
New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
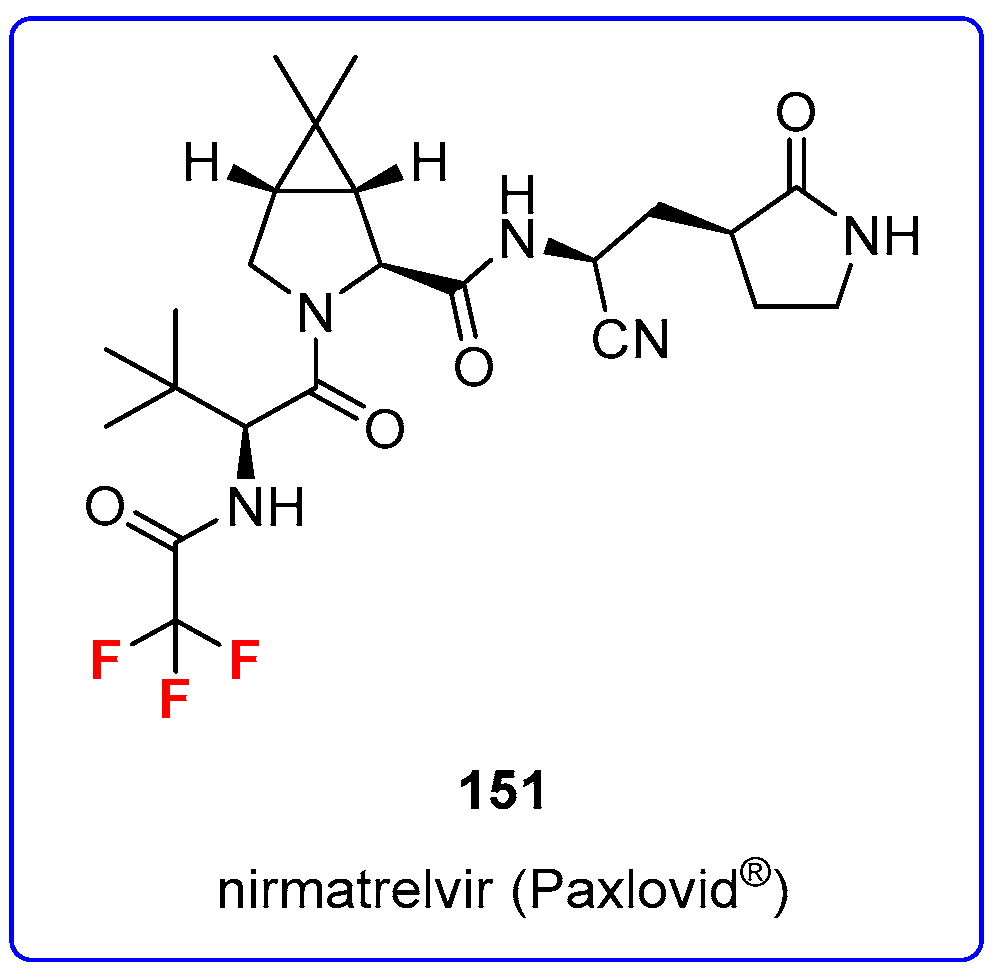
2. Fluorine and Chlorine in Medicinal Chemistry
3. New Halogen-Containing Drugs
3.1. Halogenated Anticancer Drugs Approved in 2021
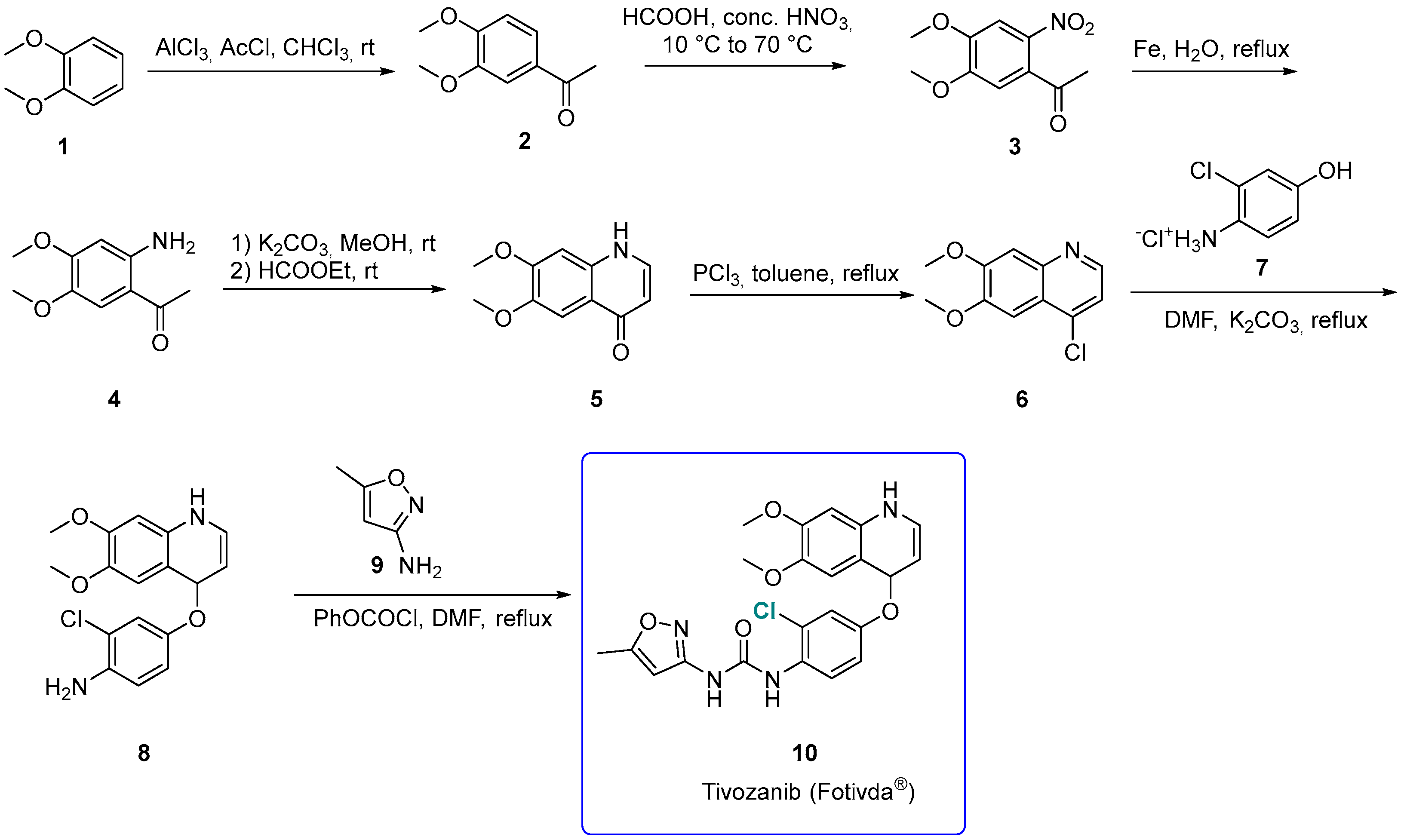
3.1.1. Tivozanib
