
Cyclodextrins Initiated Ring-Opening Polymerization of Lactide Using 4-Dimethylaminopyridine (DMAP) as Catalyst: Study of DMAP/β-CD Inclusion Complex and Access to New Structures

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
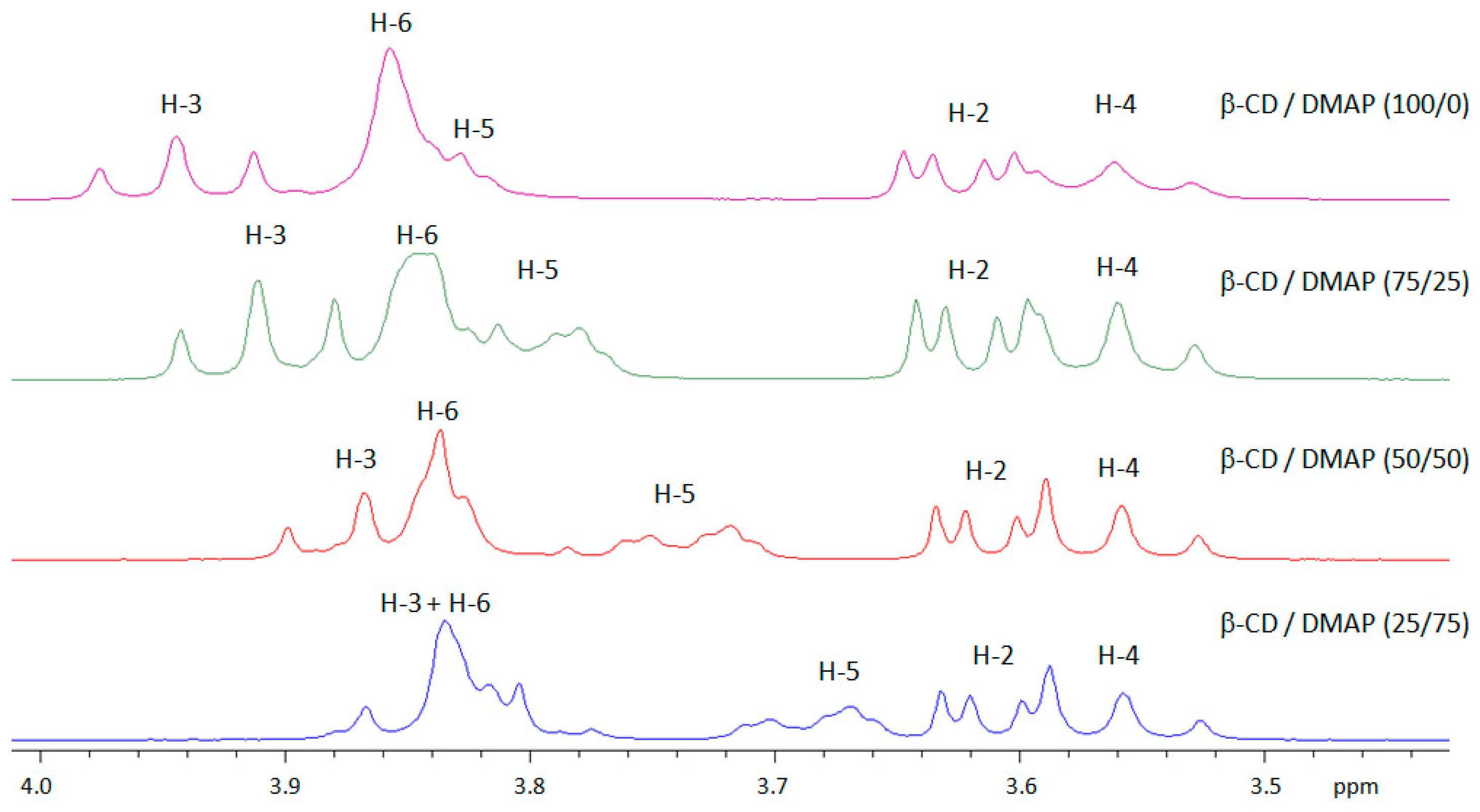
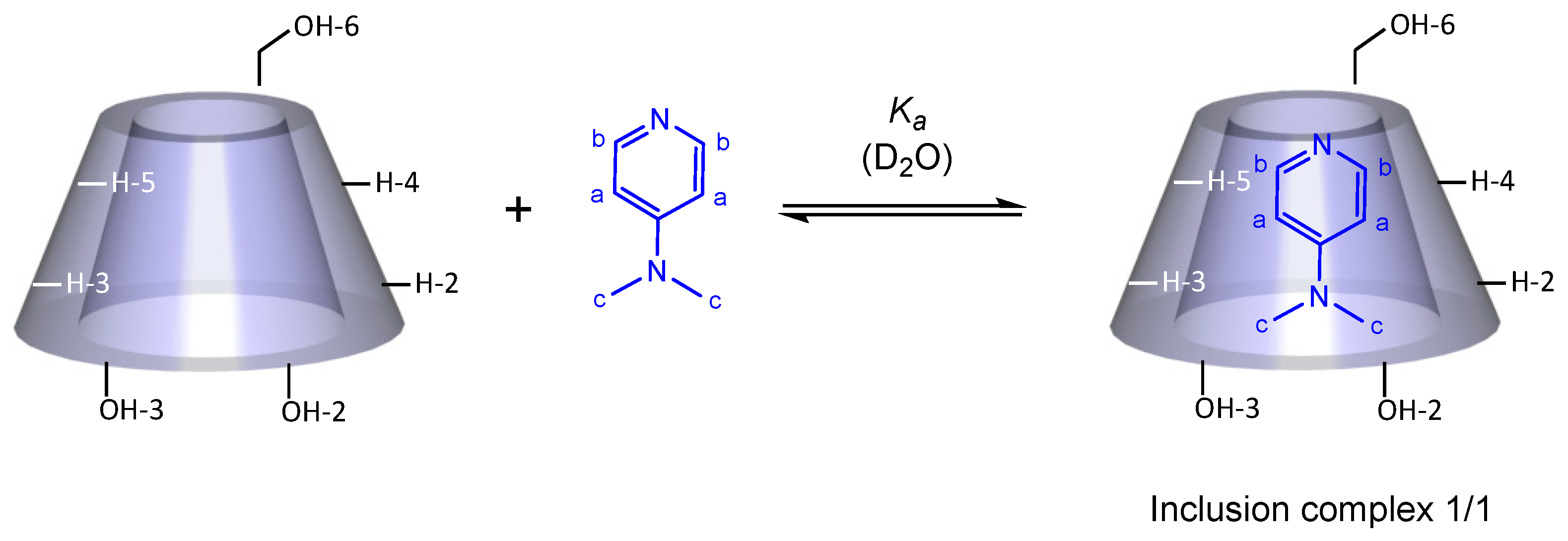
2.1. Ability of DMAP and Rac-Lactide to Form an Inclusion Complex with β-CD
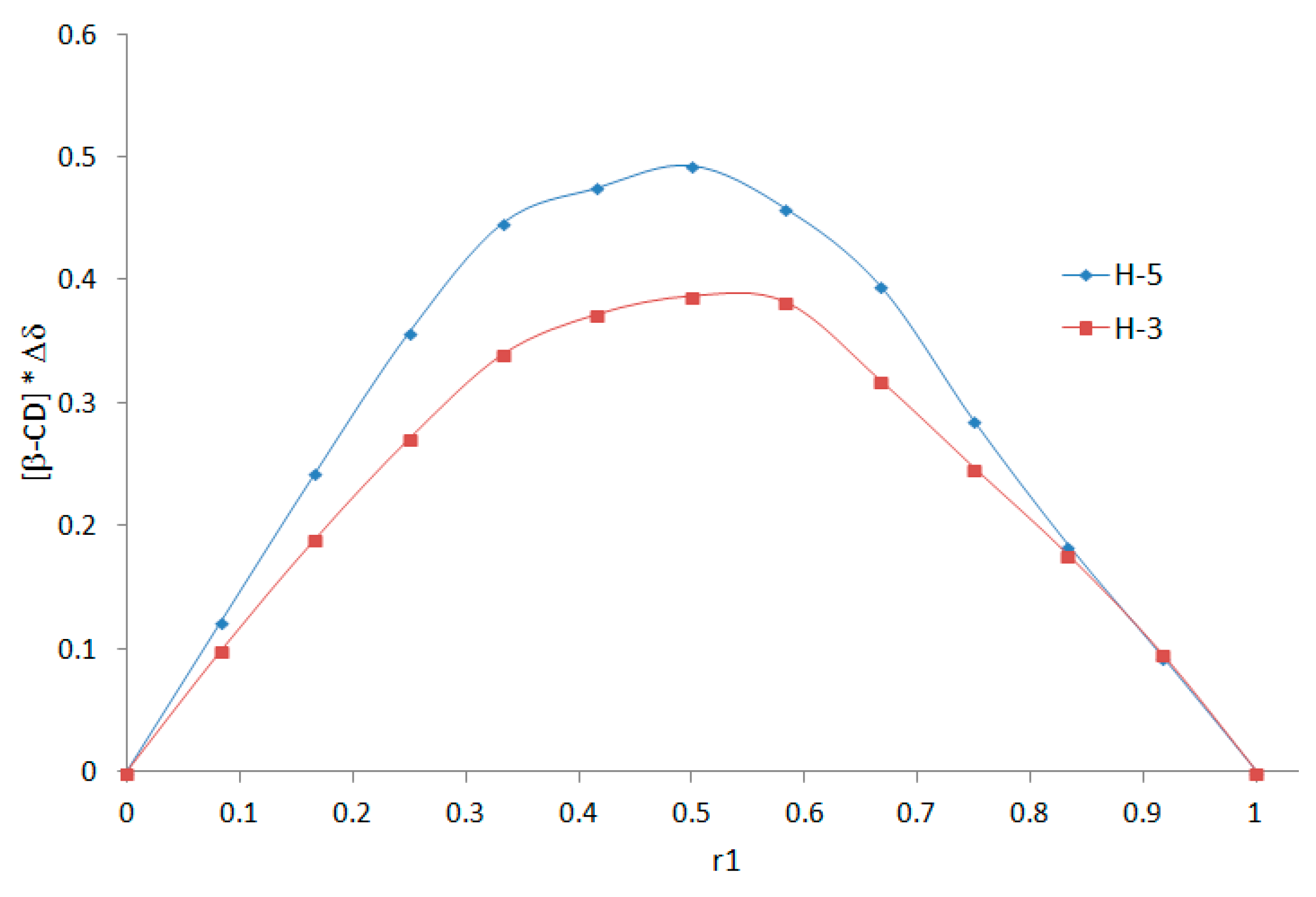
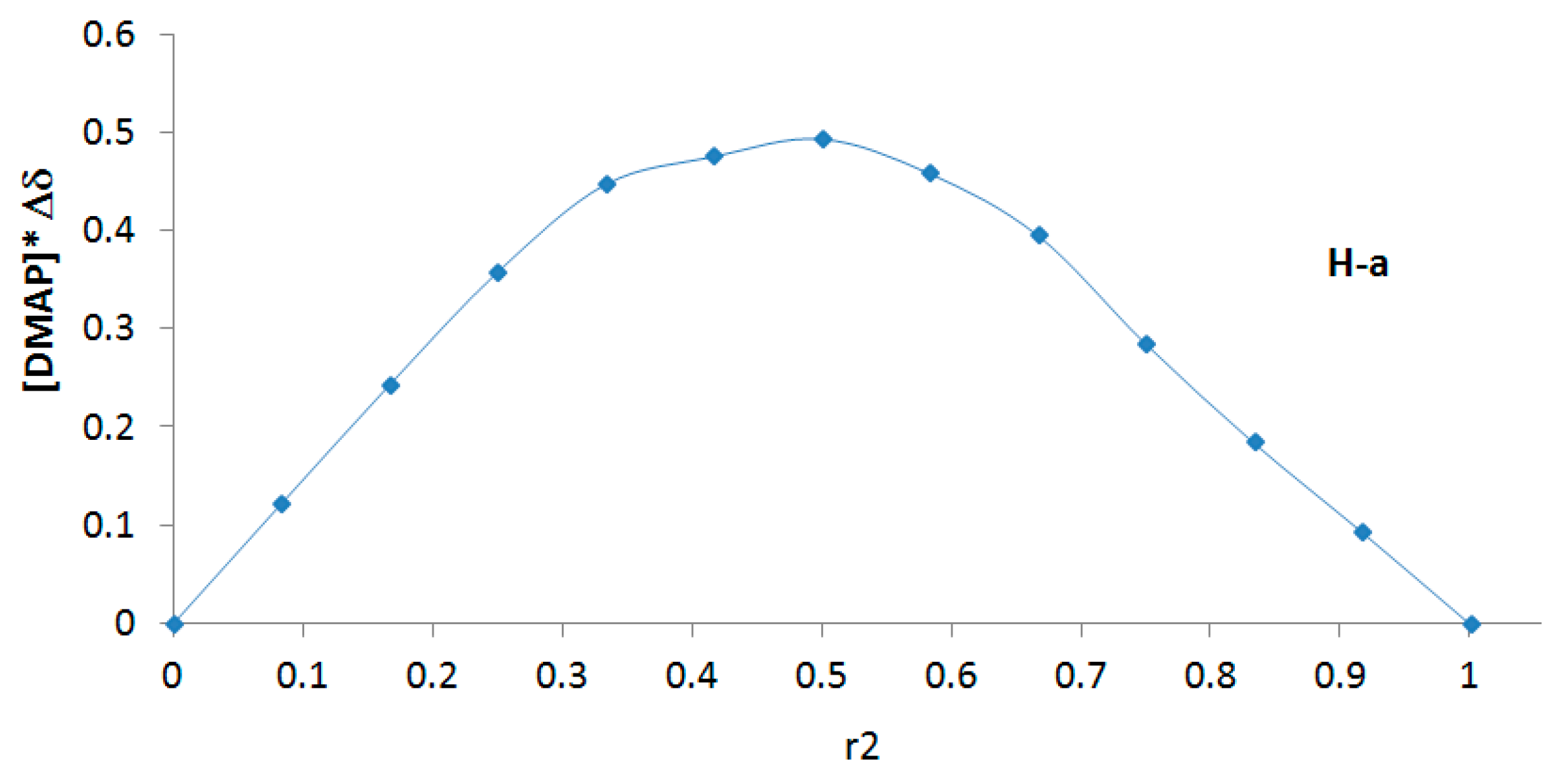
2.2. Determination of the DMAP/β-CD Inclusion Complex Stoichiometry by the Job Plots Method
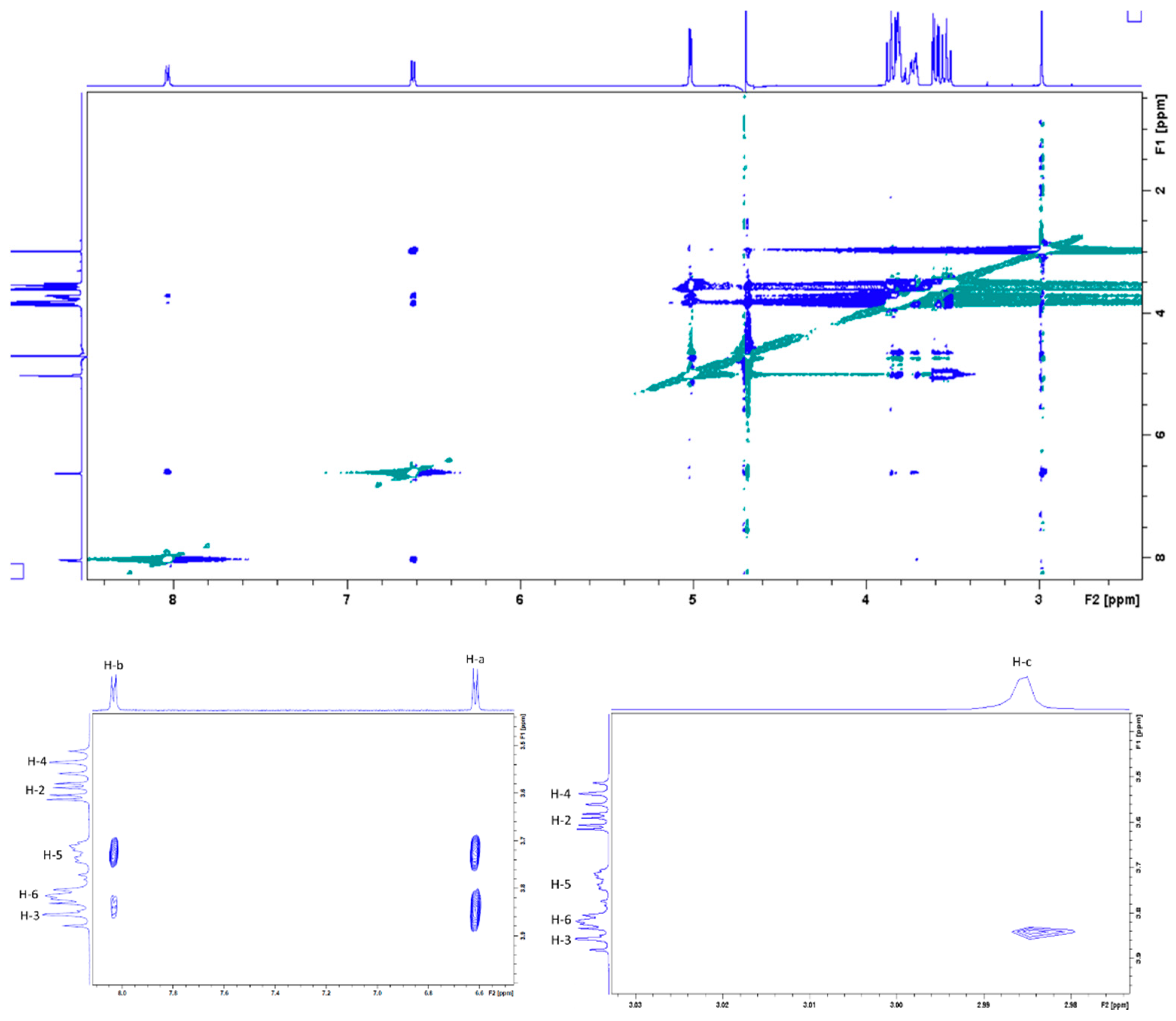
2.3. Characterization of the DMAP/β-CD Inclusion Complex Geometry
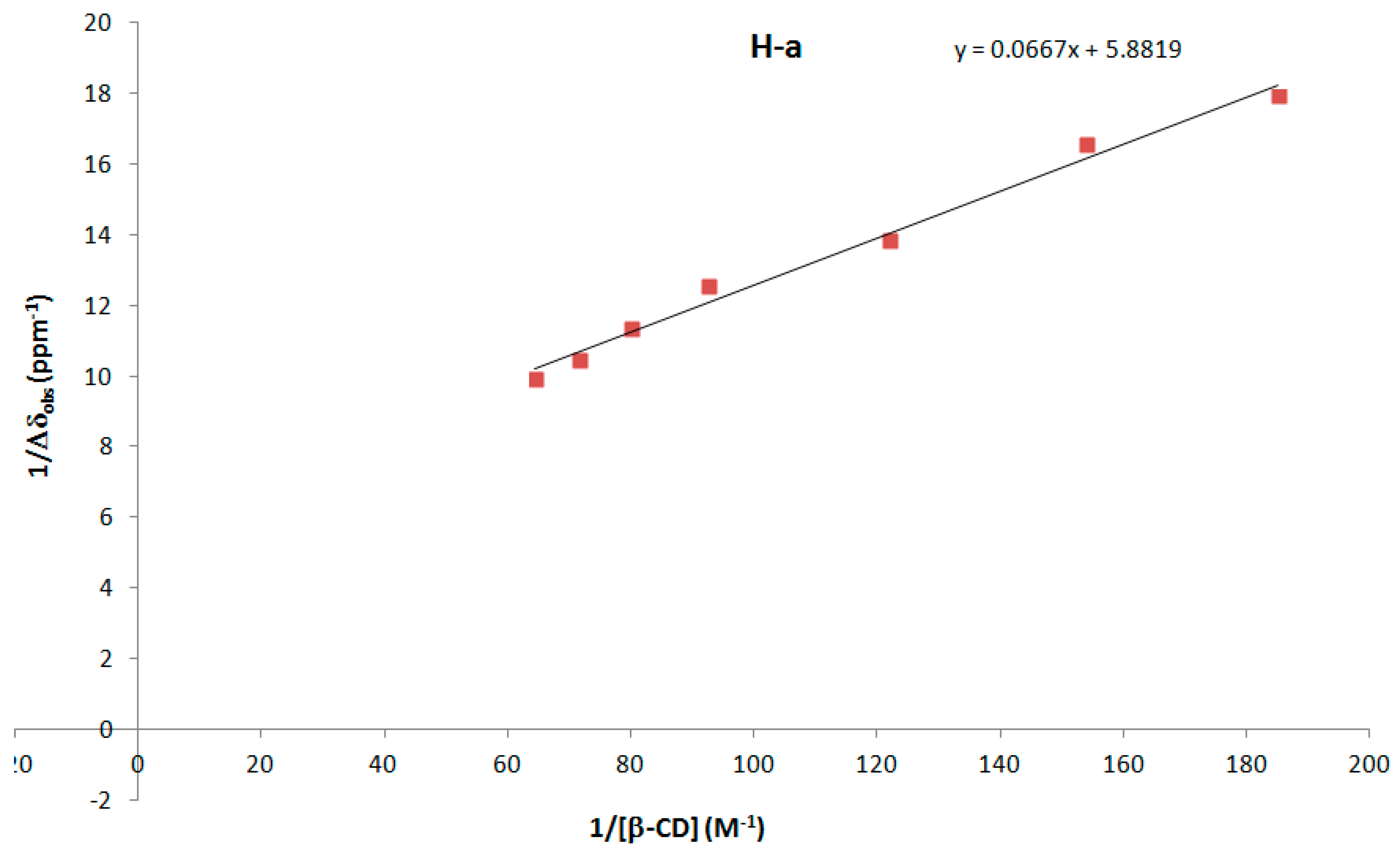
2.4. Estimation of the Association Constant (Ka) of DMAP/β-CD Inclusion Complex
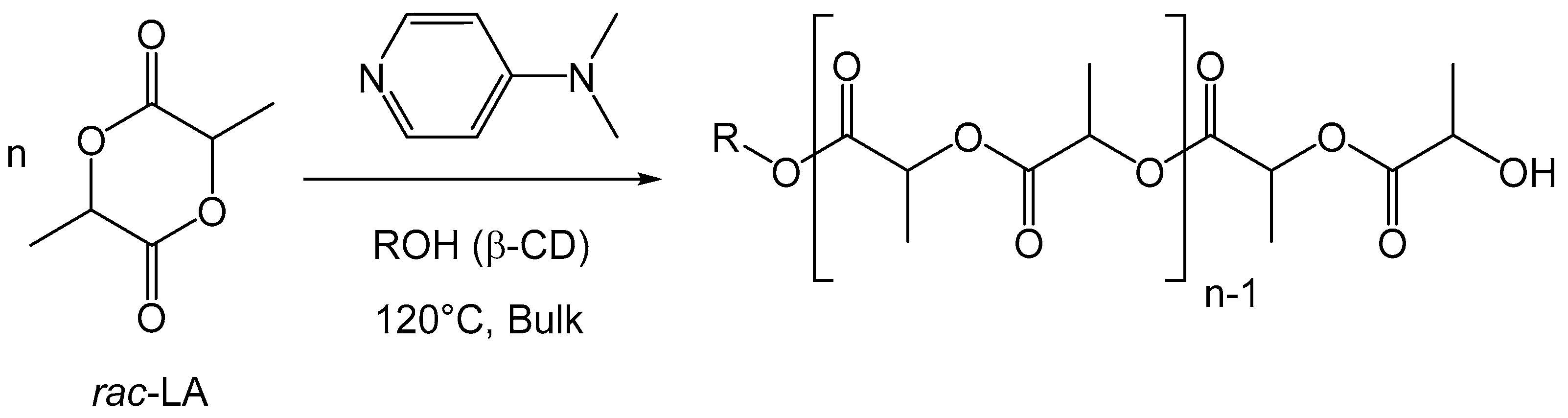
2.5. Polymerization of Rac-Lactide Using the DMAP/β-CD Inclusion Complex as the Initiator
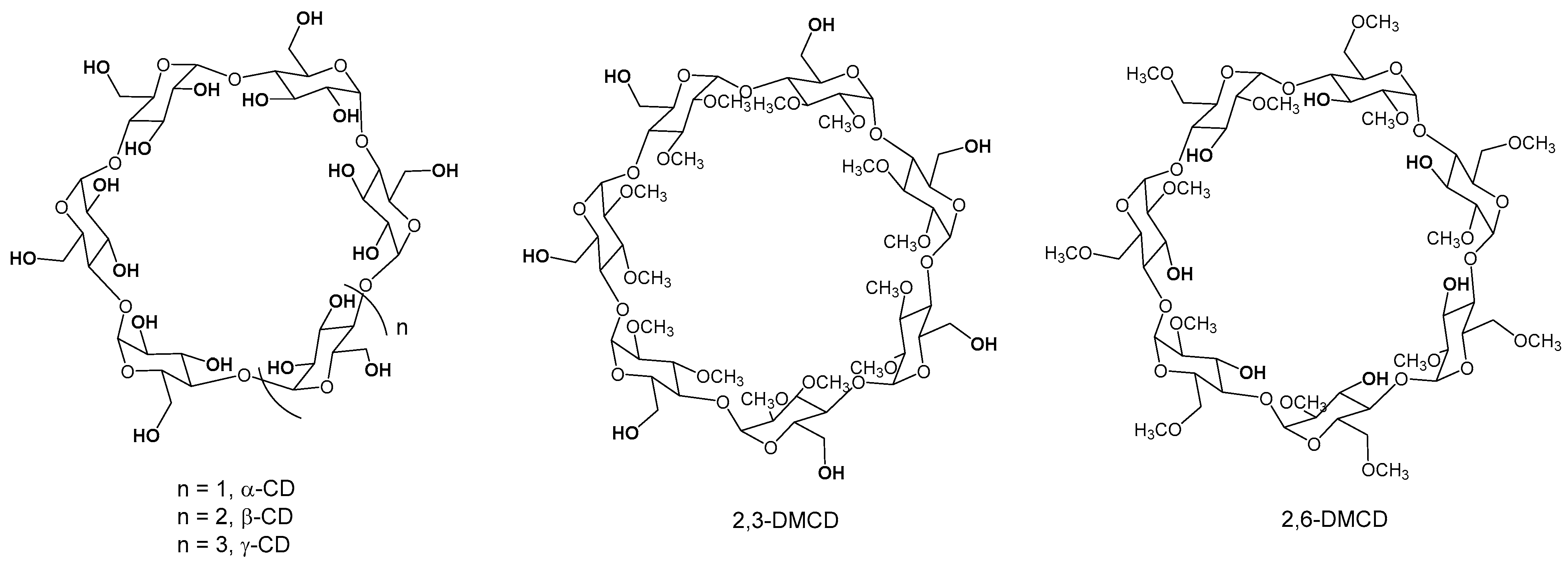
2.6. Polymerization of Rac-Lactide Using Other Native and O-Methylated Cyclodextrins as Initiators
3. Materials and Methods
3.1. Materials
3.2. Synthesis
3.2.1. Preparation of Heptakis-(2I–VII,3I–VII-di-O-methyl)-β-Cyclodextrin (2,3-DMCD)
3.2.2. General Procedure for Rac-Lactide (LA) Polymerization Initiated by CD
3.3. Characterization of the PLA Functionalized CD
3.3.1. Nuclear Magnetic Resonance (NMR) Analysis
3.3.2. Size Exclusion Chromatography (SEC) Analysis
3.4. Synthesis and Characterization of Inclusion Complexes
