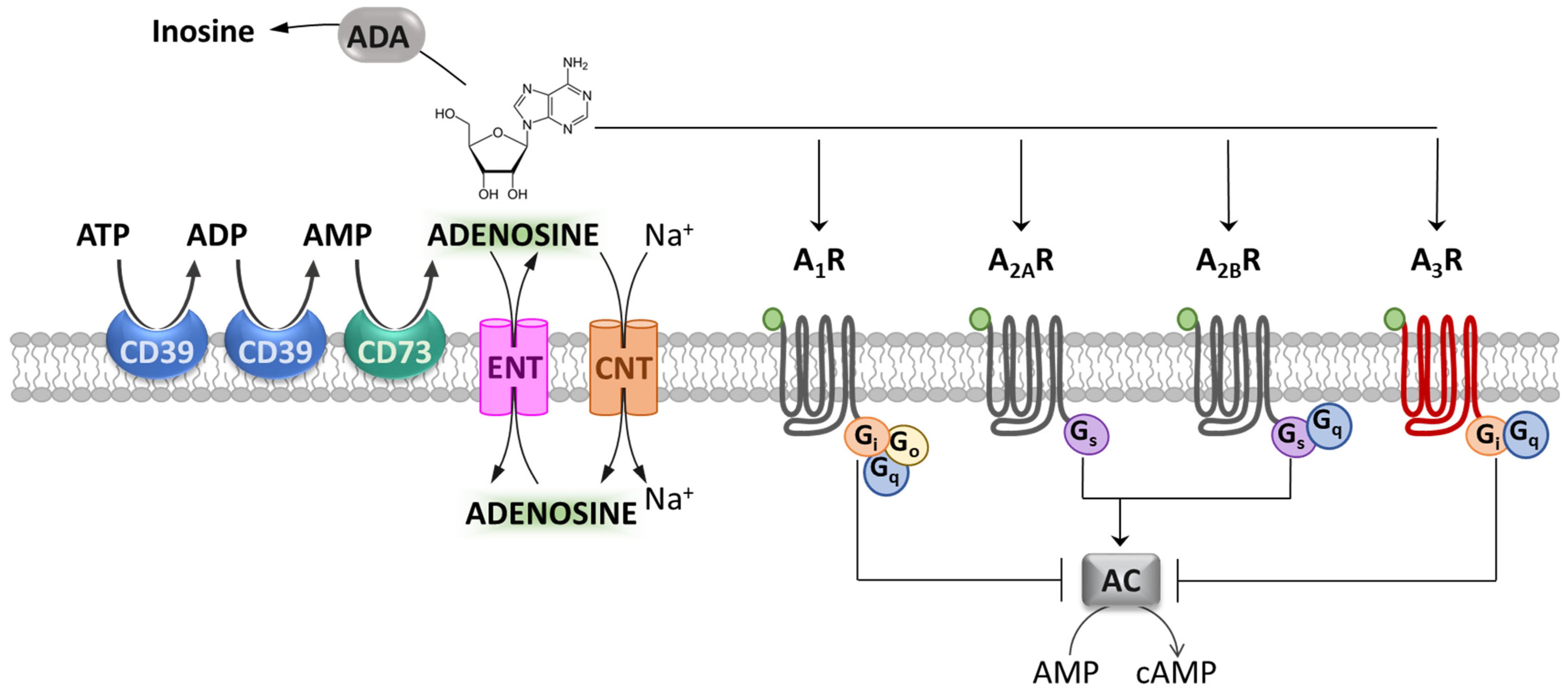{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Adenosine and Its Receptors
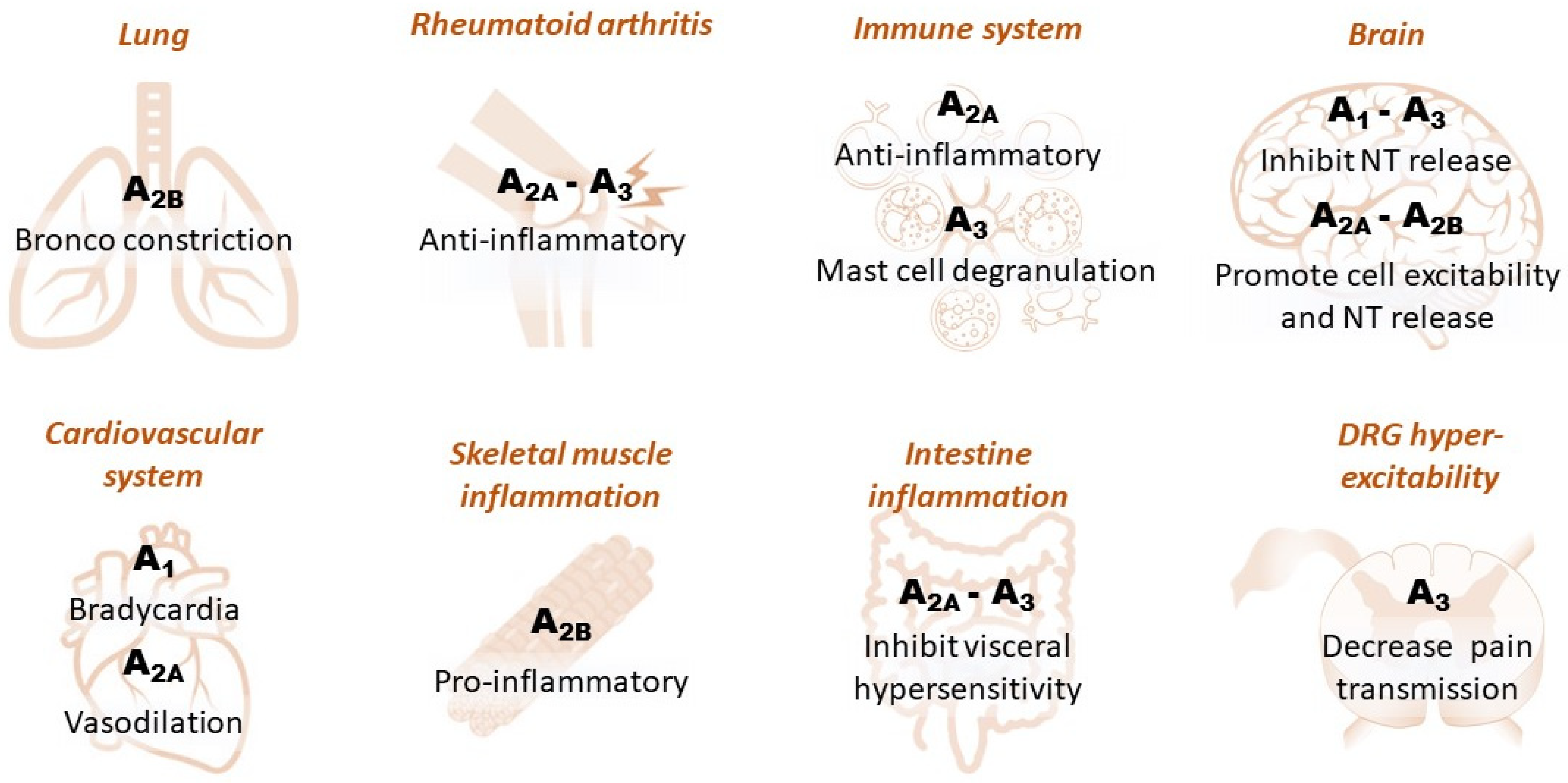
1.1. Adenosine Receptors in Peripheral Tissues
1.2. Adenosine Receptors in the CNS
2. Therapeutic Potential of A3R Ligands
2.1. A3R Ligands as Therapeutic Tools in Brain Ischemia
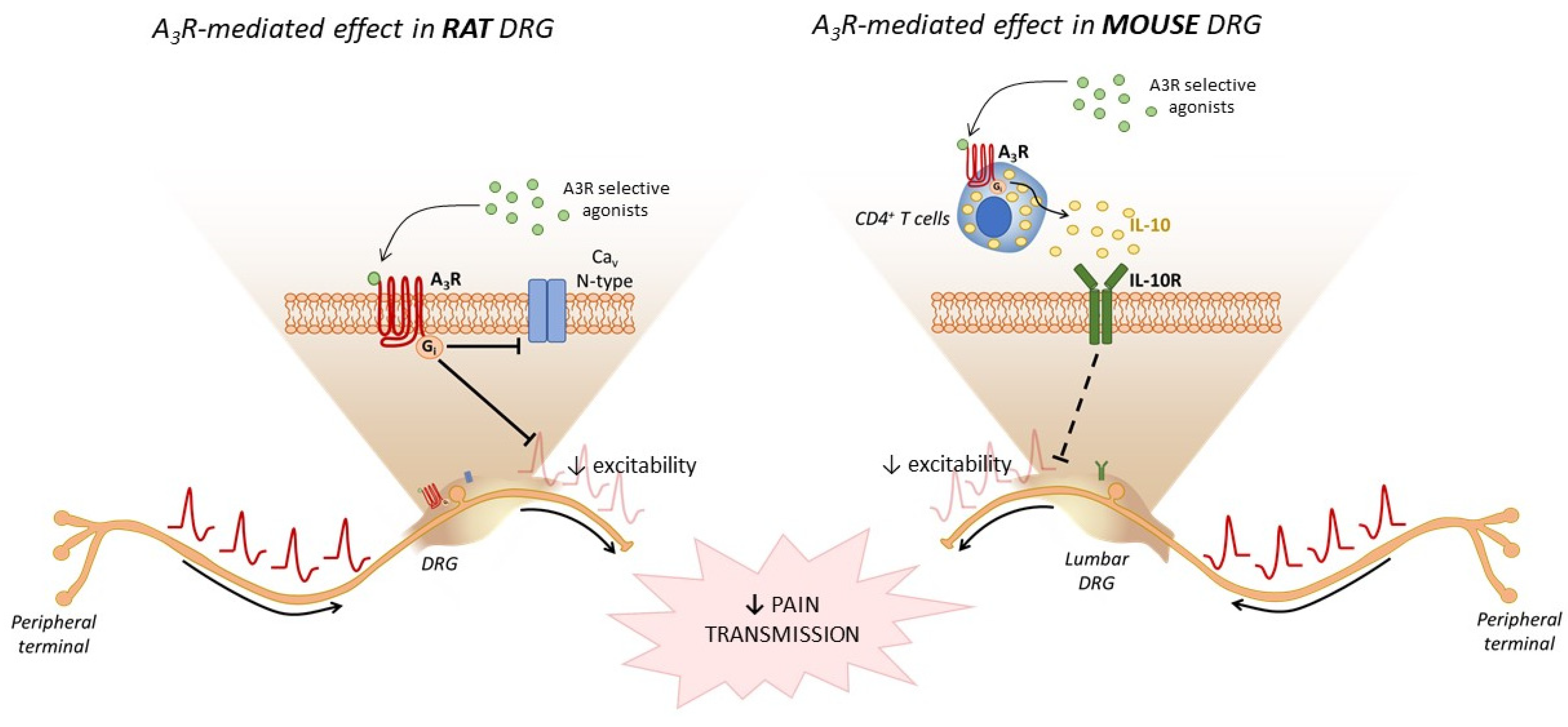
2.2. A3R Ligands as Therapeutic Tools in Chronic Pain

3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Verkhratsky, A. Early evolutionary history (from bacteria to hemichordata) of the omnipresent purinergic signalling: A tribute to Geoff Burnstock inquisitive mind. Biochem. Pharmacol. 2021, 187, 114261. [Google Scholar] [CrossRef]
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J. Neurophysiol. 2012, 107, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Coppi, E.; Cellai, L.; Maraula, G.; Dettori, I.; Melani, A.; Pugliese, A.M.; Pedata, F. Role of adenosine in oligodendrocyte precursor maturation. Front. Cell. Neurosci. 2015, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Verkhrasky, A.; Krishtal, O.A.; Burnstock, G. Purinoceptors on neuroglia. Mol. Neurobiol. 2009, 39, 190–208. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Linden, J. Cloned adenosine A3 receptors: Pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306. [Google Scholar] [CrossRef]
- Koscsó, B.; Csóka, B.; Pacher, P.; Haskó, G. Investigational A3 adenosine receptor targeting agents. Expert Opin. Investig. Drugs 2011, 20, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell. Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Colucci, R.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, S.A.; Beal, P.R.; Yao, S.Y.M.; King, A.E.; Cass, C.E.; Young, J.D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004, 447, 735–743. [Google Scholar] [CrossRef]
- Gray, J.H.; Owen, R.P.; Giacomini, K.M. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. 2004, 447, 728–734. [Google Scholar] [CrossRef]
- Pedata, F.; Corsi, C.; Melani, A.; Bordoni, F.; Latini, S. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann. N. Y. Acad. Sci. 2001, 939, 74–84. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Dunwiddie, T.V.; Bergman, B.; Lindström, K. Levels of adenosine and adenine nucleotides in slices of rat hippocampus. Brain Res. 1984, 295, 127–136. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Diao, L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J. Pharmacol. Exp. Ther. 1994, 268, 537–545. [Google Scholar]
- Al-Zaiti, S.S.; Magdic, K.S. Paroxysmal Supraventricular Tachycardia: Pathophysiology, Diagnosis, and Management. Crit. Care Nurs. Clin. N. Am. 2016, 28, 309–316. [Google Scholar] [CrossRef]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 adenosine receptor expression in rheumatoid arthritis: Upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res. Ther. 2011, 13, R197. [Google Scholar] [CrossRef] [Green Version]
