
Practical Synthesis of Phosphinic Dipeptides by Tandem Esterification of Aminophosphinic and Acrylic Acids under Silylating Conditions

 , ,
, ,
Abstract
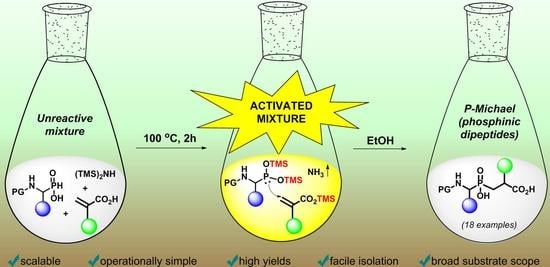
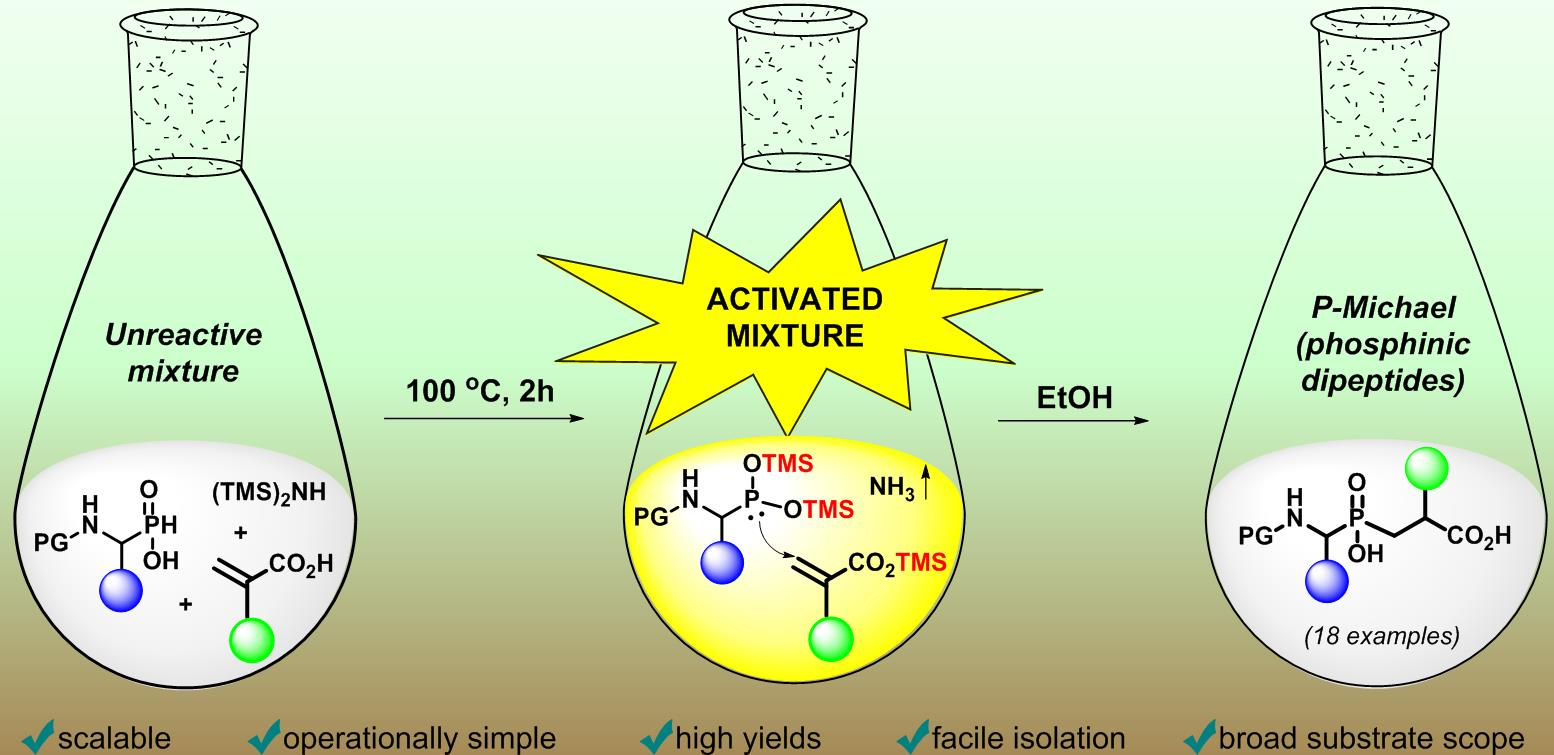
:
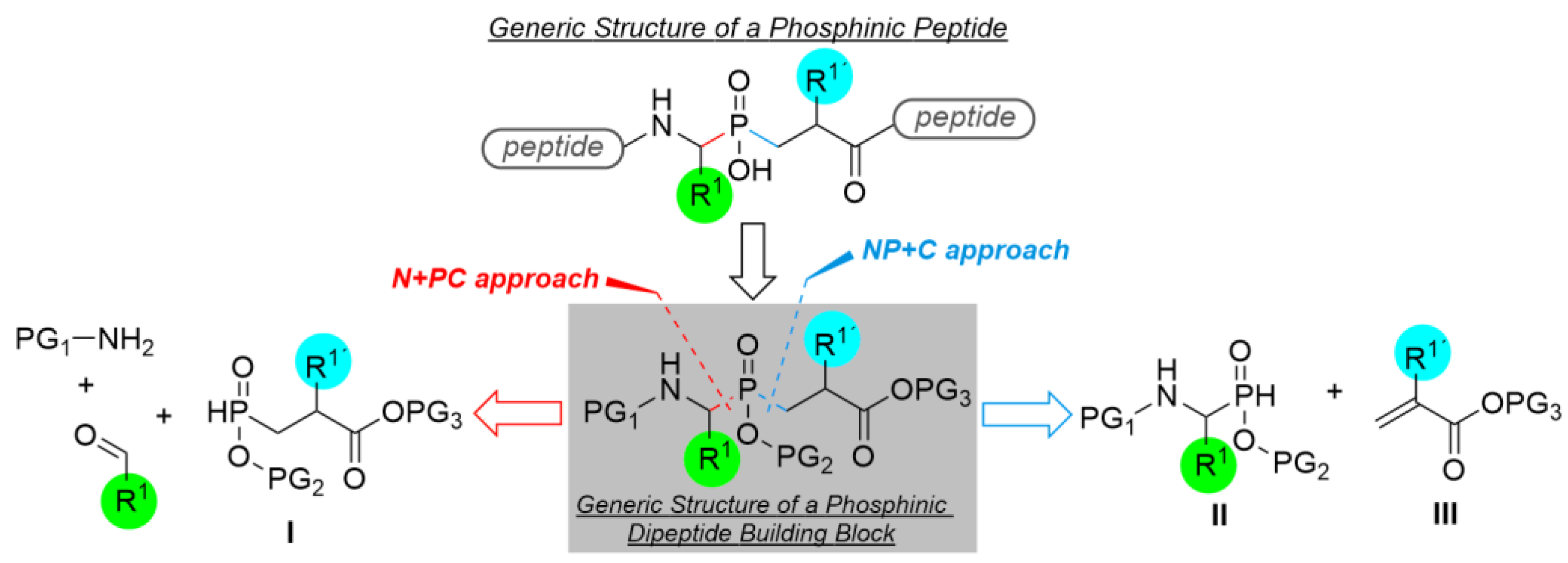
1. Introduction
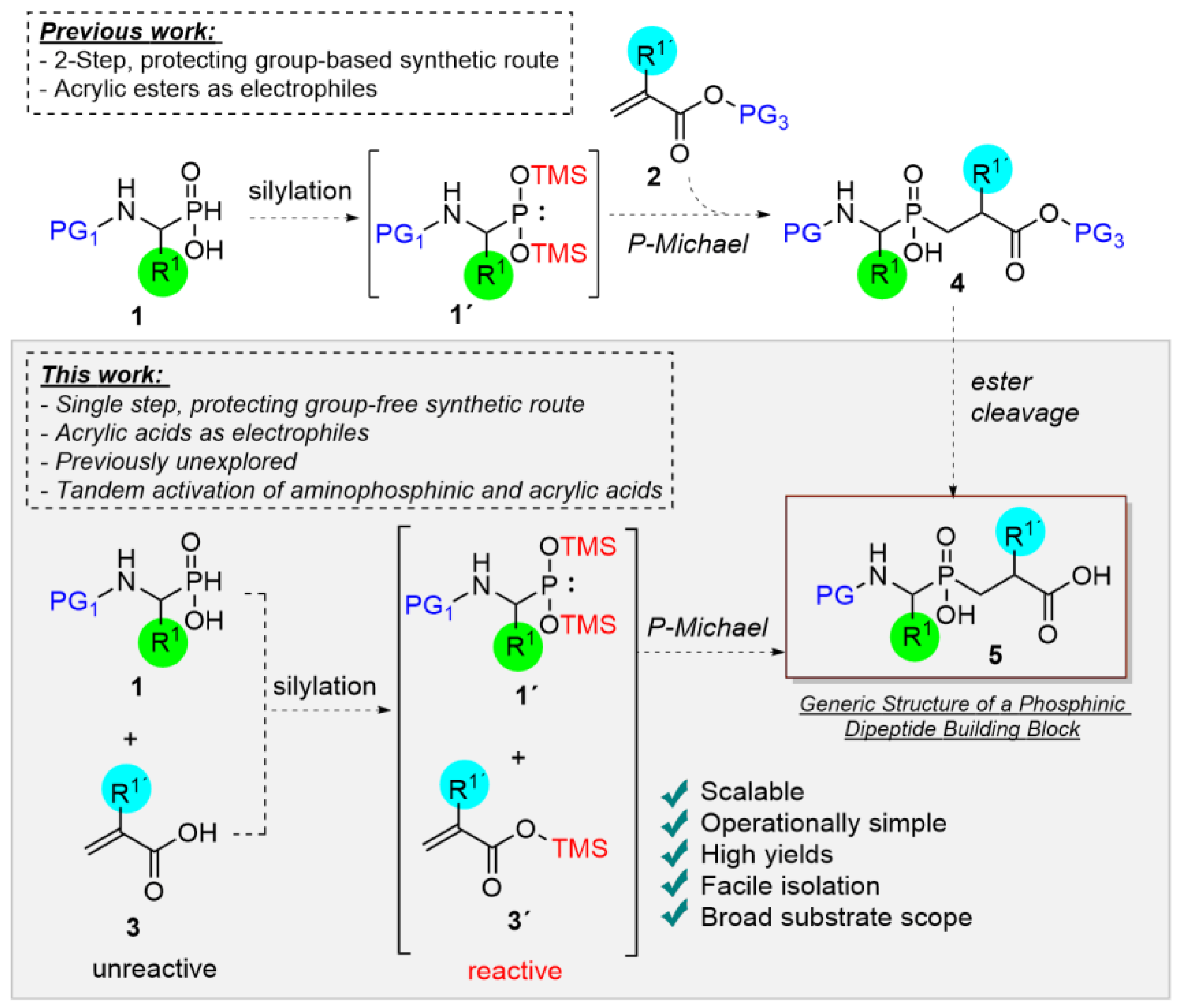
2. Results and Discussion
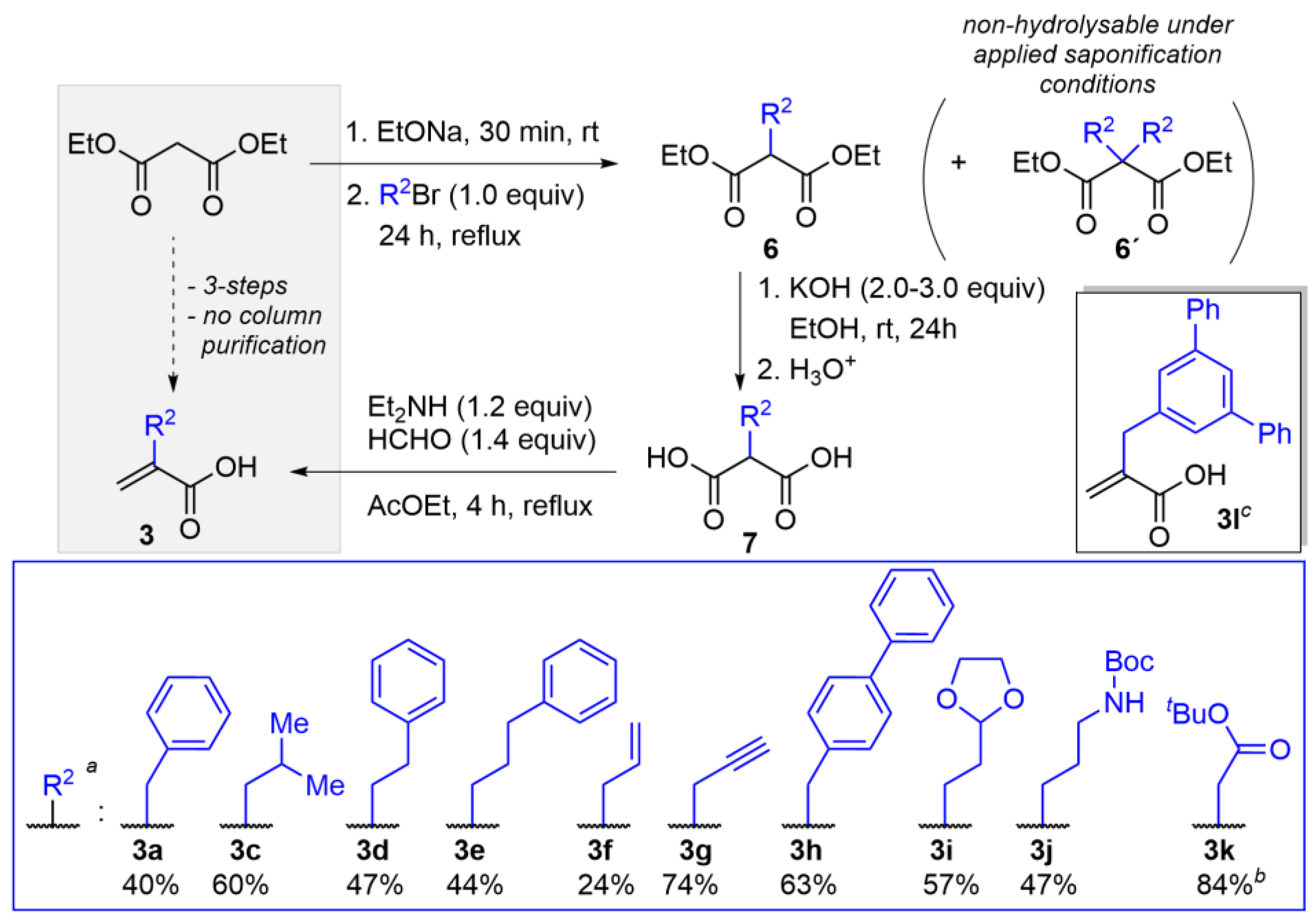
2.1. Preparation of Acrylic Acids of Type 3
2.2. Optimization
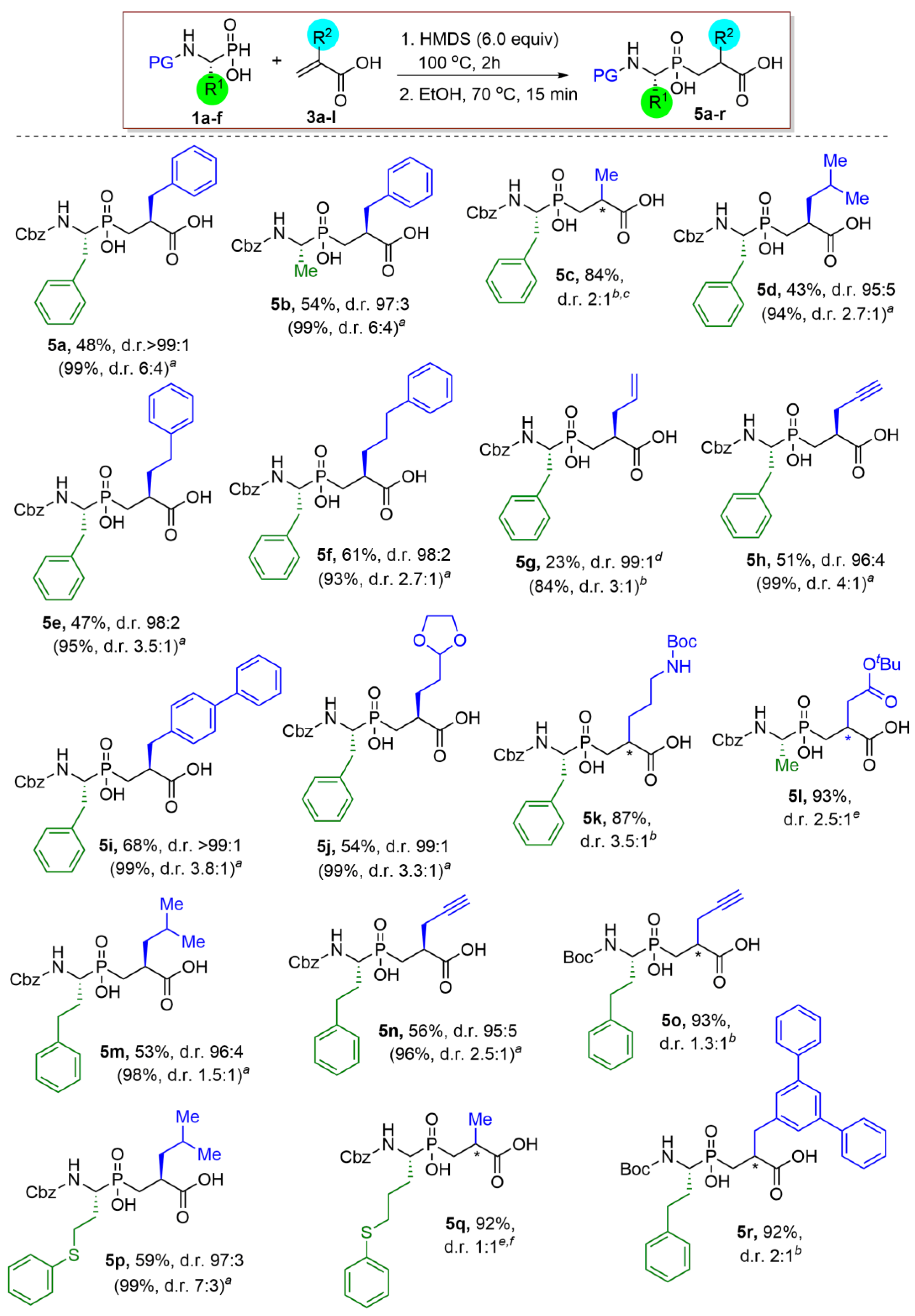
2.3. Substrate Scope
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the HMDS-Mediated P-C Bond-Forming Reaction between Phosphinic and Acrylic Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| HMDS | 1,1,1,3,3,3-hexamethyldisilazane |
| BSA | N,O-bis(trimethylsilyl)acetamide |
| TMSCl | chlorotrimethylsilane |
References
- Talma, M.; Maślanka, M.; Mucha, A. Recent developments in the synthesis and applications of phosphinic peptide analogs. Bioorg. Med. Chem. Lett. 2019, 29, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Abdou, M.M.; O’Neill, P.M.; Amigues, E.; Matziari, M. Phosphinic acids: Current status and potential for drug discovery. Drug Discov. Today 2018, 24, 916–929. [Google Scholar] [CrossRef]