3.4.1. General Procedure for Formation of Inclusion Complexes (DMAP/β-CD or Adamantane/β-CD)
- DMAP/β-CD Inclusion Complex (see Figure S2 in SI)
- Adamantane/β-CD Inclusion Complex (see Figure S3 in SI)
3.4.2. Determination of the DMAP/β-CD Inclusion Complex Stoichiometry by Job Plot Method
3.4.3. Characterization of the DMAP/β-CD Inclusion Complex Geometry
3.4.4. Estimation of the Association Constant (Ka) of DMAP/β-CD Inclusion Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dodziuk, H. (Ed.) Front matter. In Cyclodextrins and Their Complexes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. I–XVII. ISBN 978-3-527-60898-0. [Google Scholar]
- Defaye, J.; García Fernández, J.-M.; Ortiz Mellet, C. Les cyclodextrines en pharmacie: Perspectives pour le ciblage d’actifs thérapeutiques et le contrôle d’interactions membranaires. Ann. Pharm. Françaises 2007, 65, 33–49. [Google Scholar] [CrossRef]
- Loftsson, T.; Duchene, D. Cyclodextrins and Their Pharmaceutical Applications. Int. J. Pharm. 2007, 329, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schwinté, P.; Ramphul, M.; Darcy, R.; O’Sullivan, J.F. Amphiphilic Cyclodextrin Complexation of Clofazimine. J. Incl. Phenom. 2003, 47, 109–112. [Google Scholar] [CrossRef]
- Wang, Z.; Ouyang, J.; Baeyens, W.R.G. Recent Developments of Enantioseparation Techniques for Adrenergic Drugs Using Liquid Chromatography and Capillary Electrophoresis: A Review. J. Chromatogr. B 2008, 862, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.