
Bispidine Platform as a Tool for Studying Amide Configuration Stability

 , , , and
, , , and
Abstract
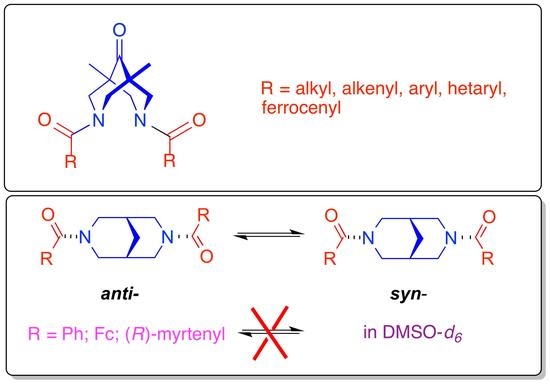
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Compounds
2.2. X-ray Studies
2.3. NMR Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Bis-amide | CDCl3 | DMSO-d6 | Data for X-ray Conformations and Solution Barriers |
|---|---|---|---|---|
| 1. | 1g | only anti * | only anti * | NMR: ΔG = 15.0 ± 0.3 kcal/mol (rotation around amide bond) * NMR: ΔG = 15.6 ± 0.3 kcal/mol (rotation around N(CO)–C2′ bond) * |
| 2. | 2 | only anti [19] | only anti [19] | X-ray: bis-anti [19] NMR: no rotation around amide bond [19] |
| 3. | 1f | only anti * | only anti * | |
| 4. | 1l | only anti [19] | only anti [19] | X-ray: anti [19] NMR: ΔG = 14.5 ± 0.2 kcal/mol [19] |
| 5. | 1e | only anti * | 0.19 * | |
| 6. | 1a | only anti * | 0.21 * | X-ray: anti [21] |
| 7. | 1b | only anti [25] | 0.25 [25] | |
| 8. | 1c | only anti [25] | 0.33 [25] | X-ray: anti [25] |
| 9. | 1b′ | only anti [25] | 0.39 [25] | |
| 10. | 1d | only anti [25] | 0.48 [25] | X-ray: anti [25] |
| 11. | 1c′ | only anti [25] | 0.54 [25] | X-ray: anti [25] |
| 12. | 1d′ | only anti [25] | 0.70 [25] | X-ray: anti [25] |
| 13. | 1i | 0.33 [27] | 1.00 * | X-ray: syn * NMR: ΔG = 16.3 ± 0.2 kcal/mol * |
| 14. | 1h | 0.33 [27] | 1.18 * | |
| 15. | 1j | 1.43 [27] | 2.33 * | |
| 16. | 1k | 1.25 [27] | 2.40 * | |
| 17. | 3 | only syn | only syn | X-ray: syn * |
2.4. QC Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vatsadze, S.Z.; Loginova, Y.D.; dos Passos Gomes, G.; Alabugin, I.V. Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy of Functional Groups. Chem.—Eur. J. 2017, 23, 3225–3245. [Google Scholar] [CrossRef]
- Cox, C.; Lectka, T. Synthetic Catalysis of Amide Isomerization Intramolecular Catalysis of Amide. Acc. Chem. Res. 2000, 33, 849–858. [Google Scholar] [CrossRef]
- Stein, R.L. Mechanism of Enzymatic and Nonenzymatic Prolyl Cis-Trans Isomerization. Adv. Protein Chem. 1993, 44, 1–24. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R.; Rush, D.J.; Keith, T.A.; Haven, N.; Avenue, W.; Haven, N. Amides. 3. Experimental and Theoretical Studies of the Effect of the Medium on the Rotational Barriers for N,N-Dimethylformamide and N,N-Dimethylacetamide. J. Am. Chem. Soc. 1995, 117, 4261–4270. [Google Scholar] [CrossRef]
- Petter, R.C.; Rao, S.J. C-N Rotational Barriers in Ferrocenecarboxamides. J. Org. Chem. 1991, 56, 2932–2934. [Google Scholar] [CrossRef]
- Otani, Y.; Nagae, O.; Naruse, Y.; Inagaki, S.; Ohno, M.; Yamaguchi, K.; Yamamoto, G.; Uchiyama, M.; Ohwada, T. An Evaluation of Amide Group Planarity in 7-Azabicyclo[2.2.1]heptane Amides. Low Amide Bond Rotation Barrier in Solution. J. Am. Chem. Soc. 2003, 125, 15191–15199. [Google Scholar] [CrossRef] [PubMed]
- Bisz, E.; Piontek, A.; Dziuk, B.; Szostak, R.; Szostak, M. Barriers to Rotation in ortho-Substituted Tertiary Aromatic Amides: Effect of Chloro-Substitution on Resonance and Distortion. J. Org. Chem. 2018, 83, 3159–3163. [Google Scholar] [CrossRef] [PubMed]