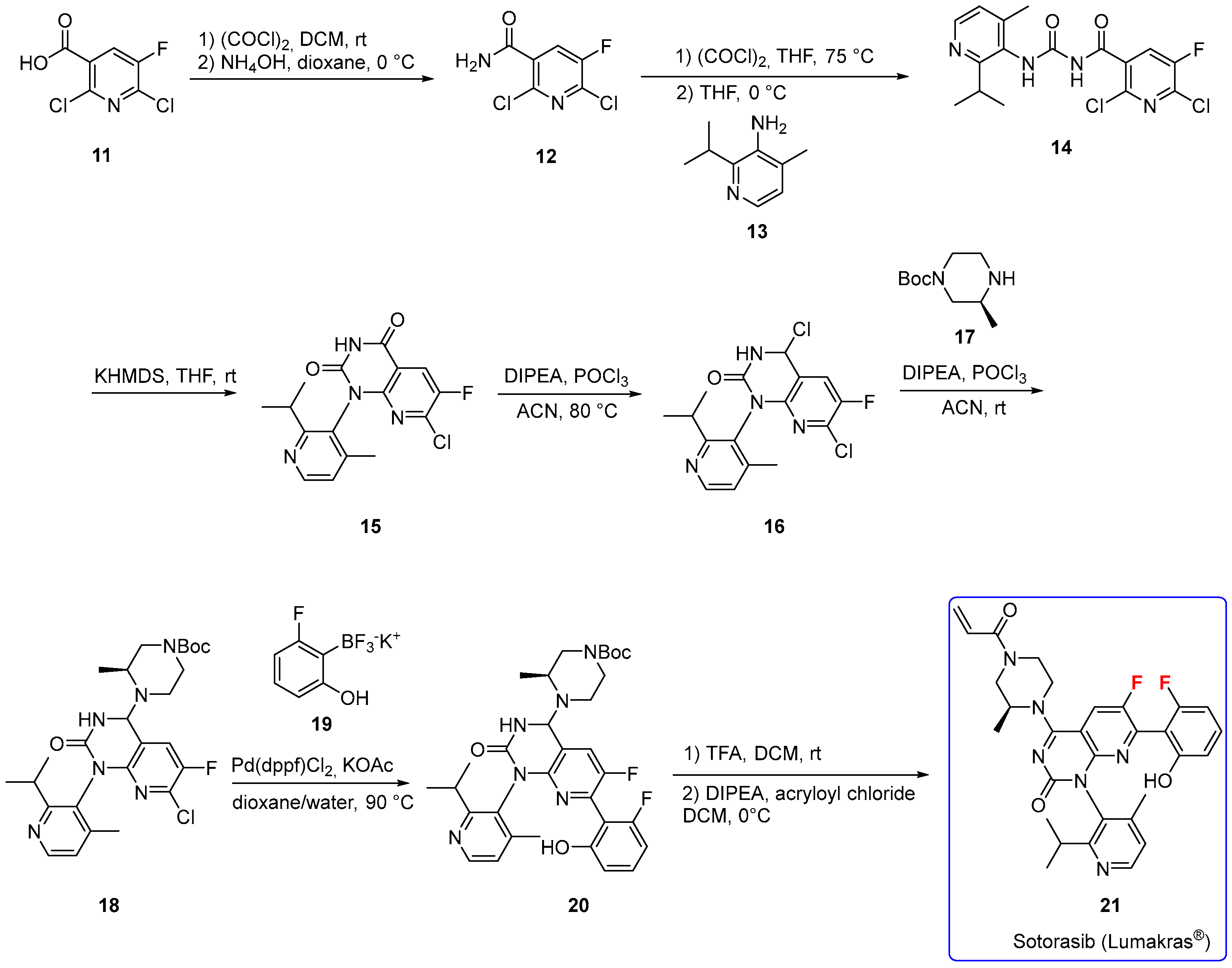
3.1.2. Sotorasib
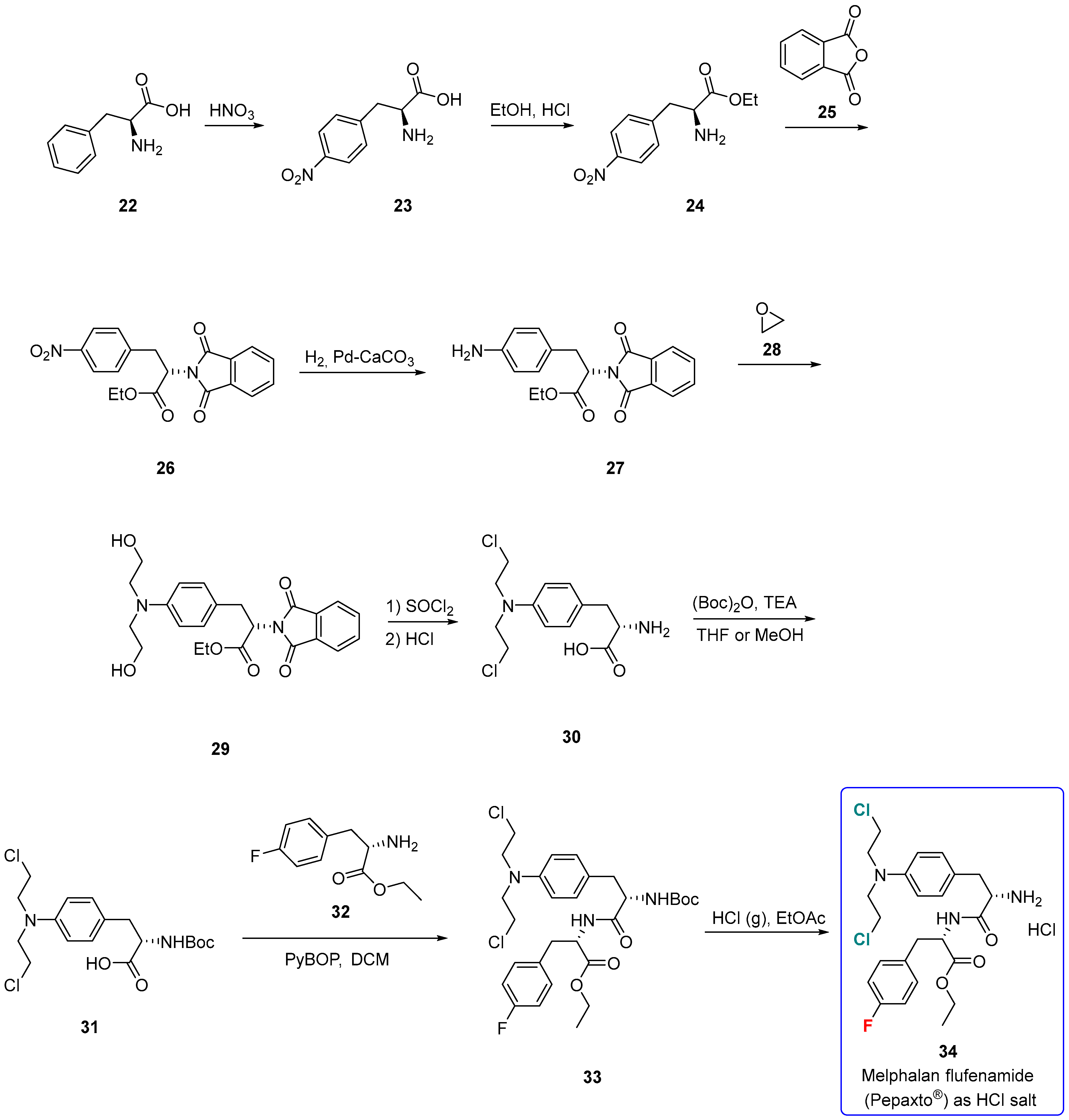
3.1.3. Melphalan Flufenamide
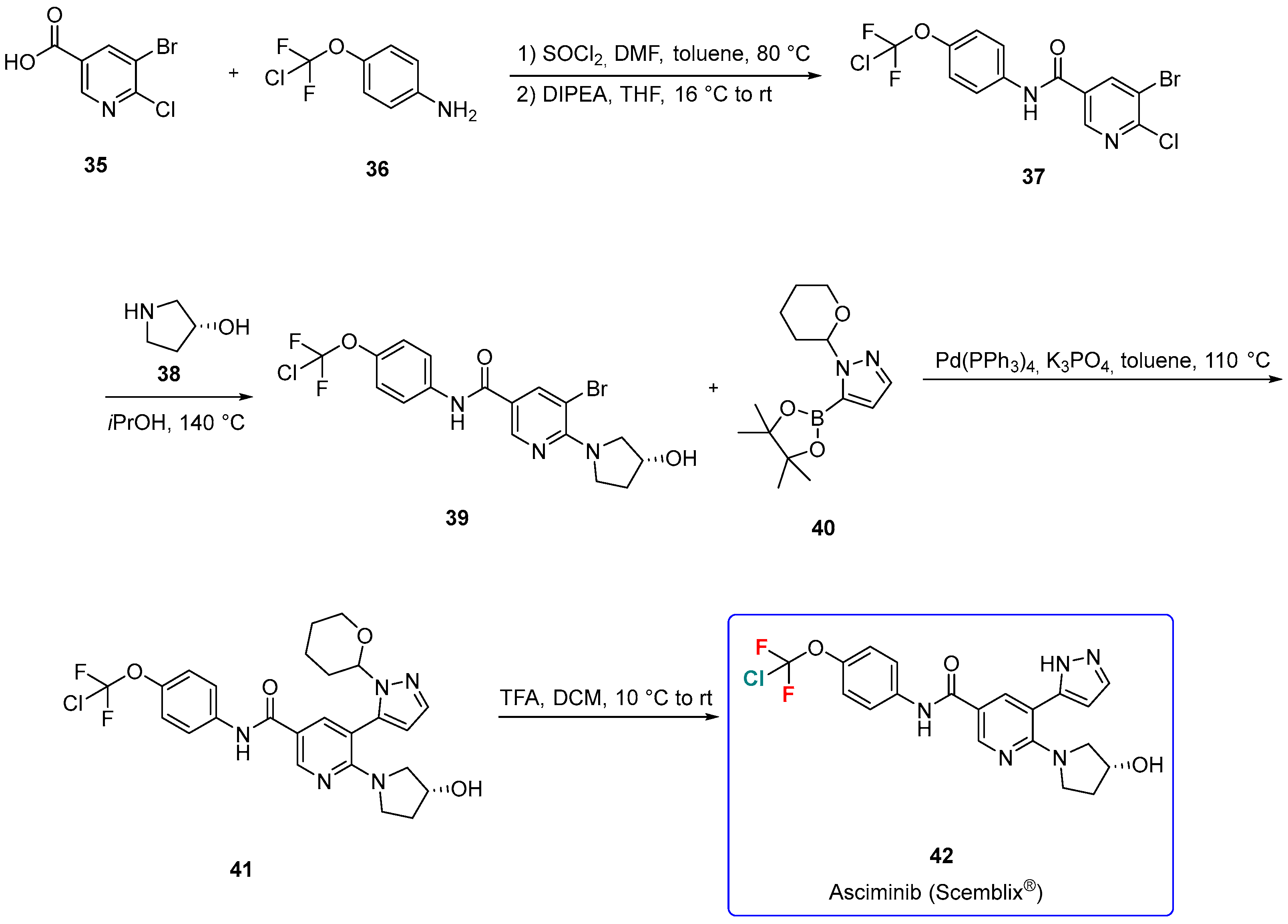
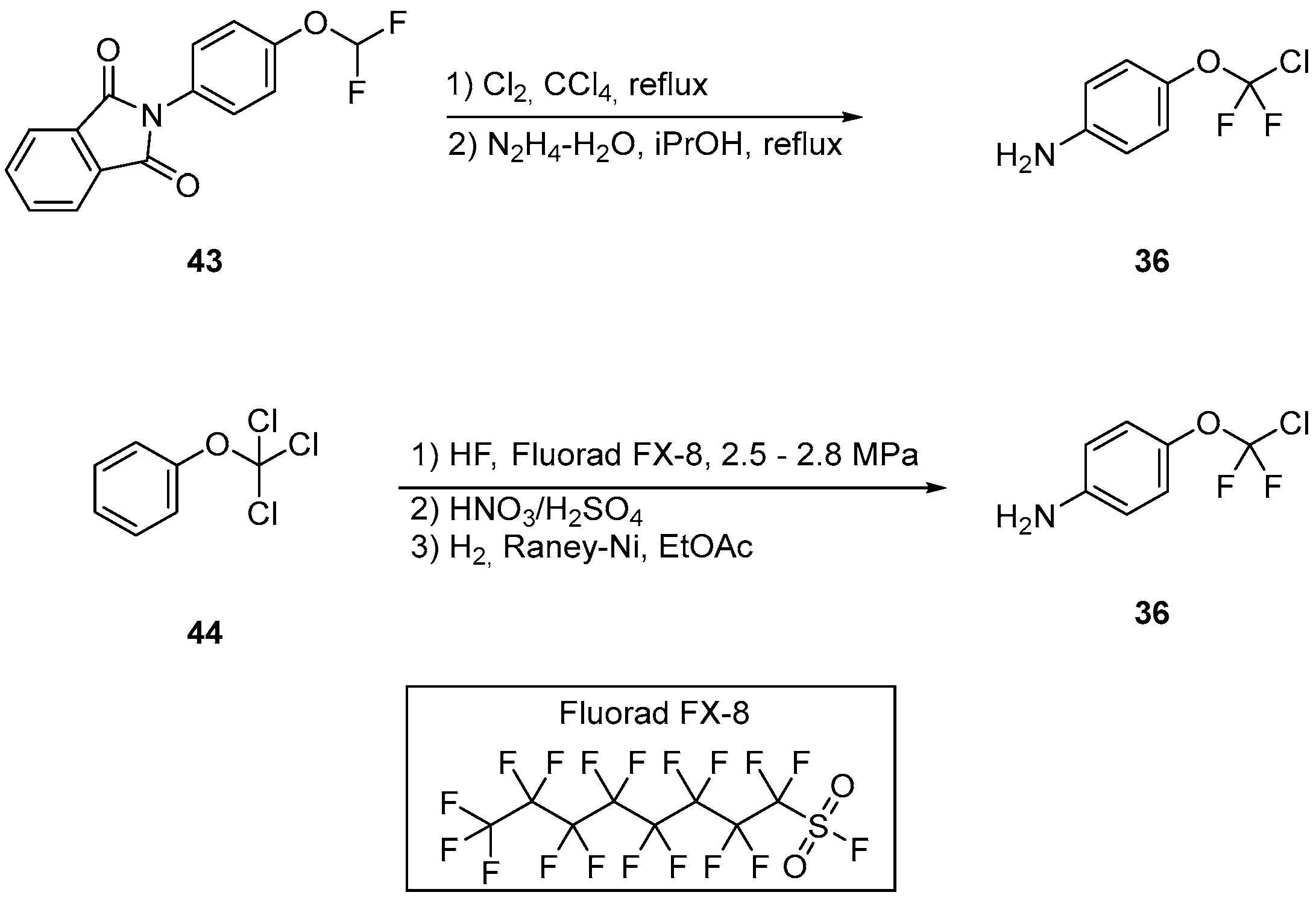
3.1.4. Asciminib
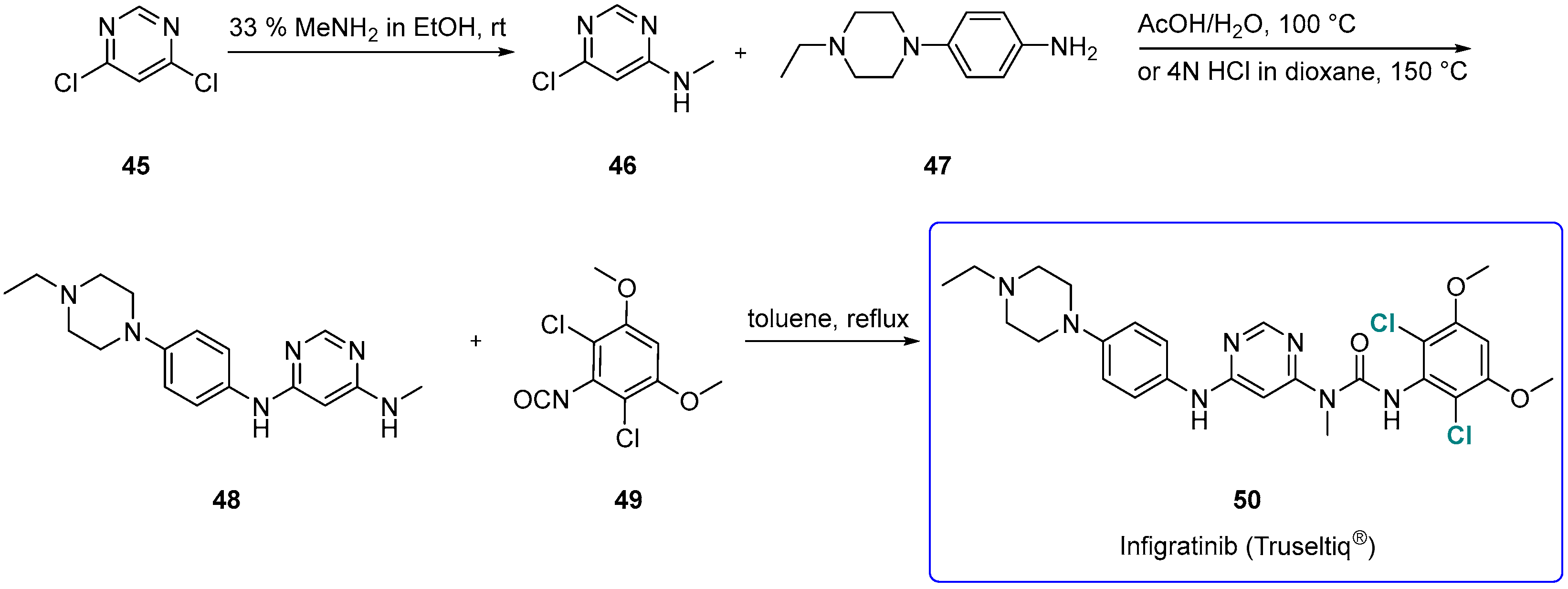
3.1.5. Infigratinib