-T.; Wang, R.-Q.; Muderawan, I.W.; Ng, S.-C. Synthesis and Application of Mono-6-(3-Methylimidazolium)-6-Deoxyperphenylcarbamoyl-β-Cyclodextrin Chloride as Chiral Stationary Phases for High-Performance Liquid Chromatography and Supercritical Fluid Chromatography. J. Chromatogr. A 2008, 1182, 136–140. [Google Scholar] [CrossRef]
- León, A.G.; Olives, A.I.; del Castillo, B.; Martín, M.A. Influence of the Presence of Methyl Cyclodextrins in High-Performance Liquid Chromatography Mobile Phases on the Separation of β-Carboline Alkaloids. J. Chromatogr. A 2008, 1192, 254–258. [Google Scholar] [CrossRef]
- Khan, A.R.; Forgo, P.; Stine, K.J.; D’Souza, V.T. Methods for Selective Modifications of Cyclodextrins. Chem. Rev. 1998, 98, 1977–1996. [Google Scholar] [CrossRef]
- Sallas, F.; Darcy, R. Amphiphilic Cyclodextrins—Advances in Synthesis and Supramolecular Chemistry. Eur. J. Org. Chem. 2008, 2008, 957–969. [Google Scholar] [CrossRef]
- Pedersen, N.R.; Kristensen, J.B.; Bauw, G.; Ravoo, B.J.; Darcy, R.; Larsen, K.L.; Pedersen, L.H. Thermolysin Catalyses the Synthesis of Cyclodextrin Esters in DMSO. Tetrahedron Asymmetry 2005, 16, 615–622. [Google Scholar] [CrossRef]
- Wang, N.; Wu, Q.; Xiao, Y.M.; Chen, C.X.; Lin, X.F. Regioselective Synthesis of Cyclodextrin Mono-Substituted Conjugates of Non-Steroidal Anti-Inflammatory Drugs at C-2 Secondary Hydroxyl by Protease in Non-Aqueous Media. Bioorg. Med. Chem. 2005, 13, 3667–3671. [Google Scholar] [CrossRef]