- Haskó, G.; Kuhel, D.G.; Chen, J.-F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabó, C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000, 14, 2065–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, R.; Junker, M.; Meurer, A.; Zaucke, F.; Straub, R.H.; Jenei-lanzl, Z. Anti-Inflammatory Effects of Endogenously Released Adenosine in Synovial Cells of Osteoarthritis and Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2021, 22, 8956. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, H.; Ogasawara, Y.; Kajiya, F. Visualisation of the Effects of Dilazep on Rat Afferent and Efferent Arterioles In Vivo. Hypertens. Res. 2008, 31, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendell, S.G.; Fan, H.; Zhang, C. G protein–coupled receptors in asthma therapy: Pharmacology and drug actions. Pharmacol. Rev. 2020, 72, 1–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, K.F.; Dent, G. Theophylline and airway inflammation. Clin. Exp. Allergy 1998, 28 (Suppl. 3), 35–41. [Google Scholar] [PubMed]
- Gessi, S.; Merighi, S.; Varani, K.; Leung, E.; Mac Lennan, S.; Borea, P.A. The A3 adenosine receptor: An enigmatic player in cell biology. Pharmacol. Ther. 2008, 117, 123–140. [Google Scholar] [CrossRef]
- Ray, P.; Torck, A.; Quigley, L.; Wangzhou, A.; Neiman, M.; Rao, C.; Lam, T.; Kim, J.Y.; Kim, T.H.; Zhang, M.Q.; et al. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research. Pain 2018, 159, 1325–1345. [Google Scholar] [CrossRef]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.S.; Richardson, P.J.; Freeman, T.C. Tissue distribution of adenosine receptor mRNAs in the rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Ramkumarh, V.; Stilesll, G.L.; Beavenll, M.A.; Mi, H. The A3 Adenosine Receptor Is the Unique Adenosine Receptor Which Facilitates Release of Allergic Mediators in Mast Cells*. J. BloLomrhL Chem. 1993, 268, 16887–16890. [Google Scholar] [CrossRef]
- Leung, C.T.; Li, A.; Banerjee, J.; Gao, Z.G.; Kambayashi, T.; Jacobson, K.A.; Civan, M.M. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 2014, 10, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andiné, P. Involvement of adenosine in ischemic and postischemic calcium regulation. Mol. Chem. Neuropathol. 1993, 18, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, R.; Lo Conte, G.; Moroni, F.; Beatrice Passani, M.; Pepeu, G. Adenosine decreases aspartate and glutamate release from rat hippocampal slices. Eur. J. Pharmacol. 1984, 104, 19–26. [Google Scholar] [CrossRef]
- Deb, P.K.; Deka, S.; Borah, P.; Abed, S.N.; Klotz, K.-N. Medicinal Chemistry and Therapeutic Potential of Agonists, Antagonists and Allosteric Modulators of A1 Adenosine Receptor: Current Status and Perspectives. Curr. Pharm. Des. 2019, 25, 2697–2715. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.L.; Ribeiro, J.A. Adenosine A2 receptor activation facilitates 45Ca2+ uptake by rat brain synaptosomes. Eur. J. Pharmacol. 1996, 310, 257–261. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.A.; Kull, B.; Fredholm, B.B.; Ribeiro, J.A. Adenosine A2A receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1 receptor inhibition. Neuroscience 2002, 112, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Melani, A.; Gianfriddo, M.; Vannucchi, M.G.; Cipriani, S.; Baraldi, P.G.; Giovannini, M.G.; Pedata, F. The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006, 1073–1074, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Fusco, I.; Cherchi, F.; Catarzi, D.; Colotta, V.; Varano, F.; Pedata, F.; Pugliese, A.M.A.M.; Coppi, E. Functional characterization of a novel adenosine A2B receptor agonist on short-term plasticity and synaptic inhibition during oxygen and glucose deprivation in the rat CA1 hippocampus. Brain Res. Bull. 2019, 151, 174–180. [Google Scholar] [CrossRef]
- Liu, Y.J.; Chen, J.; Li, X.; Zhou, X.; Hu, Y.M.; Chu, S.F.; Peng, Y.; Chen, N.H. Research progress on adenosine in central nervous system diseases. CNS Neurosci. Ther. 2019, 25, 899–910. [Google Scholar] [CrossRef]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Lana, D.; Giovannini, M.G.; Pedata, F.; Pugliese, A.M. A2b adenosine receptors: When outsiders may become an attractive target to treat brain ischemia or demyelination. Int. J. Mol. Sci. 2020, 21, 9697. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A 3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The selective antagonism of adenosine A2B receptors reduces the synaptic failure and neuronal death induced by oxygen and glucose deprivation in rat CA1 hippocampus in vitro. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppi, E.; Cherchi, F.; Fusco, I.; Dettori, I.; Gaviano, L.; Magni, G.; Catarzi, D.; Colotta, V.; Varano, F.; Rossi, F.; et al. Adenosine A2B receptors inhibit K+ currents and cell differentiation in cultured oligodendrocyte precursor cells and modulate sphingosine-1-phosphate signaling pathway. Biochem. Pharmacol. 2020, 177, 113956. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Giacomelli, L.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. Targeting A3 and A2A adenosine receptors in the fight against cancer. Expert Opin. Ther. Targets 2019, 23, 669–678. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [Green Version]
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J. Rheumatol. 2008, 35, 41–48. [Google Scholar] [CrossRef]
- Luongo, L.; Salvemini, D. Targeting metabotropic adenosine receptors for neuropathic pain: Focus on A2A. Brain. Behav. Immun. 2018, 69, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Kleindorfer, D.; Lindsell, C.J.; Brass, L.; Koroshetz, W.; Broderick, J.P. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke 2008, 39, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Diller, K.R.; Zhu, L. Hypothermia Therapy for Brain Injury. Annu. Rev. Biomed. Eng. 2009, 11, 135–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Klutz, A.M.; Tosh, D.K.; Ivanov, A.A.; Preti, D.; Baraldi, P.G. Medicinal Chemistry of the A3 Adenosine Receptor: Agonists, Antagonists, and Receptor Engineering. Handb. Exp. Pharmacol. 