- Drakenberg, T.; Dahlqvist, K.I.; Forsén, S. The barrier to internal rotation in amides. IV. N,N-dimethylamides; substituent and solvent effects. J. Phys. Chem. 1972, 76, 2178–2183. [Google Scholar] [CrossRef]
- Da Silva, C.O.; Mennucci, B.; Vreven, T. Combining microsolvation and polarizable continuum studies: New insights in the rotation mechanism of amides in water. J. Phys. Chem. 2003, 107, 6630–6637. [Google Scholar] [CrossRef]
- Lei, P.; Meng, G.; Shi, S.; Ling, Y.; An, J.; Szostak, R.; Szostak, M. Suzuki-Miyaura cross-coupling of amides and esters at room temperature: Correlation with barriers to rotation around C-N and C-O bonds. Chem. Sci. 2017, 8, 6525–6530. [Google Scholar] [CrossRef] [Green Version]
- Zefirov, N.S.; Palyulin, V.A. Conformational Analysis of Bicyclo[3.3.1]nonanes and Their Hetero Analogs. Top. Stereochem. 1991, 20, 171–230. [Google Scholar] [CrossRef]
- Palyulin, V.A.; Emets, S.V.; Chertkov, V.A.; Kasper, C.; Schneider, H.J. Conformational switching of 3,7-diacyl-3,7-diazabicyclo[3.3.1]nonanes by metal binding and by solvent changes. Eur. J. Org. Chem. 1999, 2, 3479–3482. [Google Scholar] [CrossRef]
- Wang, Z.; Islam, M.J.; Vukotic, V.N.; Revington, M.J. Conformational Study of N,N′-Diacyl Bispidines and Dioxo Bis-bispidines: Planar Chirality and Molecular Switching. J. Org. Chem. 2016, 81, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- Levinger, S.; Sharabi-Ronen, Y.; Mainfeld, A.; Albeck, A. Structural and spatial considerations in the N,N′-diacyl- and bis(alkoxycarbonyl)bispidinone series. J. Org. Chem. 2008, 73, 7793–7796. [Google Scholar] [CrossRef]
- Veremeeva, P.N.; Grishina, I.V.; Zaborova, O.V.; Averin, A.D.; Palyulin, V.A. Synthesis of 3,7-diacyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane derivatives as promising lipid bilayer modifiers. Tetrahedron 2019, 75, 4444–4450. [Google Scholar] [CrossRef]
- McCabe, P.H.; Milne, N.J.; Sim, G.A. Conformational control in the 3,7-diazabicyclo[3.3.1]nonane system. J. Chem. Soc. Chem. Commun. 1985, 10, 625–626. [Google Scholar] [CrossRef]
- Palyulin, V.A.; Emets, S.V.; Potekhin, K.A.; Lysov, A.E.; Chertkov, V.A.; Zefirov, N.S. The Synthesis and Crystal and Molecular Structures of 3,7-Diacetyl- and 3,7-Bis(thioacetyl)-1,5-dimethyl-3,7- diazabicyclo[3.3.1]nonanes. Dokl. Chem. 2000, 375, 782–785. [Google Scholar] [CrossRef]
- Sharma, S.; Thorat, S.H.; Gonnade, R.G.; Jasinski, J.P.; Butcher, R.; Haridas, V. Engineering Molecular Topology: A Pseudopeptidic Macrocyclic Figure-Eight Motif. Eur. J. Org. Chem. 2017, 2017, 1120–1124. [Google Scholar] [CrossRef]
- Medved’ko, A.V.; Krut’ko, D.P.; Gaisen, S.V.; Churakov, A.V.; Minyaev, M.E.; Moiseeva, A.A.; Lemenovsky, D.A.; Yu, H.; Wang, L.; Vatsadze, S.Z. First examples of bispidine-ferrocene cyclophanes. J. Organomet. Chem. 2021, 949, 121945. [Google Scholar] [CrossRef]
- Sharma, S.; Gopalakrishna, M.V.S.; Venugopalan, P.; Suresh, C.H.; Haridas, V. Stackabilization: Self-assembling bispidinophanes. Tetrahedron 2015, 71, 8302–8306. [Google Scholar] [CrossRef]