- Choisnard, L.; Gèze, A.; Putaux, J.-L.; Wong, Y.-S.; Wouessidjewe, D. Nanoparticles of β-Cyclodextrin Esters Obtained by Self-Assembling of Biotransesterified β-Cyclodextrins. Biomacromolecules 2006, 7, 515–520. [Google Scholar] [CrossRef] [PubMed]
- van de Manakker, F.; Vermonden, T.; van Nostrum, C.F.; Hennink, W.E. Cyclodextrin-Based Polymeric Materials: Synthesis, Properties, and Pharmaceutical/Biomedical Applications. Biomacromolecules 2009, 10, 3157–3175. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.E. Nanostructuring and Functionalizing Polymers with Cyclodextrins. Polymer 2008, 49, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Phuphuak, Y.; Miao, Y.; Zinck, P.; Chirachanchai, S. Balancing Crystalline and Amorphous Domains in PLA through Star-Structured Polylactides with Dual Plasticizer/Nucleating Agent Functionality. Polymer 2013, 54, 7058–7070. [Google Scholar] [CrossRef]
- Miao, Y.; Zinck, P. Ring-Opening Polymerization of Cyclic Esters Initiated by Cyclodextrins. Polym. Chem. 2012, 3, 1119. [Google Scholar] [CrossRef]
- Normand, M.; Kirillov, E.; Carpentier, J.-F.; Guillaume, S.M. Cyclodextrin-Centered Polyesters: Controlled Ring-Opening Polymerization of Cyclic Esters from β-Cyclodextrin-Diol. Macromolecules 2012, 45, 1122–1130. [Google Scholar] [CrossRef]
- Takashima, Y.; Osaki, M.; Harada, A. Cyclodextrin-Initiated Polymerization of Cyclic Esters in Bulk: Formation of Polyester-Tethered Cyclodextrins. J. Am. Chem. Soc. 2004, 126, 13588–13589. [Google Scholar] [CrossRef]
- Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Polymerization of Lactones Initiated by Cyclodextrins: Effects of Cyclodextrins on the Initiation and Propagation Reactions. Macromolecules 2007, 40, 3154–3158. [Google Scholar] [CrossRef]
- Galia, A.; Scialdone, O.; Spanò, T.; Valenti, M.G.; Grignard, B.; Lecomte, P.; Monflier, E.; Tilloy, S.; Rousseau, C. Ring Opening Polymerization of ε-Caprolactone in the Presence of Wet β-Cyclodextrin: Effect of the Operative Pressure and of Water Molecules in the β-Cyclodextrin Cavity. RSC Adv. 2016, 6, 90290–90299. [Google Scholar] [CrossRef]
- Shen, J.; Hao, A.; Du, G.; Zhang, H.; Sun, H. A Convenient Preparation of 6-Oligo(Lactic Acid)Cyclomaltoheptaose as Kinetically Degradable Derivative for Controlled Release of Amoxicillin. Carbohydr. Res. 2008, 343, 2517–2522. [Google Scholar] [CrossRef] [PubMed]
- Peptu, C.; Balan-Porcarasu, M.; Šišková, A.; Škultéty, Ľ.; Mosnáček, J. Cyclodextrins Tethered with Oligolactides—Green Synthesis and Structural Assessment. Beilstein J. Org. Chem. 2017, 13, 779–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Rousseau, C.; Mortreux, A.; Martin, P.; Zinck, P. Access to New Carbohydrate-Functionalized Polylactides via Organocatalyzed Ring-Opening Polymerization. Polymer 2011, 52, 5018–5026. [Google Scholar] [CrossRef]
- Takashima, Y.; Kawaguchi, Y.; Nakagawa, S.; Harada, A. Inclusion Complex Formation and Hydrolysis of Lactones by Cyclodextrins. Chem. Lett. 2003, 32, 1122–1123. [Google Scholar] [CrossRef]