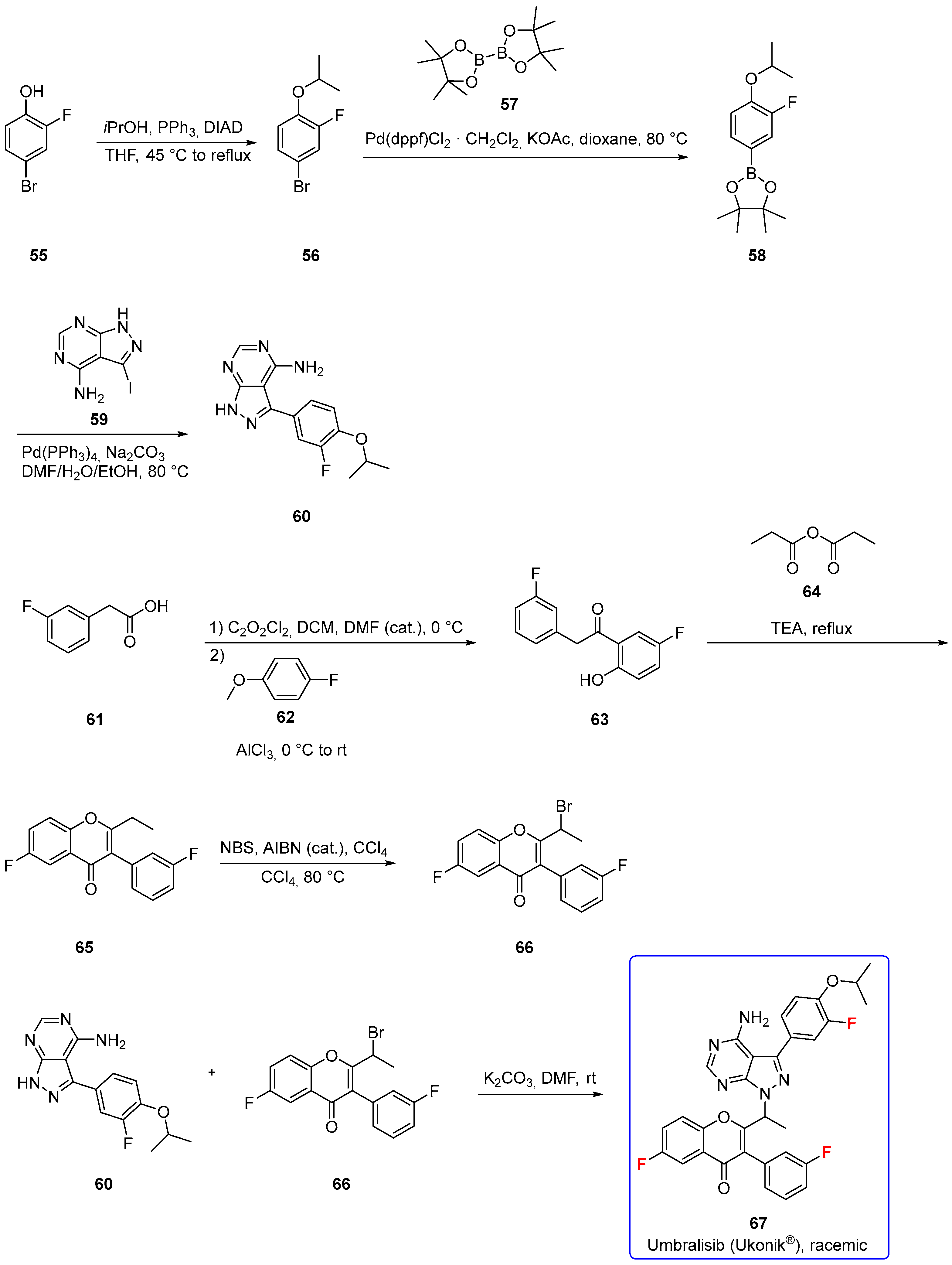
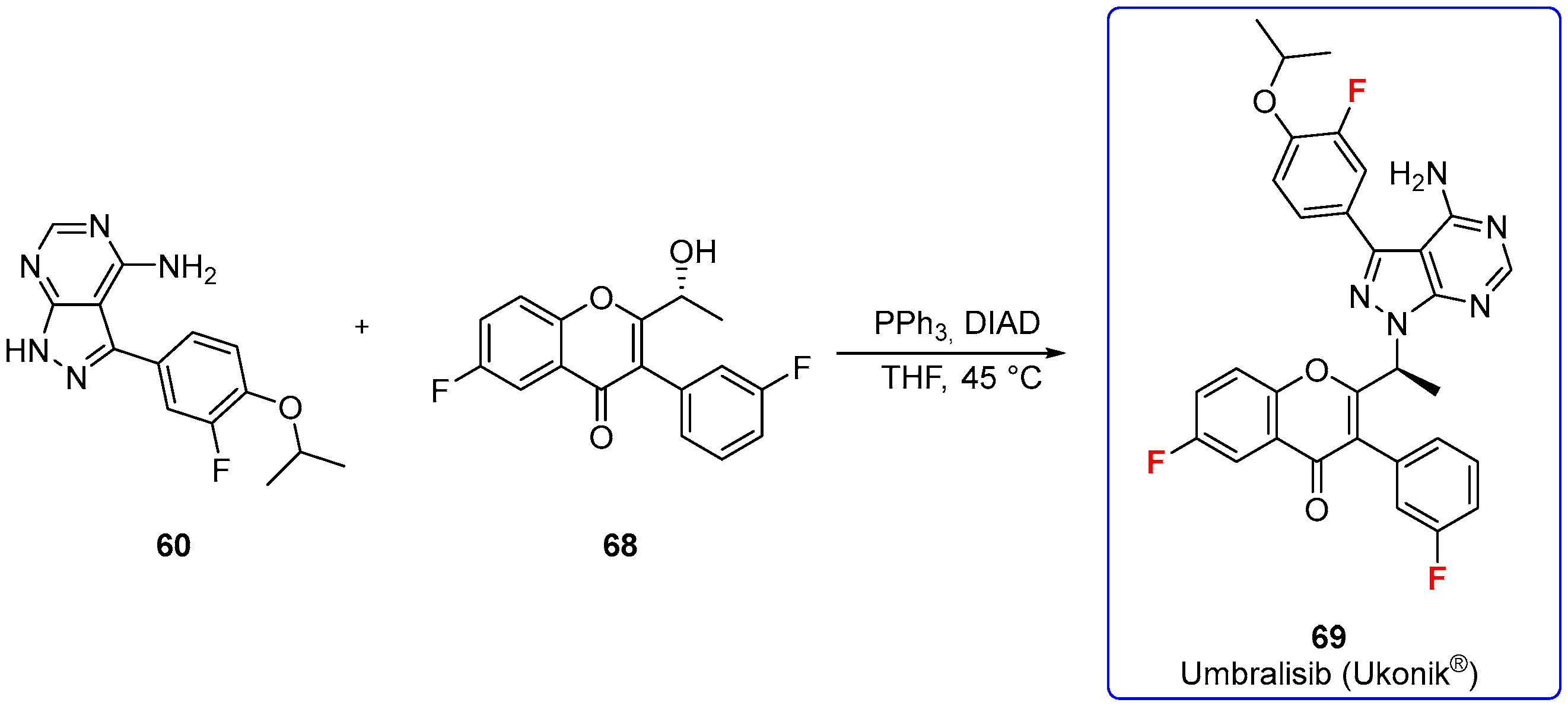
3.1.6. Umbralisib
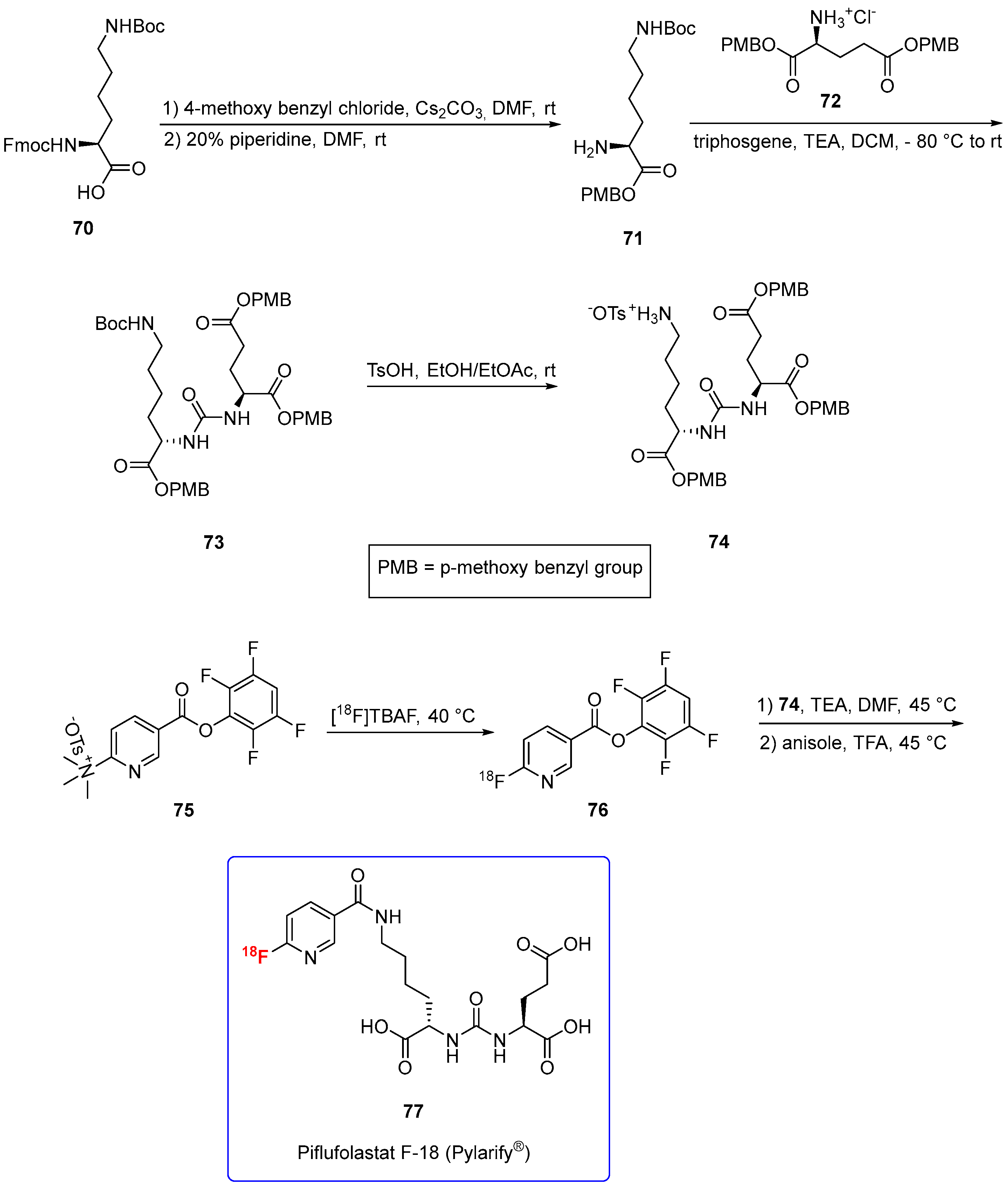
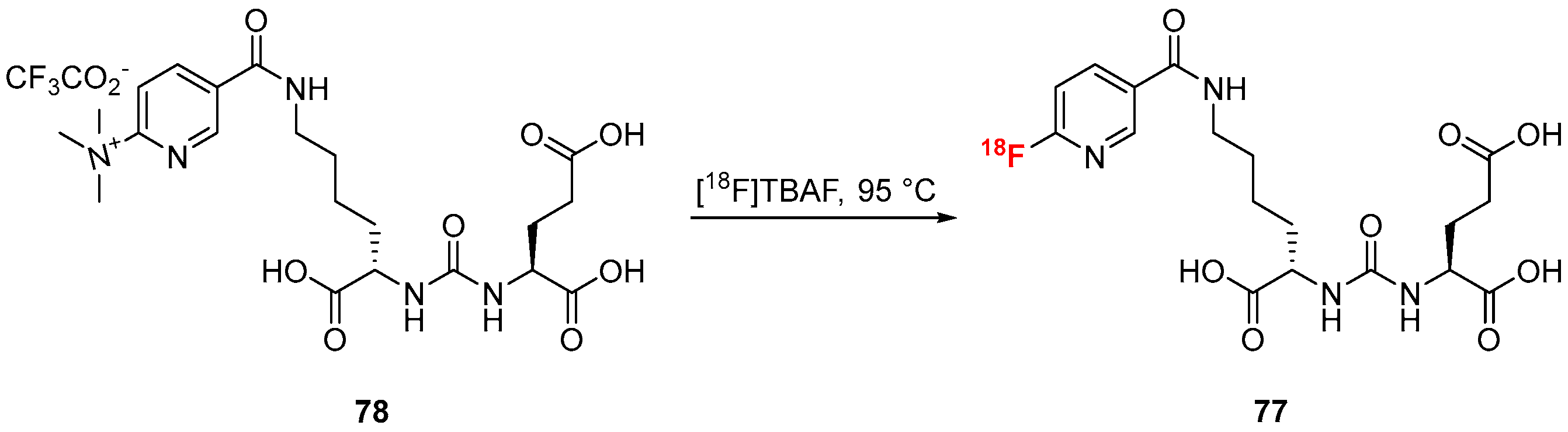
3.1.7. Piflufolastat F-18
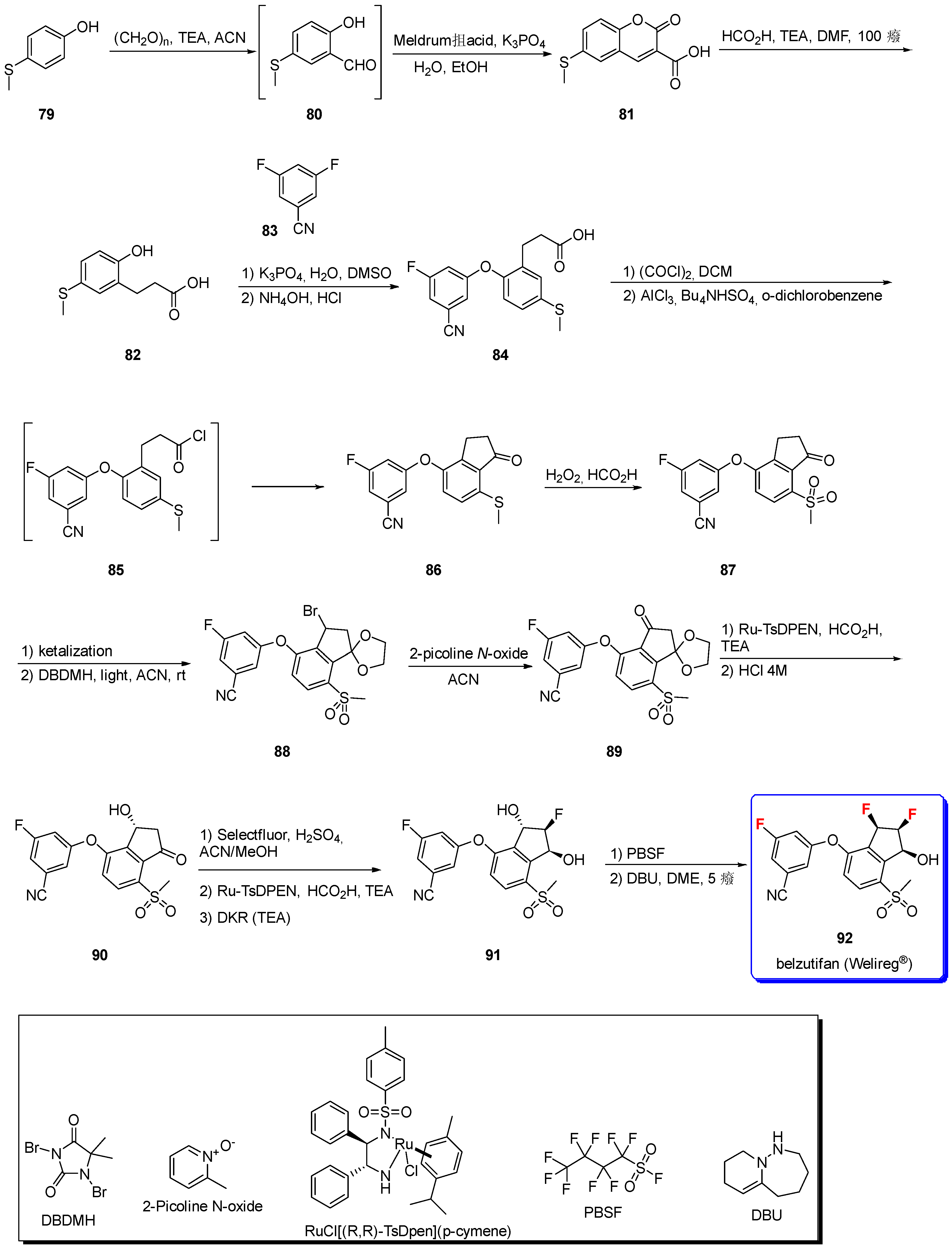
3.1.8. Belzutifan
3.2. Halogenated Antiviral Drugs Approved in 2021