- Levina, O.I.; Potekhin, K.A.; Kurkutova, E.N.; Struchkov, Y.T.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Crystal and molecular structure of 3,7-diacetyl-1,5-diphenyl-3,7-diazabicyclo [3,3,1] nonane-9-one. Dokl. Akad. Nauk SSSR 1985, 281, 1367. [Google Scholar]
- Haridas, V.; Sadanandan, S.; Sharma, Y.K.; Chinthalapalli, S.; Shandilya, A. Bispidine as a secondary structure nucleator in peptides. Tetrahedron Lett. 2012, 53, 623–626. [Google Scholar] [CrossRef]
- Comba, P.; Pritzkow, H.; Schiek, W. A very rigid bis-bispidine tetraazamacrocycle and its unusual copper(II) complex. Angew. Chem. Int. Ed. 2001, 40, 2465–2468. [Google Scholar] [CrossRef]
- Palyulin, V.A.; Emets, S.V.; Potekhin, K.A.; Lysov, A.E.; Sumskaya, Y.G.; Zefirov, N.S. Synthesis and Crystal and Molecular Structure of 3,7-Dithenoyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane. Dokl. Akad. Nauk SSSR 2001, 380, 639. [Google Scholar] [CrossRef]
- Churakov, A.V.; Medved’ko, A.V.; Prikhodchenko, P.V.; Krut’ko, D.P.; Vatsadze, S.Z. First example of peroxosolvate of iodine-containing organic molecule. Mendeleev Commun. 2021, 31, 352–355. [Google Scholar] [CrossRef]
- Tomassoli, I.; Gündisch, D. Bispidine as a Privileged Scaffold. Curr. Top. Med. Chem. 2016, 16, 1314–1342. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakov, D.; Baev, D.; Kalinin, M.; Dalinger, A.; Chirkova, V.; Belenkaya, S.; Khvostov, A.; Krut’ko, D.; Medved’ko, A.; Volosnikova, E.; et al. Design and Evaluation of Bispidine-Based SARS-CoV-2 Main Protease Inhibitors. ACS Med. Chem. Lett. 2021. [Google Scholar] [CrossRef]
- Haridas, V.; Rajgokul, K.S.; Sadanandan, S.; Agrawal, T.; Sharvani, V.; Gopalakrishna, M.V.S.; Bijesh, M.B.; Kumawat, K.L.; Basu, A.; Medigeshi, G.R. Bispidine-Amino Acid Conjugates Act as a Novel Scaffold for the Design of Antivirals That Block Japanese Encephalitis Virus Replication. PLoS Negl. Trop. Dis. 2013, 7, e2005. [Google Scholar] [CrossRef]
- Miyahara, Y.; Goto, K.; Inazu, T. Synthesis and properties of a novel tetraazamacrocycle containing two bispidine units. Chem. Lett. 2000, 29, 620–621. [Google Scholar] [CrossRef]
- Miyahara, Y.; Goto, K.; Inazu, T. A novel hindered macrocyclic tetramine containing two bispidine units. A new type of proton sponge. Tetrahedron Lett. 2001, 42, 3097–3099. [Google Scholar] [CrossRef]
- Islam, M.J.; Miller, E.J.; Gordner, J.S.; Patel, D.; Wang, Z. Effective synthesis of bis-bispidinone and facile difunctionalization of highly rigid macrocyclic tetramines. Tetrahedron Lett. 2013, 54, 2133–2136. [Google Scholar] [CrossRef]
- Singh, H.; Chenna, A.; Gangwar, U.; Borah, J.; Goel, G. Bispidine as β-strand nucleator: From β-arch to nano cages and vesicles. Chem. Sci. 2021, 12, 15757–15764. [Google Scholar] [CrossRef]
- Veremeeva, P.N.; Bovina, E.M.; Grishina, I.V.; Lapteva, V.L.; Palyulin, V.A.; Zefirov, N.S. Synthesis of amphiphilic diacyl derivatives of 3,7-diazabicyclo[3.3.1]nonan-9-one. Mendeleev Commun. 2018, 28, 25–26. [Google Scholar] [CrossRef]