- Hao, X.; Liang, C.; Jian-Bin, C. Preparation and Spectroscopic Studies of an Inclusion Complex of Adenine with β-Cyclodextrin in Solution and in the Solid State. Analyst 2002, 127, 834–837. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Benítez, D.; Yoon, I.; Stoddart, J.F. Inclusion Behavior of β-Cyclodextrin with Bipyridine Molecules: Factors Governing Host-Guest Inclusion Geometries. Chem.-Asian J. 2009, 4, 446–456. [Google Scholar] [CrossRef]
- Zhao, Y. Self-Assembly Behavior of Inclusion Complex Formed by b-Cyclodextrin with a-Aminopyridine. Sci. China Ser. B 2004, 47, 200. [Google Scholar] [CrossRef]
- Acuña-Rougier, C.; Jullian, C.; Olea-Azar, C. NMR as a Tool for Simultaneous Study of Diasteroisomeric Inclusion Complexes, Part 2: Complexes Formed by Racemic Mixture of 4′-Hydroxyflavanone and Two Cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 157–166. [Google Scholar] [CrossRef]
- Pirnau, A.; Floare, C.G.; Bogdan, M. The Complexation of Flurbiprofen with β-Cyclodextrin: A NMR Study in Aqueous Solution. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 113–120. [Google Scholar] [CrossRef]
- Morin, N.; Chilouet, A.; Millet, J.; Rouland, J.-C. Bifonazole-β-Cyclodextrin Inclusion Complexes. Thermal Analysis and X-Ray Powder Diffraction Study. J. Therm. Anal. Calorim. 2000, 62, 187–201. [Google Scholar] [CrossRef]