- Georgiadis, D.; Dive, V. Phosphinic Peptides as Potent Inhibitors of Zinc-Metalloproteases. Top. Curr. Chem. 2015, 360, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A. Synthesis and modifications of phosphinic dipeptide analogues. Molecules 2012, 17, 13530–13568. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the alpha-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Dive, V.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Beau, F.; Cuniasse, P.; Yiotakis, A. Phosphinic peptides as zinc metalloproteinase inhibitors. Cell. Mol. Life Sci. 2004, 61, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Synthetic Methods of Phosphonopeptides. Molecules 2020, 25, 5894. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Synthesis of Phosphinopeptides and Phosphinodepsipeptides. Asian J. Org. Chem. 2021, 10, 287–295. [Google Scholar] [CrossRef]
- Xu, J. Synthetic strategies of phosphonodepsipeptides. Beilstein J. Org. Chem. 2021, 17, 461–484. [Google Scholar] [CrossRef]
- Nury, C.; Czarny, B.; Cassar-Lajeunesse, E.; Georgiadis, D.; Bregant, S.; Dive, V. A Pan Photoaffinity Probe for Detecting Active Forms of Matrix Metalloproteinases. ChemBioChem 2013, 14, 107–114. [Google Scholar] [CrossRef]
- Bordenave, T.; Helle, M.; Beau, F.; Georgiadis, D.; Tepshi, L.; Bernes, M.; Ye, Y.; Levenez, L.; Poquet, E.; Nozach, H.; et al. Synthesis and In Vitro and In Vivo Evaluation of MMP-12 Selective Optical Probes. Bioconjug. Chem. 2016, 27, 2407–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavian, M.; Bordenave, T.; Georgiadis, D.; Beau, F.; Zhang, J.; Golestani, R.; Toczek, J.; Jung, J.