2009, 193, 123. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.O.; Xiao-duo, J.; Siddiqi, S.M.; Jacobson, K.A.; Olah, M.E.; Stiles, G.L. 2-Substitution of N6-benzyladenosine-5’-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994, 37, 3614–3621. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Ji, X.D.; Li, A.H.; Melman, N.; Siddiqui, M.A.; Shin, K.J.; Marquez, V.E.; Ravi, R.G. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J. Med. Chem. 2000, 43, 2196–2203. [Google Scholar] [CrossRef] [Green Version]
- Volpini, R.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Lammi, C.; Marucci, G.; Ramadori, A.T.; Klotz, K.N.; Cristalli, G. Synthesis and biological evaluation of 2-alkynyl-N6-methyl- 5′-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor. J. Med. Chem. 2009, 52, 7897–7900. [Google Scholar] [CrossRef]
- Tosh, D.K.; Deflorian, F.; Phan, K.; Gao, Z.G.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Structure-Guided Design of A3 Adenosine Receptor-Selective Nucleosides: Combination of 2-Arylethynyl and Bicyclo[3.1.0]hexane Substitutions. J. Med. Chem. 2012, 55, 4847. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Paoletta, S.; Phan, K.; Gao, Z.G.; Jacobson, K.A. Truncated nucleosides as A3 adenosine receptor ligands: Combined 2-arylethynyl and bicyclohexane substitutions. ACS Med. Chem. Lett. 2012, 3, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, I.M.; Jacobson, M.A.; Basile, A.; Jacobson, K.A. Behavioral characterization of mice lacking the A3 adenosine receptor: Sensitivity to hypoxic neurodegeneration. Cell. Mol. Neurobiol. 2003, 23, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, S.; Lewerenz, A.; Nieber, K. Activation of A_3 receptors by endogenous adenosine inhibits synaptic transmission during hypoxia in rat cortical neurons - IOS Press. Restor. Neurol. Neurosci. 2003, 21, 55–63. [Google Scholar]
- Von Lubitz, D.K.J.E.; Lin, R.C.S.; Popik, P.; Carter, M.F.; Jacobson, K.A. Adenosine A3 receptor stimulation and cerebral ischemia. Eur. J. Pharmacol. 1994, 263, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Colotta, V.; Lenzi, O.; Catarzi, D.; Varano, F.; Squarcialupi, L.; Costagli, C.; Galli, A.; Ghelardini, C.; Pugliese, A.M.; Maraula, G.; et al. 3-Hydroxy-1H-quinazoline-2,4-dione derivatives as new antagonists at ionotropic glutamate receptors: Molecular modeling and pharmacological studies. Eur. J. Med. Chem. 2012, 54, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, A.M.; Coppi, E.; Spalluto, G.; Corradetti, R.; Pedata, F. A3 adenosine receptor antagonists delay irreversible synaptic failure caused by oxygen and glucose deprivation in the rat CA1 hippocampus in vitro. Br. J. Pharmacol. 2006, 147, 524–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese, A.M.; Coppi, E.; Volpini, R.; Cristalli, G.; Corradetti, R.; Jeong, L.S.; Jacobson, K.A.; Pedata, F. Role of adenosine A3 receptors on CA1 hippocampal neurotransmission during oxygen-glucose deprivation episodes of different duration. Biochem. Pharmacol. 