- Shaver, M.P.; Wagner, B.D.; Tennekone, G.K. Cyclodextrin-Based Biodegradable Polymer Stars: Synthesis and Fluorescence Studies. Green Mater. 2014, 2, 31–42. [Google Scholar] [CrossRef]
- Thakur, K.A.; Kean, R.T.; Hall, E.S.; Kolstad, J.J.; Lindgren, T.A.; Doscotch, M.A.; Siepmann, J.I.; Munson, E.J. High-Resolution 13C and 1H Solution NMR Study of Poly (Lactide). Macromolecules 1997, 30, 2422–2428. [Google Scholar] [CrossRef]
- Boger, J.; Corcoran, R.J.; Lehn, J.-M. Cyclodextrin Chemistry. Selective Modification of All Primary Hydroxyl Groups of α- and β-Cyclodextrins. HCA 1978, 61, 2190–2218. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | δfree (ppm) | δc (ppm) | Δδc 1 (ppm) |
|---|---|---|---|
| H-1 | 5.0472 | 5.0356 | 0.012 |
| H-2 | 3.6237 | 3.611 | 0.013 |
| H-3 | 3.9446 | 3.868 | 0.077 |
| H-4 | 3.5611 | 3.5583 | 0.003 |
| H-5 | 3.8175 | 3.7086 | 0.109 |
| H-6 | 3.8572 | 3.8368 | 0.021 |
| H-a | 6.674 | 6.6371 | 0.037 |
| H-b | 8.0420 | 8.0478 | −0.006 |
| H-c | 2.9921 | 3.0078 | −0.016 |
| Entry | Init. 1 | M/ROH | Time (min) | Conv. 2 (%) | DP/OH 3 Calc. | DP/OH 4 Final | Init Eff. 5 % | DM 6 |
|---|---|---|---|---|---|---|---|---|
| 1 7 | DIC | 30 | 60 | 4 | - | - | - | |
| 2 | DIC | 10 | 30 | 94 | 9.4 | 14 | 67 | 1.10 |
| 3 8 | β-CD | 10 | 30 | 97 | 9.7 | 10 | 97 | 1.09 |
| 4 | DIC | 2 | 10 | 83 | 1.65 | 2.7 | 61 | 1.13 |
| 5 | DIC | 20 | 60 | 96 | 19.2 | 24.1 | 80 | 1.14 |
| 6 | AIC | 2 | 10 | 47 | 0.95 | 1.2 | 79 | 1.14 |
| 7 | α-CD | 10 | 10 | 97 | 9.6 | 11.2 | 86 | 1.07 |