-J.; Ye, Y.; Kim, H.-Y.; et al. Optical imaging of MMP-12 active form in inflammation and aneurysm. Sci. Rep. 2016, 6, 38345. [Google Scholar] [CrossRef] [Green Version]
- Covaleda, G.; Gallego, P.; Vendrell, J.; Georgiadis, D.; Lorenzo, J.; Dive, V.; Aviles, F.X.; Reverter, D.; Devel, L. Synthesis and Structural/Functional Characterization of Selective M14 Metallocarboxypeptidase Inhibitors Based on Phosphinic Pseudopeptide Scaffold: Implications on the Design of Specific Optical Probes. J. Med. Chem. 2019, 62, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, M.; Bruyat, P.; Malgorn, C.; Doladilhe, M.; Cassar-Lajeunesse, E.; Fruchart Gaillard, C.; De Souza, M.; Beau, F.; Thai, R.; Correia, I.; et al. Ligand-Directed Modification of Active Matrix Metalloproteases: Activity-based Probes with no Photolabile Group. Angew. Chem. Int. Ed. 2021, 60, 18272–18279. [Google Scholar] [CrossRef]
- Yiotakis, A.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Dive, V. Phosphinic peptides: Synthetic approaches and biochemical evaluation as Zn-metalloprotease inhibitors. Curr. Org. Chem. 2004, 8, 1135–1158. [Google Scholar] [CrossRef]
- Kokkala, P.; Rajeshkumar, T.; Mpakali, A.; Stratikos, E.; Vogiatzis, K.D.; Georgiadis, D. A Carbodiimide-Mediated P–C Bond-Forming Reaction: Mild Amidoalkylation of P-Nucleophiles by Boc-Aminals. Org. Lett. 2021, 23, 1726–1730. [Google Scholar] [CrossRef] [PubMed]