2007, 74, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Vissiennon, Z.; Eschke, D.; Nieber, K. Adenosine A1 and A3 receptors mediate inhibition of synaptic transmission in rat cortical neurons. Neuropharmacology 2001, 40, 85–95. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Diao, L.; Kim, H.O.; Jiang, J.L.; Jacobson, K.A. Activation of hippocampal adenosine A3 receptors produces a desensitization of A1 receptor-mediated responses in rat hippocampus. J. Neurosci. 1997, 17, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Fleming, K.M.; Mogul, D.J. Adenosine A3 receptors potentiate hippocampal calcium current by a PKA-dependent/PKC-independent pathway. Neuropharmacology 1997, 36, 353–362. [Google Scholar] [CrossRef]
- Macek, T.A.; Schaffhauser, H.; Jeffrey Conn, P. Protein Kinase C and A3 Adenosine Receptor Activation Inhibit Presynaptic Metabotropic Glutamate Receptor (mGluR) Function and Uncouple mGluRs from GTP-Binding Proteins. J. Neurosci. 1998, 18, 6138–6146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costenla, A.R.; Lopes, L.V.; De Mendonça, A.; Ribeiro, J.A. A functional role for adenosine A3 receptors: Modulation of synaptic plasticity in the rat hippocampus. Neurosci. Lett. 2001, 302, 53–57. [Google Scholar] [CrossRef]
- Laudadio, M.A.; Psarropoulou, C. The A3 adenosine receptor agonist 2-Cl-IB-MECA facilitates epileptiform discharges in the CA3 area of immature rat hippocampal slices. Epilepsy Res. 2004, 59, 83–94. [Google Scholar] [CrossRef]
- George, P.M.; Steinberg, G.K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, J.; Doerr, M.; Hu, R.; Sun, P.Z. Refined Ischemic Penumbra Imaging with Tissue pH and Diffusion Kurtosis Magnetic Resonance Imaging. Transl. Stroke Res. 2021, 12, 742–753. [Google Scholar] [CrossRef]
- Hurford, R.; Sekhar, A.; Hughes, T.A.T.; Muir, K.W. Diagnosis and management of acute ischaemic stroke. Pract. Neurol. 2020, 20, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Liston, T.E.; Hinz, S.; Müller, C.E.; Holstein, D.M.; Wendling, J.; Melton, R.J.; Campbell, M.; Korinek, W.S.; Suresh, R.R.; Sethre-Hofstad, D.A.; et al. Nucleotide P2Y1 receptor agonists are in vitro and in vivo prodrugs of A1/A3 adenosine receptor agonists: Implications for roles of P2Y1 and A1/A3 receptors in physiology and pathology. Purinergic Signal. 2020, 16, 543–559. [Google Scholar] [CrossRef]
- Liston, T.E.; Hama, A.; Boltze, J.; Poe, R.B.; Natsume, T.; Hayashi, I.; Takamatsu, H.; Korinek, W.S.; Lechleiter, J.D. Adenosine A1R/A3R (Adenosine A1 and A3 Receptor) Agonist AST-004 Reduces Brain Infarction in a Nonhuman Primate Model of Stroke. Stroke 2022, 53, 238–248. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet. Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338, 1–18. [Google Scholar] [CrossRef]
- Yoon, M.H.; Bae, H.B.; Choi, J. Il Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth. Analg. 2005, 101, 1417–1421. [Google Scholar] [CrossRef]