| 8 | α-CD | 30 | 20 | 99 | 19.8 | 20.3 | 98 | 1.09 |
| 9 | γ-CD | 2 | 10 | 97 | 1.9 | 2.2 | 88 | 1.18 |
| 10 | γ-CD | 10 | 30 | 96 | 9.6 | 10.8 | 89 | 1.09 |
| 11 | 2,6-DM | 30 | 60 | 99 | 29.8 | ca. 80 | 37 | 1.49 |
| 12 | 2,3-DM | 10 | 30 | 96 | 9.6 | 9.6 | 100 | 1.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meimoun, J.; Phuphuak, Y.; Miyamachi, R.; Miao, Y.; Bria, M.; Rousseau, C.; Nogueira, G.; Valente, A.; Favrelle-Huret, A.; Zinck, P. Cyclodextrins Initiated Ring-Opening Polymerization of Lactide Using 4-Dimethylaminopyridine (DMAP) as Catalyst: Study of DMAP/β-CD Inclusion Complex and Access to New Structures. Molecules 2022, 27, 1083. https://doi.org/10.3390/molecules27031083
Meimoun J, Phuphuak Y, Miyamachi R, Miao Y, Bria M, Rousseau C, Nogueira G, Valente A, Favrelle-Huret A, Zinck P. Cyclodextrins Initiated Ring-Opening Polymerization of Lactide Using 4-Dimethylaminopyridine (DMAP) as Catalyst: Study of DMAP/β-CD Inclusion Complex and Access to New Structures. Molecules. 2022; 27(3):1083. https://doi.org/10.3390/molecules27031083
Chicago/Turabian StyleMeimoun, Julie, Yupin Phuphuak, Remi Miyamachi, Yong Miao, Marc Bria, Cyril Rousseau, Guilherme Nogueira, Andreia Valente, Audrey Favrelle-Huret, and Philippe Zinck. 2022. "Cyclodextrins Initiated Ring-Opening Polymerization of Lactide Using 4-Dimethylaminopyridine (DMAP) as Catalyst: Study of DMAP/β-CD Inclusion Complex and Access to New Structures" Molecules 27, no. 3: 1083. https://doi.org/10.3390/molecules27031083