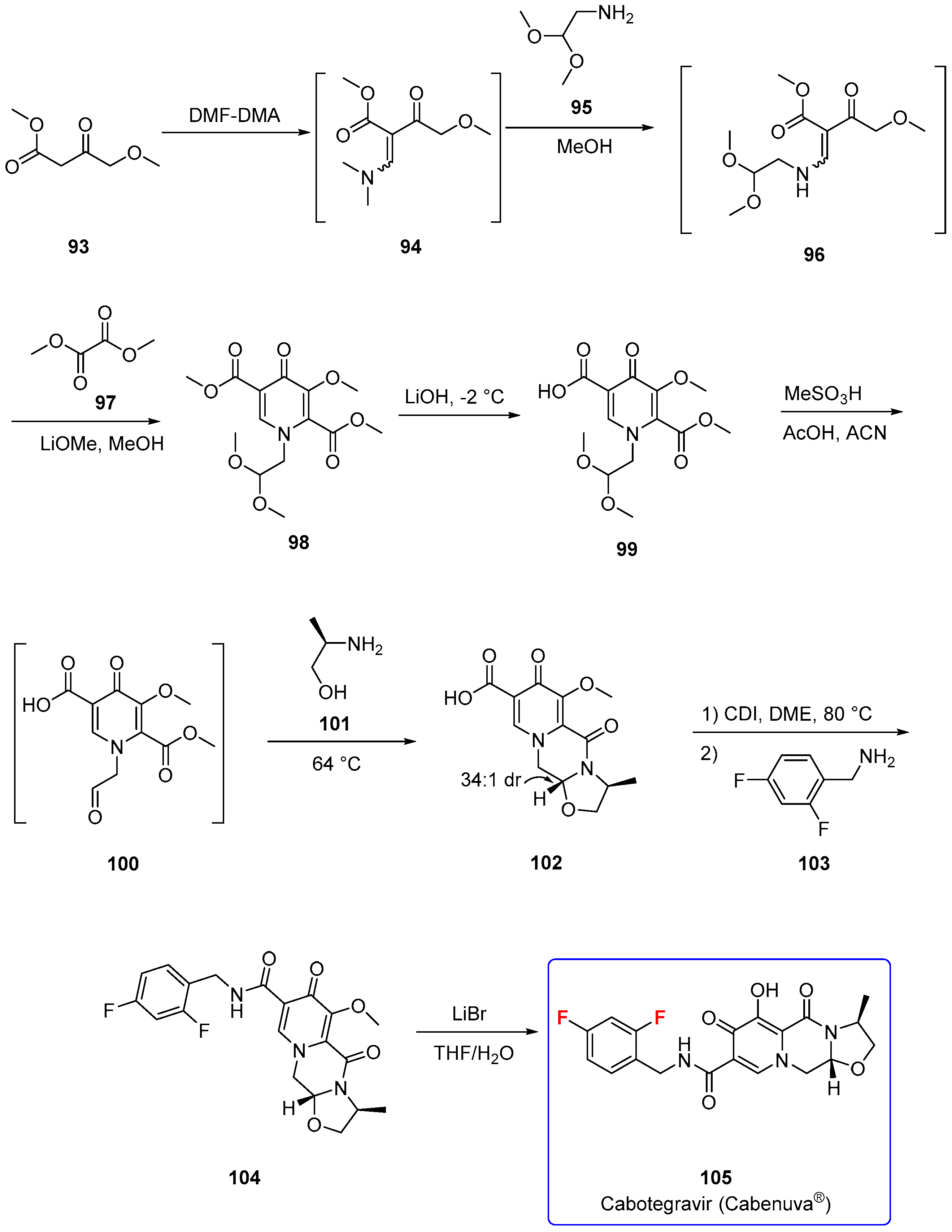
3.2.1. Cabotegavir
3.2.2. Maribavir
3.3. Halogenated Drugs Approved in 2021 for the Treatment of Multiple Sclerosis
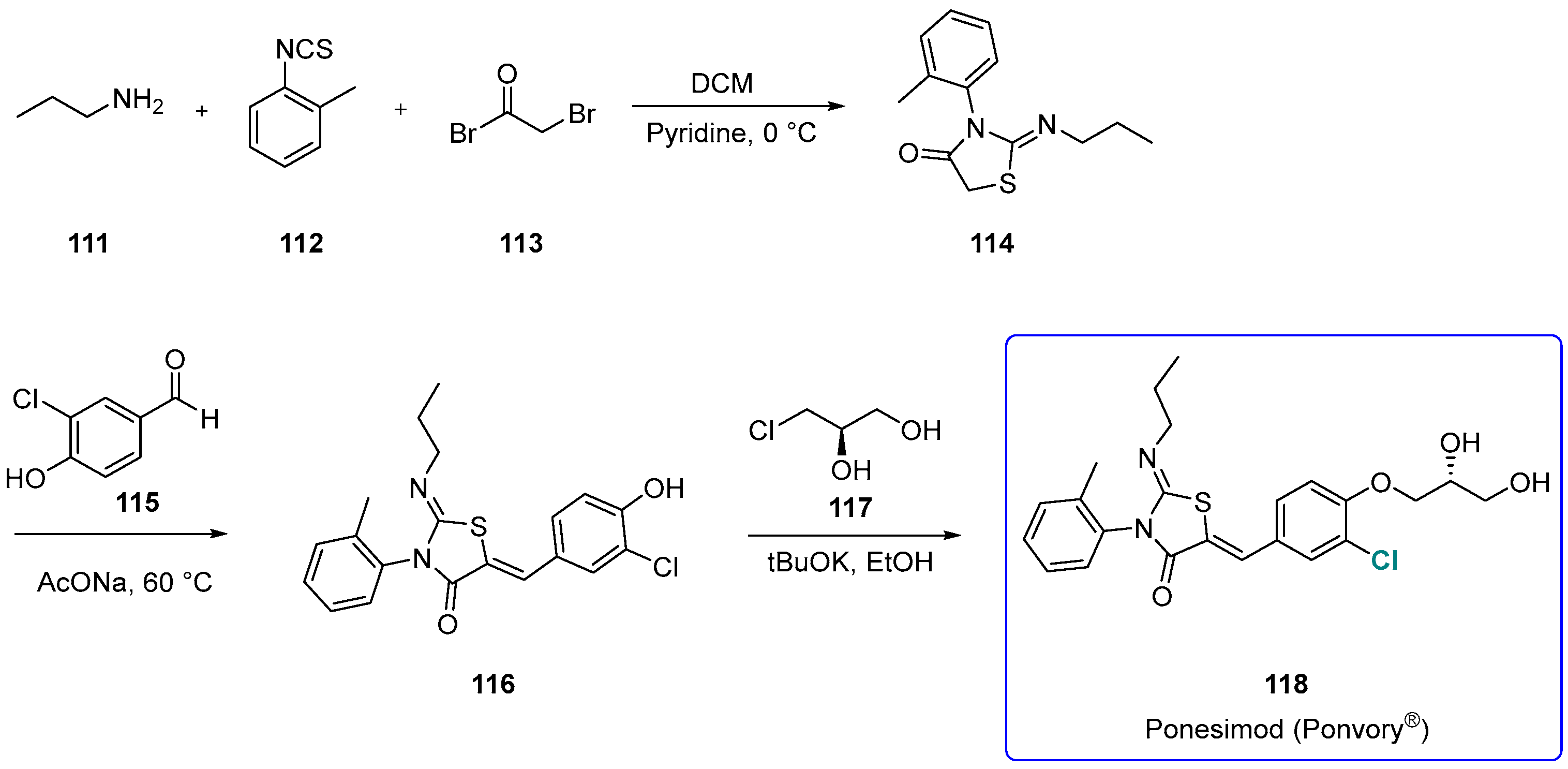
Ponesimod
3.4. Halogenated Drugs Approved in 2021 for the Treatment of Migraines