- Viveros-Ceballos, J.L.; Ordóñez, M.; Sayago, F.J.; Cativiela, C. Stereoselective Synthesis of α-Amino-C-phosphinic Acids and Derivatives. Molecules 2016, 21, 1141. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Yuan, C. Enantioselective synthesis of H-phosphinic acids bearing natural amino acid residues. J. Org. Chem. 2013, 78, 6962–6974. [Google Scholar] [CrossRef]
- Baylis, E.K.; Campbell, C.D.; Dingwall, J.G. 1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J. Chem. Soc. Perkin Trans. 1 1984, 2845–2853. [Google Scholar] [CrossRef]
- Kokkala, P.; Mpakali, A.; Mauvais, F.X.; Papakyriakou, A.; Daskalaki, I.; Petropoulou, I.; Kavvalou, S.; Papathanasopoulou, M.; Agrotis, S.; Fonsou, T.M.; et al. Optimization and Structure-Activity Relationships of Phosphinic Pseudotripeptide Inhibitors of Aminopeptidases That Generate Antigenic Peptides. J. Med. Chem. 2016, 59, 9107–9123. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Weglarz-Tomczak, E.; Berlicki, L.; Pawelczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Makaritis, A.; Georgiadis, D.; Dive, V.; Yiotakis, A. Diastereoselective solution and multipin-based combinatorial array synthesis of a novel class of potent phosphinic metalloprotease inhibitors. Chem. Eur. J. 2003, 9, 2079–2094. [Google Scholar] [CrossRef] [PubMed]