- Taiwo, Y.O.; Levine, J.D. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 1990, 38, 757–762. [Google Scholar] [CrossRef]
- Karlsten, R.; Gordh, T.; Post, C. Local Antinociceptive and Hyper algesic Effects in the Formalin Test after Peripheral Administration of Adenosine Analogues in Mice. Pharmacol. Toxicol. 1992, 70, 434–438. [Google Scholar] [CrossRef]
- Doak, G.J.; Sawynok, J. Complex role of peripheral adenosine in the genesis of the response to subcutaneous formalin in the rat. Eur. J. Pharmacol. 1995, 281, 311–318. [Google Scholar] [CrossRef]
- Taiwo, Y.O.; Levine, J.D. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience 1991, 44, 131–135. [Google Scholar] [CrossRef]
- Khasar, S.G.; Wang, J.F.; Taiwo, Y.O.; Heller, P.H.; Green, P.G.; Levine, J.D. Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat: A G-protein βγ subunit-mediated effect? Neuroscience 1995, 67, 189–195. [Google Scholar] [CrossRef]
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Kwilasz, A.J.; Ellis, A.; Wieseler, J.; Loram, L.; Favret, J.; McFadden, A.; Springer, K.; Falci, S.; Rieger, J.; Maier, S.F.; et al. Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A 2A receptor agonists. Brain. Behav. Immun. 2018, 69, 470–479. [Google Scholar] [CrossRef]
- Falsini, M.; Catarzi, D.; Varano, F.; Ceni, C.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Lucarini, E.; et al. Antioxidant-Conjugated 1,2,4-Triazolo[4,3-a]pyrazin-3-one Derivatives: Highly Potent and Selective Human A2A Adenosine Receptor Antagonists Possessing Protective Efficacy in Neuropathic Pain. J. Med. Chem. 2019, 62, 8511–8531. [Google Scholar] [CrossRef]
- Magni, G.; Riccio, D.; Ceruti, S. Tackling Chronic Pain and Inflammation through the Purinergic System. Curr. Med. Chem. 2018, 25, 3830–3865. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Adebiyi, M.G.; Luo, J.; Sun, K.; Le, T.T.T.; Zhang, Y.; Wu, H.; Zhao, S.; Karmouty-Quintana, H.; Liu, H.; et al. Sustained Elevated Adenosine via ADORA2B Promotes Chronic Pain through Neuro-immune Interaction. Cell Rep. 2016, 16, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Kotańska, M.; Szafarz, M.; Mika, K.; Dziubina, A.; Bednarski, M.; Müller, C.E.; Sapa, J.; Kieć-Kononowicz, K. PSB 603 - a known selective adenosine A2B receptor antagonist - has anti-inflammatory activity in mice. Biomed. Pharmacother. 2021, 135, 111164. [Google Scholar] [CrossRef]
- Zhang, L.; Paine, C.; Dip, R. Selective regulation of nuclear orphan receptors 4A by adenosine receptor subtypes in human mast cells. J. Cell Commun. Signal. 2010, 4, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Abad, A.S.S.; Falanji, F.; Ghanbarabadi, M.; Rad, A.; Nazemi, S.; Pejhan, A.; Amin, B. Assessment of anti-nociceptive effect of allopurinol in a neuropathic pain model. Brain Res. 2019, 1720, 146238. [Google Scholar] [CrossRef]
- Puntambekar, P. Direct Interaction of Adenosine with the TRPV1 Channel Protein. J. Neurosci. 2004, 24, 3663–3671. [Google Scholar] [CrossRef]