Atogepant
3.5. Halogenated Drugs Approved in 2021 for the Treatment of Vasculitis
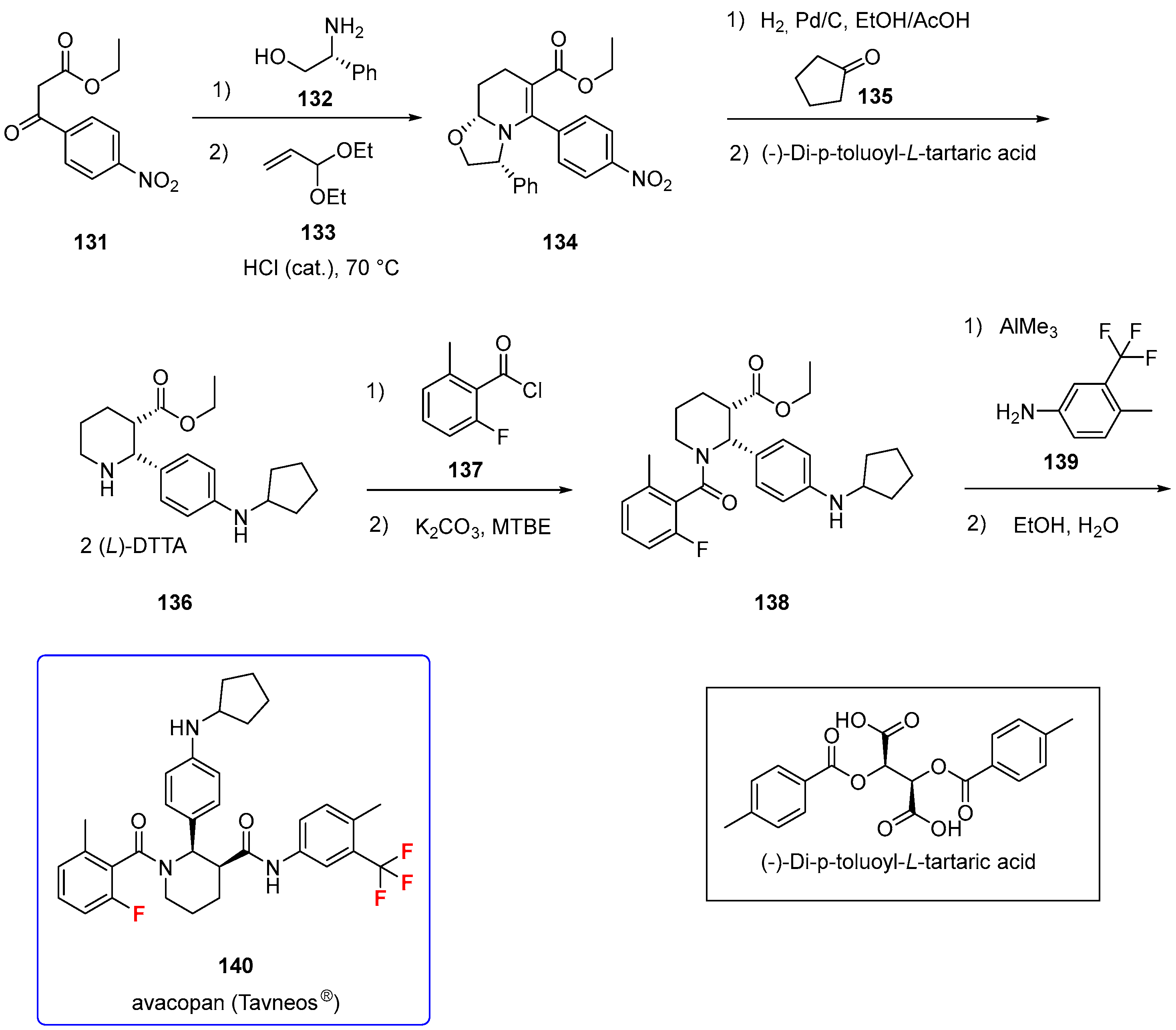
Avacopan
3.6. Halogenated Drugs Approved in 2021 for the Treatment of Cardiovascular Disease
Vericiguat
4. Conclusions and Future Perspectives
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- New Drug Therapy Approvals 2021. Available online: https://www.fda.gov/media/155227/download (accessed on 15 February 2022).
- Novel Drug Approvals for 2020|FDA. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020 (accessed on 15 February 2022).