- Jullien, N.; Makritis, A.; Georgiadis, D.; Beau, F.; Yiotakis, A.; Dive, V. Phosphinic tripeptides as dual angiotensin-converting enzyme C-domain and endothelin-converting enzyme-1 inhibitors. J. Med. Chem. 2010, 53, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Mores, A.; Matziari, M.; Beau, F.; Cuniasse, P.; Yiotakis, A.; Dive, V. Development of potent and selective phosphinic peptide inhibitors of angiotensin-converting enzyme 2. J. Med. Chem. 2008, 51, 2216–2226. [Google Scholar] [CrossRef]
- Matziari, M.; Bauer, K.; Dive, V.; Yiotakis, A. Synthesis of the phosphinic analogue of thyrotropin releasing hormone. J. Org. Chem. 2008, 73, 8591–8593. [Google Scholar] [CrossRef]
- Matralis, A.N.; Xanthopoulos, D.; Huot, G.; Lopes-Paciencia, S.; Cole, C.; de Vries, H.; Ferbeyre, G.; Tsantrizos, Y.S. Molecular tools that block maturation of the nuclear lamin A and decelerate cancer cell migration. Bioorg. Med. Chem. 2018, 26, 5547–5554. [Google Scholar] [CrossRef]
- Skinner-Adams, T.S.; Lowther, J.; Teuscher, F.; Stack, C.M.; Grembecka, J.; Mucha, A.; Kafarski, P.; Trenholme, K.R.; Dalton, J.P.; Gardiner, D.L. Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds. J. Med. Chem. 2007, 50, 6024–6031. [Google Scholar] [CrossRef]