- Chang, C.-H.; Chang, Y.-S.; Hsieh, Y.-L. Transient receptor potential vanilloid subtype 1 depletion mediates mechanical allodynia through cellular signal alterations in small-fiber neuropathy. Pain Reports 2021, 6, e922. [Google Scholar] [CrossRef]
- Haddad, M. Adenosine A2B Receptors-Mediated Induction of Interleukin-6 in Skeletal Muscle Cells. Turk. J. Pharm. Sci. 2017, 14, 19. [Google Scholar] [CrossRef]
- Chen, Z.; Janes, K.; Chen, C.; Doyle, T.; Bryant, L.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Controlling murine and rat chronic pain through A 3 adenosine receptor activation. FASEB J. 2012, 26, 1855–1865. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Giancotti, L.A.; Lauro, F.; Mufti, F.; Salvemini, D. Treatment of chronic neuropathic pain: Purine receptor modulation. Pain 2020, 161, 1425–1441. [Google Scholar] [CrossRef]
- Tosh, D.K.; Finley, A.; Paoletta, S.; Moss, S.M.; Gao, Z.-G.; Gizewski, E.T.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. In Vivo Phenotypic Screening for Treating Chronic Neuropathic Pain: Modification of C 2-Arylethynyl Group of Conformationally Constrained A 3 Adenosine Receptor Agonists. J. Med. Chem. 2014, 57, 9901–9914. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Padia, J.; Salvemini, D.; Jacobson, K.A. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 2015, 11, 371. [Google Scholar] [CrossRef] [Green Version]
- Wahlman, C.; Doyle, T.M.; Little, J.W.; Luongo, L.; Janes, K.; Chen, Z.; Esposito, E.; Tosh, D.K.; Cuzzocre, S.; Jacobson, K.A.; et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 2018, 159, 1025–1034. [Google Scholar] [CrossRef]
- Suresh, R.R.; Jain, S.; Chen, Z.; Tosh, D.K.; Ma, Y.; Podszun, M.C.; Rotman, Y.; Salvemini, D.; Jacobson, K.A. Design and in vivo activity of A 3 adenosine receptor agonist prodrugs. Purinergic Signal. 2020, 16, 367–377. [Google Scholar] [CrossRef]
- Little, J.W.; Ford, A.; Symons-Liguori, A.M.; Chen, Z.; Janes, K.; Doyle, T.; Xie, J.; Luongo, L.; Tosh, D.K.; Maione, S.; et al. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. [Google Scholar] [CrossRef]
- Stockstill, K.; Wahlman, C.; Braden, K.; Chen, Z.; Yosten, G.L.; Tosh, D.K.; Jacobson, K.A.; Doyle, T.M.; Samson, W.K.; Salvemini, D. Sexually dimorphic therapeutic response in bortezomib-induced neuropathic pain reveals altered pain physiology in female rodents. Pain 2020, 161, 177–184. [Google Scholar] [CrossRef]
- Coppi, E.; Cherchi, F.; Fusco, I.; Failli, P.; Vona, A.; Dettori, I.; Gaviano, L.; Lucarini, E.; Jacobson, K.A.K.A.; Tosh, D.K.D.K.; et al. Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 2019, 160, 1103–1118. [Google Scholar] [CrossRef]
- MacDonald, R.L.; Skerritt, J.H.; Werz, M.A. Adenosine agonists reduce voltage-dependent calcium conductance of mouse sensory neurones in cell culture. J. Physiol. 1986, 370, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Hannon, H.E.; Atchison, W.D. Omega-conotoxins as experimental tools and therapeutics in pain management. Mar. Drugs 2013, 11, 680–699. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert Opin. Investig. Drugs 2000, 9, 2403–2410. [Google Scholar] [CrossRef]