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Martínez, F.J.; Barrajón-Catalán, E.; Micol, V. Tackling Antibiotic Resistance with Compounds of Natural Origin: A Comprehensive Review. Biomedicines 2020, 8, 405. [Google Scholar] [CrossRef]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated Compounds from Marine Algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.S.; Fujimori, D.G.; Walsh, C.T. Halogenation Strategies in Natural Product Biosynthesis. Chem. Biol. 2008, 15, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V.A.; Han, J. Fluorine-Containing Pharmaceuticals Approved by the FDA in 2020: Synthesis and Biological Activity. Chin. Chem. Lett. 2021, 32, 3342–3354. [Google Scholar] [CrossRef]
- Carvalho, M.F.; Oliveira, R.S. Natural Production of Fluorinated Compounds and Biotechnological Prospects of the Fluorinase Enzyme. Crit. Rev. Biotechnol. 2017, 37, 880–897. [Google Scholar] [CrossRef] [Green Version]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. Chem. Med. Chem. 2007, 2, 1100–1115. [Google Scholar] [CrossRef]
- Rowley, M.; Hallett, D.J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T.J.; Patel, S.; Marwood, R.; Patel, S.; Thomas, S.; et al. 3-(4-Fluoropiperidin-3-Yl)-2-Phenylindoles as High Affinity, Selective, and Orally Bioavailable H5-HT2A Receptor Antagonists. J. Med. Chem. 2001, 44, 1603–1614. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The Role of Fluorine in Medicinal Chemistry: Review Article. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Eskandari, K.; Lesani, M. Does Fluorine Participate in Halogen Bonding? Chem. Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, D.B.; Karukurichi, K.R.; de la Salud-Bea, R.; Nelson, D.L.; McCune, C.D. Use of Fluorinated Functionality in Enzyme Inhibitor Development: Mechanistic and Analytical Advantages. J. Fluor. Chem. 2008, 129, 731–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Song, M.; Ali, A.I.M.; Mudassir, S.M.; Ge, H. Recent Advances in the Application of Selectfluor as a “Fluorine-free” Functional Reagent in Organic Synthesis. Chem. Asian J. 2020, 15, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ma, R.; Fu, Q.; Chen, R.; Wang, Z.; Wang, L.; Ma, Y. Selective Electrophilic Di- and Monofluorinations for the Synthesis of 4-Difluoromethyl and 4-Fluoromethyl Quinazolin(Thi)Ones by a Selectfluor-Triggered Multi-Component Reaction. Org. Chem. Front. 2022. [Google Scholar] [CrossRef]
- Cotman, A.E.; Guérin, T.; Kovačević, I.; Benedetto Tiz, D.; Durcik, M.; Fulgheri, F.; Možina, Š.; Secci, D.; Sterle, M.; Ilaš, J.; et al. Practical Synthesis and Application of Halogen-Doped Pyrrole Building Blocks. ACS Omega 2021, 6, 9723–9730. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Durón, S.G.; Burkart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic Insight and Applications. Angew. Chem. Int. Ed. 2005, 44, 192–212. [Google Scholar] [CrossRef]
- Fang, W.Y.; Ravindar, L.; Rakesh, K.P.; Manukumar, H.M.; Shantharam, C.S.; Alharbi, N.S.; Qin, H.L. Synthetic Approaches and Pharmaceutical Applications of Chloro-Containing Molecules for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 173, 117–153. [Google Scholar] [CrossRef]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; Junior, W.F.d.A.; Leite, A.C.L. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- A Proclamation on the 50th Anniversary of the National Cancer Act of 1971. Available online: https://www.whitehouse.gov/briefing-room/presidential-actions/2021/12/22/a-proclamation-on-the-50th-anniversary-of-the-national-cancer-act-of-1971/ (accessed on 5 February 2022).
- Anonymous Fotivda. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/fotivda (accessed on 24 January 2022).
- Westerman, M.E.; Wood, C.G. Editorial Commentary: Tivozanib versus Sorafenib in Patients with Advanced Renal Cell Carcinoma (TIVO-3): A Phase 3, Multicentre, Randomised, Controlled, Open-Label Study. Ann. Transl. Med. 2020, 8, 1037. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, Q.; Wu, P.; He, C.; Yin, L.; Xu, F.; Yin, Z.; Yue, G.; Zou, Y.; Li, L.; et al. The Synthesis Review of the Approved Tyrosine Kinase Inhibitors for Anticancer Therapy in 2015-2020. Bioorg. Chem. 2021, 113, 105011. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Qin, Y. Synthesis of Tivozanib. Chin. J. Pharm. 2013, 44, 541–544. [Google Scholar]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Lanman, B.A.; Chen, J.; Reed, A.B.; Cee, V.J.; Liu, L.; Kopecky, D.J.; Lopez, P.; Wurz, R.P.; Nguyen, T.T.; Booker, S.; et al. Kras G12c Inhibitors and Methods of Using the Same. WO/2018/217651, 29 November 2018. [Google Scholar]
- Parsons, A.T.; Beaver, M. Improved Synthesis of Kras G12c Inhibitor Compound. WO/2021/097212, 20 May 2021. [Google Scholar]
- Dhillon, S. Melphalan Flufenamide (Melflufen): First Approval. Drugs 2021, 81, 963–969. [Google Scholar] [CrossRef]
- Wahlstrom, N.H.; Wennerberg, J.A. Process for Preparation of Nitrogen Mustard Derivatives. WO2016180740A1, 17 November 2016. [Google Scholar]
- Lehmann, F.; Wennerberg, J. Melflufen: A Journey from Discovery to Multi-Kilogram Production. In Complete Accounts of Integrated Drug Discovery and Development: Recent Examples from the Pharmaceutical Industry Volume 3; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2020; Volume 1369, pp. 157–177. ISBN 978-0-8412-9864-4. [Google Scholar]
- FDA. FDA Approves Asciminib for Philadelphia Chromosome-Positive Chronic Myeloid Leukemia. 2021. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-asciminib-philadelphia-chromosome-positive-chronic-myeloid-leukemia (accessed on 15 February 2022).
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [Green Version]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The Allosteric Inhibitor ABL001 Enables Dual Targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Makitruk, V.L.; Nuzha, Y.O.; Petko, K.I.; Fialkov, Y.A.; Shalamai, A.S.; Yagupol’s’kii, L.M. 2-Amino-6-(trifluoromethoxy)benzothiazole (Borizole) and admixtures accompanying it. Zhurnal Org. Farmatsevtichnoi Khimii 2007, 5, 28–33. [Google Scholar]
- Fu, L.; Wang, A.; He, B.; Sun, J.; Song, C. Preparation Method for 4-(Chlorodifluoromethoxy)Aniline. CN104119238A, 29 October 2014. [Google Scholar]
- Kang, C. Infigratinib: First Approval. Drugs 2021, 81, 1355–1360. [Google Scholar] [CrossRef]
- Brameld, K.A.; Owens, T.D.; Verner, E.; Venetsanakos, E.; Bradshaw, J.M.; Phan, V.T.; Tam, D.; Leung, K.; Shu, J.; LaStant, J.; et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-Yl)Propyl)-6-(2,6-Dichloro-3,5-Dimethoxyphenyl)-2-(Methylamino)Pyrido[2,3-d]Pyrimidin-7(8H)-One (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 2017, 60, 6516–6527. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-Dimethoxy-Phenyl)-1-{6-[4-(4-Ethyl-Piperazin-1-Yl)-Phenylamino]-Pyrimidin-4-Yl}-1-Methyl-Urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Samaniego, F.; Jurczak, W.; Ghosh, N.; Derenzini, E.; Reeves, J.A.; Knopińska-Posłuszny, W.; Cheah, C.Y.; Phillips, T.; Lech-Maranda, E.; et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients with Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2021, 39, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Umbralisib-TG Therapeutics. Available online: https://www.tgtherapeutics.com/our-pipeline/umbralisib/ (accessed on 7 February 2022).
- Burris, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a Novel PI3Kδ and Casein Kinase-1ε Inhibitor, in Relapsed or Refractory Chronic Lymphocytic Leukaemia and Lymphoma: An Open-Label, Phase 1, Dose-Escalation, First-in-Human Study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef]
- Weiss, M.; Miskin, H.; Sportelli, P.; Vakkalanka, S.K.V.S. Combination of Anti-Cd20 Antibody and Pi3 Kinase Selective Inhibitor. WO/2014/071125, 8 May 2014. [Google Scholar]
- Keam, S.J. Piflufolastat F 18: Diagnostic First Approval. Mol. Diagn. Ther. 2021, 25, 647–656. [Google Scholar] [CrossRef]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.C.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and Evaluation of Technetium-99m- and Rhenium-Labeled Inhibitors of the Prostate-Specific Membrane Antigen (PSMA). J. Med. Chem. 2008, 51, 4504–4517. [Google Scholar] [CrossRef] [Green Version]
- Dasilva, J.; Dornan, M. Process for the Preparation of (18F)DCFPYL. CA3038601A1, 8 August 2020. [Google Scholar]
- FDA Approves Belzutifan for Cancers Associated with von Hippel-Lindau Disease. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belzutifan-cancers-associated-von-hippel-lindau-disease (accessed on 24 February 2022).
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D.; et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-Hydroxy-7-Methylsulfonylindan-4-Yl]Oxy-5-Fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Tan, L.; Chen, L.; Dalby, S.M.; DiRocco, D.A.; Duan, J.; Feng, M.; Gong, G.; Guo, H.; Hethcox, J.C.; et al. Manufacturing Process Development for Belzutifan, Part 1: A Concise Synthesis of the Indanone Starting Material. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Doušová, H.; Horák, R.; Růžičková, Z.; Šimůnek, P. An Intramolecular C–N Cross-Coupling of β-Enaminones: A Simple and Efficient Way to Precursors of Some Alkaloids of Galipea Officinalis. Beilstein J. Org. Chem. 2015, 11, 884–892. [Google Scholar] [CrossRef] [Green Version]
- Bottecchia, C.; Lévesque, F.; McMullen, J.P.; Ji, Y.; Reibarkh, M.; Peng, F.; Tan, L.; Spencer, G.; Nappi, J.; Lehnherr, D.; et al. Manufacturing Process Development for Belzutifan, Part 2: A Continuous Flow Visible-Light-Induced Benzylic Bromination. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Chen, Z.; Salehi Marzijarani, N.; Quirie, S.; Pirrone, G.F.; Dalby, S.M.; Wang, T.; Kim, J.; Peng, F.; Fine, A.J. Manufacturing Process Development for Belzutifan, Part 3: Completing a Streamlined through-Process with a Safe and Scalable Oxidation. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Salehi Marzijarani, N.; Fine, A.J.; Dalby, S.M.; Gangam, R.; Poudyal, S.; Behre, T.; Ekkati, A.R.; Armstrong, B.M.; Shultz, C.S.; Dance, Z.E.X.; et al. Manufacturing Process Development for Belzutifan, Part 4: Nitrogen Flow Criticality for Transfer Hydrogenation Control. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Wang, T.; Phillips, E.M.; Dalby, S.M.; Sirota, E.; Axnanda, S.; Shultz, C.S.; Patel, P.; Waldman, J.H.; Alwedi, E.; Wang, X.; et al. Manufacturing Process Development for Belzutifan, Part 5: A Streamlined Fluorination–Dynamic Kinetic Resolution Process. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Pirnot, M.; Stone, K.; Wright, T.J.; Lamberto, D.J.; Schoell, J.; Lam, Y.; Zawatzky, K.; Wang, X.; Dalby, S.M.; Fine, A.J.; et al. Manufacturing Process Development for Belzutifan, Part 6: Ensuring Scalability for a Deoxyfluorination Reaction. Org. Process Res. Dev. 2021. [Google Scholar] [CrossRef]
- Cattaneo, D.; Gervasoni, C. Pharmacokinetics and Pharmacodynamics of Cabotegravir, a Long-Acting HIV Integrase Strand Transfer Inhibitor. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cherukupalli, S.; Xie, M.; Wang, W.; Jiang, X.; Jia, R.; Pannecouque, C.; de Clercq, E.; Kang, D.; Zhan, P.; et al. Contemporary Medicinal Chemistry Strategies for the Discovery and Development of Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem. 2022. [Google Scholar] [CrossRef]
- Hughes, D.L. Review of Synthetic Routes and Final Forms of Integrase Inhibitors Dolutegravir, Cabotegravir, and Bictegravir. Org. Process Res. Dev. 2019, 23, 716–729. [Google Scholar] [CrossRef]
- Wang, H.; Kowalski, M.D.; Lakdawala, A.S.; Vogt, F.G.; Wu, L. An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor. Org. Lett. 2015, 17, 564–567. [Google Scholar] [CrossRef]
- LIVTENCITYTM (Maribavir). Available online: https://www.livtencity.com/ (accessed on 28 January 2022).