- Yiotakis, A.; Vassiliou, S.; Jiráček, J.; Dive, V. Protection of the hydroxyphosphinyl function of phosphinic dipeptides by adamantyl. Application to the solid-phase synthesis of phosphinic peptides. J. Org. Chem. 1996, 61, 6601–6605. [Google Scholar] [CrossRef]
- Thottathil, J.K.; Ryono, D.E.; Przybyla, C.A.; Moniot, J.L.; Neubeck, R. Preparation of phosphinic acids: Michael additions of phosphonous acids/esters to conjugated systems. Tetrahedron Lett. 1984, 25, 4741–4744. [Google Scholar] [CrossRef]
- Chen, H.; Noble, F.; Roques, B.P.; Fournié-Zaluski, M.C. Long lasting antinociceptive properties of enkephalin degrading enzyme (NEP and APN) inhibitor prodrugs. J. Med. Chem. 2001, 44, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Raguin, O.; Fournié-Zaluski, M.C.; Romieu, A.; Pèlegrin, A.; Chatelet, F.; Pélaprat, D.; Barbet, J.; Roques, B.P.; Gruaz-Guyon, A. A labeled neutral endopeptidase inhibitor as a potential tool for tumor diagnosis and prognosis. Angew. Chem. Int. Ed. 2005, 44, 4058–4061. [Google Scholar] [CrossRef]
- Selvam, C.; Oueslati, N.; Lemasson, I.A.; Brabet, I.; Rigault, D.; Courtiol, T.; Cesarini, S.; Triballeau, N.; Bertrand, H.O.; Goudet, C.; et al. A virtual screening hit reveals new possibilities for developing group III metabotropic glutamate receptor agonists. J. Med. Chem. 2010, 53, 2797–2813. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Noble, F.; Mothé, A.; Meudal, H.; Coric, P.; Danascimento, S.; Roques, B.P.; George, P.; Fournié-Zaluski, M.C. Phosphinic derivatives as new dual enkephalin-degrading enzyme inhibitors: Synthesis, biological properties, and antinociceptive activities. J. Med. Chem. 2000, 43, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.K.K.; Kim, S.; Zard, S.Z. Free-Radical Variant for the Synthesis of Functionalized 1,5-Diketones. Org. Lett. 2013, 15, 4818–4821. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Smith, A.B.; Ducry, L.; Corbett, R.M.; Hirschmann, R. Intramolecular Hydrogen-Bond Participation in Phosphonylammonium Salt Formation. Org. Lett. 2000, 2, 3887–3890. [Google Scholar] [CrossRef] [PubMed]
- Maben, Z.; Arya, R.; Georgiadis, D.; Stratikos, E.; Stern, L.J. Conformational dynamics linked to domain closure and substrate binding explain the ERAP1 allosteric regulation mechanism. Nat. Commun. 2021, 12, 5302. [Google Scholar] [CrossRef]
- Zervoudi, E.; Saridakis, E.; Birtley, J.R.; Seregin, S.S.; Reeves, E.; Kokkala, P.; Aldhamen, Y.A.; Amalfitano, A.; Mavridis, I.M.; James, E.; et al. Rationally designed inhibitor targeting antigentrimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. USA 2013, 110, 19890–19895. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Acrylic Acid (Equiv) | Reaction Conditions 1 | Conversion of 1a, % (yield to 5a, %) 2 |
|---|---|---|---|
| 1 | 3a (1.3) | BSA (4 equiv), CH2Cl2, rt, 48 h | 53 (41) |