- Brookes, M.E.; Eldabe, S.; Batterham, A. Ziconotide Monotherapy: A Systematic Review of Randomised Controlled Trials. Curr. Neuropharmacol. 2016, 15, 217–231. [Google Scholar] [CrossRef]
- Castro, J.; Harrington, A.M.; Hughes, P.A.; Martin, C.M.; Ge, P.; Shea, C.M.; Jin, H.; Jacobson, S.; Hannig, G.; Mann, E.; et al. Linaclotide inhibits colonic nociceptors and relieves abdominal pain via guanylate cyclase-C and extracellular cyclic guanosine 3’,5’-monophosphate. Gastroenterology 2013, 145, 1334–1346. [Google Scholar] [CrossRef]
- Chey, W.D.; Lembo, A.J.; Lavins, B.J.; Shiff, S.J.; Kurtz, C.B.; Currie, M.G.; MacDougall, J.E.; Jia, X.D.; Shao, J.Z.; Fitch, D.A.; et al. Linaclotide for irritable bowel syndrome with constipation: A 26-week, randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety. Am. J. Gastroenterol. 2012, 107, 1702–1712. [Google Scholar] [CrossRef]
- Lucarini, E.; Coppi, E.; Micheli, L.; Parisio, C.; Vona, A.; Cherchi, F.; Pugliese, A.M.; Pedata, F.; Failli, P.; Palomino, S.; et al. Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 2020, 161, 2179–2190. [Google Scholar] [CrossRef]
- Janes, K.; Esposito, E.; Doyle, T.; Cuzzocrea, S.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain 2014, 155, 2560–2567. [Google Scholar] [CrossRef] [Green Version]
- Janes, K.; Wahlman, C.; Little, J.W.; Doyle, T.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain. Behav. Immun. 2015, 44, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.; Squillace, S.; Lauro, F.; Giancotti, L.A.; Coppi, E.; Cherchi, F.; Di Cesare Mannelli, L.; Ghelardini, C.; Kolar, G.; Wahlman, C.; et al. Adenosine A3 agonists reverse neuropathic pain via T cell–mediated production of IL-10. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppi, E.; Cherchi, F.; Venturini, M.; Lucarini, E.; Corradetti, R.; Di Cesare Mannelli, L.; Ghelardini, C.; Pedata, F.; Pugliese, A.M. Therapeutic Potential of Highly Selective A3 Adenosine Receptor Ligands in the Central and Peripheral Nervous System. Molecules 2022, 27, 1890. https://doi.org/10.3390/molecules27061890
Coppi E, Cherchi F, Venturini M, Lucarini E, Corradetti R, Di Cesare Mannelli L, Ghelardini C, Pedata F, Pugliese AM. Therapeutic Potential of Highly Selective A3 Adenosine Receptor Ligands in the Central and Peripheral Nervous System. Molecules. 2022; 27(6):1890. https://doi.org/10.3390/molecules27061890
Chicago/Turabian StyleCoppi, Elisabetta, Federica Cherchi, Martina Venturini, Elena Lucarini, Renato Corradetti, Lorenzo Di Cesare Mannelli, Carla Ghelardini, Felicita Pedata, and Anna Maria Pugliese. 2022. "Therapeutic Potential of Highly Selective A3 Adenosine Receptor Ligands in the Central and Peripheral Nervous System" Molecules 27, no. 6: 1890. https://doi.org/10.3390/molecules27061890