- Mozhaitsev, E.S.; Ponomarev, K.Y.; Patrusheva, O.S.; Medvedko, A.V.; Dalinger, A.I.; Rogachev, A.D.; Komarova, N.I.; Korchagina, D.V.; Suslov, E.V.; Volcho, K.P.; et al. Conjugates of Bispidine and Monoterpenoids as Ligands of Metal Complex Catalysts for the Henry Reaction. Russ. J. Org. Chem. 2020, 56, 1969–1981. [Google Scholar] [CrossRef]
- Minasyan, G.G.; Agadzhanyan, K.E.; Admyan, G.G. Synthesis and conversions of polyhedral compounds 17. Conversion of 1,3-diaza- and 1,3,5-triazaadamantanes to nitrogen-containing pentacyclic compounds. Chem. Heterocycl. Compd. 1994, 30, 94–98. [Google Scholar] [CrossRef]
- Lavrov, M.I.; Veremeeva, P.N.; Karlov, D.S.; Zamoyski, V.L.; Grigoriev, V.V.; Palyulin, V.A. Tricyclic derivatives of bispidine as AMPA receptor allosteric modulators. Mendeleev Commun. 2019, 29, 619–621. [Google Scholar] [CrossRef]
- Kotlyarova, A.A.; Ponomarev, K.Y.; Morozova, E.A.; Korchagina, D.V.; Suslov, E.V.; Pavlova, A.V.; Tolstikova, T.G.; Volcho, K.P.; Salakhutdinov, N.F. The effect of 3,7-diazabicyclo3.3.1nonanes containing monoterpenoid moieties on the physical activity of mice. J. Res. Pharm. 2020, 24, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Goddard, R.; Pörschke, K.R. Degradation of dichloromethane by bispidine. J. Phys. Org. Chem. 2012, 25, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2007. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Karnoukhova, V.A.; Dmitrienko, A.O.; Lyssenko, K.A. Toward a Rigorous Definition of a Strength of Any Interaction between Bader’s Atomic Basins. J. Phys. Chem. 2017, 121, 4517–4522. [Google Scholar] [CrossRef]
- Medved’ko, A.V.; Egorova, B.V.; Komarova, A.A.; Rakhimov, R.D.; Krut’Ko, D.P.; Kalmykov, S.N.; Vatsadze, S.Z. Copper-Bispidine Complexes: Synthesis and Complex Stability Study. ACS Omega 2016, 1, 854–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruker. APEX-III; Bruker AXS Inc.: Madison, WI, USA, 2019. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A. Platon squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Spek, A. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krut’ko, D.P.; Medved’ko, A.V.; Lyssenko, K.A.; Churakov, A.V.; Dalinger, A.I.; Kalinin, M.A.; Gudovannyy, A.O.; Ponomarev, K.Y.; Suslov, E.V.; Vatsadze, S.Z. Bispidine Platform as a Tool for Studying Amide Configuration Stability. Molecules 2022, 27, 430. https://doi.org/10.3390/molecules27020430
Krut’ko DP, Medved’ko AV, Lyssenko KA, Churakov AV, Dalinger AI, Kalinin MA, Gudovannyy AO, Ponomarev KY, Suslov EV, Vatsadze SZ. Bispidine Platform as a Tool for Studying Amide Configuration Stability. Molecules. 2022; 27(2):430. https://doi.org/10.3390/molecules27020430
Chicago/Turabian StyleKrut’ko, Dmitry P., Alexey V. Medved’ko, Konstantin A. Lyssenko, Andrei V. Churakov, Alexander I. Dalinger, Mikhail A. Kalinin, Alexey O. Gudovannyy, Konstantin Y. Ponomarev, Eugeny V. Suslov, and Sergey Z. Vatsadze. 2022. "Bispidine Platform as a Tool for Studying Amide Configuration Stability" Molecules 27, no. 2: 430. https://doi.org/10.3390/molecules27020430