| 2 | 3a′ (1.3) | BSA (4 equiv), CH2Cl2, rt, 48 h | 90 (89) 3 |
| 3 | 3a (1.3) | TMSCl (7 equiv), i Pr2EtN (7 equiv), CH2Cl2, rt, 48 h | 86 (81) |
| 4 | 3a (1.2) | HMDS (6 equiv), 100 °C, 2 h | 100 (99) |
| 5 | 3a (1.2) | HMDS (6 equiv), 70 °C, 2 h | 72 (70) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokkala, P.; Voreakos, K.; Lelis, A.; Patiniotis, K.; Skoulikas, N.; Devel, L.; Ziotopoulou, A.; Kaloumenou, E.; Georgiadis, D. Practical Synthesis of Phosphinic Dipeptides by Tandem Esterification of Aminophosphinic and Acrylic Acids under Silylating Conditions. Molecules 2022, 27, 1242. https://doi.org/10.3390/molecules27041242
Kokkala P, Voreakos K, Lelis A, Patiniotis K, Skoulikas N, Devel L, Ziotopoulou A, Kaloumenou E, Georgiadis D. Practical Synthesis of Phosphinic Dipeptides by Tandem Esterification of Aminophosphinic and Acrylic Acids under Silylating Conditions. Molecules. 2022; 27(4):1242. https://doi.org/10.3390/molecules27041242
Chicago/Turabian StyleKokkala, Paraskevi, Kostas Voreakos, Angelos Lelis, Konstantinos Patiniotis, Nikolaos Skoulikas, Laurent Devel, Angeliki Ziotopoulou, Eleni Kaloumenou, and Dimitris Georgiadis. 2022. "Practical Synthesis of Phosphinic Dipeptides by Tandem Esterification of Aminophosphinic and Acrylic Acids under Silylating Conditions" Molecules 27, no. 4: 1242. https://doi.org/10.3390/molecules27041242