- Kozak, I.; McCutchan, J.A.; Freeman, W.R. Chapter 81—HIV-Associated Infections. In Retina, 5th ed.; Ryan, S.J., Sadda, S.R., Hinton, D.R., Schachat, A.P., Sadda, S.R., Wilkinson, C.P., Wiedemann, P., Schachat, A.P., Eds.; W.B. Saunders: London, UK, 2013; pp. 1441–1472. ISBN 978-1-4557-0737-9. [Google Scholar]
- Freeman, S. Process for Preparing Substituted Benzimidazole Compounds. WO2001077083A1, 18 October 2001. [Google Scholar]
- Kappos, L.; Fox, R.J.; Burcklen, M.; Freedman, M.S.; Havrdová, E.K.; Hennessy, B.; Hohlfeld, R.; Lublin, F.; Montalban, X.; Pozzilli, C.; et al. Ponesimod Compared with Teriflunomide in Patients with Relapsing Multiple Sclerosis in the Active-Comparator Phase 3 OPTIMUM Study. JAMA Neurol. 2021, 78, 558–567. [Google Scholar] [CrossRef]
- Petersen, G.; Wittmann, R.; Arndt, V.; Göpffarth, D. Epidemiology of multiple sclerosis in Germany: Regional differences and drug prescription in the claims data of the statutory health insurance. Nervenarzt 2014, 85, 990–998. [Google Scholar] [CrossRef]
- Wiendl, H.; Gold, R.; Berger, T.; Derfuss, T.; Linker, R.; Mäurer, M.; Aktas, O.; Baum, K.; Berghoff, M.; Bittner, S.; et al. Multiple Sclerosis Therapy Consensus Group (MSTCG): Position Statement on Disease-Modifying Therapies for Multiple Sclerosis (White Paper). Ther. Adv. Neurol. Disord. 2021, 14, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Bolli, M.H.; Abele, S.; Binkert, C.; Bravo, R.; Buchmann, S.; Bur, D.; Gatfield, J.; Hess, P.; Kohl, C.; Mangold, C.; et al. 2-Imino-Thiazolidin-4-One Derivatives as Potent, Orally Active S1P1 Receptor Agonists. J. Med. Chem. 2010, 53, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- Herse, C. Procédé De Préparation De (2z,5z)-5-(3-Chloro-4-((r)-2,3-Dihydroxypropoxy)benzylidene)-2-(propylimino)-3-|(o-Tolyl)thiazolidin-4-One Et Intermédiaire Utilisé Dans Ledit Procédé. WO/2014/027330, 20 February 2014. [Google Scholar]
- Abele, S.; Bolli, S.; Schmidt, G. New Process for the Preparation of 2-Imino-Thiazolidin-4-One Derivatives. WO-2008062376-A3, 10 July 2008. [Google Scholar]
- Burch, R.; Rizzoli, P.; Loder, E. The Prevalence and Impact of Migraine and Severe Headache in the United States: Updated Age, Sex, and Socioeconomic-Specific Estimates from Government Health Surveys. Headache. 2021, 61, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Atogepant. Available online: https://go.drugbank.com/drugs/DB16098 (accessed on 28 January 2022).
- Russo, A.F. Calcitonin Gene-Related Peptide (CGRP): A New Target for Migraine. Ann. Rev. Pharmacol. Toxicol. 2015, 55, 533–552. [Google Scholar] [CrossRef] [Green Version]
- Dubowchik, G.M.; Conway, C.M.; Xin, A.W. Blocking the CGRP Pathway for Acute and Preventive Treatment of Migraine: The Evolution of Success. J. Med. Chem. 2020, 63, 6600–6623. [Google Scholar] [CrossRef]
- Martelletti, P.; Cipolla, F.; Capi, M.; Curto, M.; Lionetto, L. Atogepant. Calcitonin Gene-Related Peptide (CGRP) Receptor Antagonist, Preventive Treatment of Migraine. Drugs Future 2020, 45, 285. [Google Scholar] [CrossRef]
- Bell, I.M.; Fraley, M.E.; Gallicchio, S.N.; Ginnetti, A.; Mitchell, H.J.; Paone, D.V.; Staas, D.D.; Wang, C.; Zartman, C.B.; Stevenson, H.E. Piperidinone Carboxamide Azaindane Cgrp Receptor Antagonists. WO2012064910A1, 12 May 2012. [Google Scholar]
- Geboes, K.; Dalle, I. Vasculitis and the Gastrointestinal Tract. Acta Gastroenterol. Belg. 2002, 65, 204–212. [Google Scholar]
- Felicetti, M.; Treppo, E.; Posarelli, C.; Ferro, F.; Bond, M.; Monti, S.; Elefante, E.; Trentin, F.; Delvino, P.; Talarico, R.; et al. One Year in Review 2020: Vasculitis. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 124), 3–14. [Google Scholar]
- Merkel, P.A.; Jayne, D.R.; Wang, C.; Hillson, J.; Bekker, P. Evaluation of the Safety and Efficacy of Avacopan, a C5a Receptor Inhibitor, in Patients with Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Treated Concomitantly with Rituximab or Cyclophosphamide/Azathioprine: Protocol for a Randomized, Double-Blind, Active-Controlled, Phase 3 Trial. JMIR Res. Protoc. 2020, 9, e16664. [Google Scholar] [CrossRef] [Green Version]
- Fan, P.; Kalisiak, J.; Krasinski, A.; Lui, R.; Powers, J.; Punna, S.; Tanaka, H.; Zhang, P. Processes and Intermediates in the Preparation of C5aR Antagonists. US20160090357A1, 31 March 2016. [Google Scholar]
- Walden, R.; Tomlinson, B. Cardiovascular Disease. In Herbal Medicine: Biomolecular and Clinical Aspects; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011; ISBN 978-1-4398-0713-2. [Google Scholar]
- Mechanism of Action (MOA) of VERQUVO® (Vericiguat). Available online: https://www.merckconnect.com/verquvo/mechanism-of-action/ (accessed on 29 January 2022).
- Zheng, W.; Wang, Z.; Jiang, X.; Zhao, Q.; Shen, J. Targeted Drugs for Treatment of Pulmonary Arterial Hypertension: Past, Present, and Future Perspectives. J. Med. Chem. 2020, 63, 15153–15186. [Google Scholar] [CrossRef]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Fey, P.; Griebenow, N.; Kroh, W.; Becker-Pelster, E.-M.; et al. Discovery of the Soluble Guanylate Cyclase Stimulator Vericiguat (BAY 1021189) for the Treatment of Chronic Heart Failure. J. Med. Chem. 2017, 60, 5146–5161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halford, B. How Pfizer Scientists Transformed an Old Drug Lead into a COVID-19 Antiviral. Chem. Eng. New 2022, 100, 3. [Google Scholar]
- Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19, FDA New Release, 22 December 2021. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19 (accessed on 15 February 2022).






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-HT2A a (nM) | pKa b | F (%) c | |
|---|---|---|---|
 | 0.99 | 10.4 | Poor |
 | 0.43 | 8.5 | 18 |
 | 0.06 | - | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetto Tiz, D.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use. Molecules 2022, 27, 1643. https://doi.org/10.3390/molecules27051643
Benedetto Tiz D, Bagnoli L, Rosati O, Marini F, Sancineto L, Santi C. New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use. Molecules. 2022; 27(5):1643. https://doi.org/10.3390/molecules27051643
Chicago/Turabian StyleBenedetto Tiz, Davide, Luana Bagnoli, Ornelio Rosati, Francesca Marini, Luca Sancineto, and Claudio Santi. 2022. "New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use" Molecules 27, no. 5: 1643. https://doi.org/10.3390/molecules27051643