
Alternative Targets for Modulators of Mitochondrial Potassium Channels


1
Laboratory of Intracellular Ion Channels, Nencki Institute of Experimental Biology, Polish Academy of Sciences, 02-093 Warsaw, Poland
2
Department of Histology, Medical University of Gdansk, 1a Debinki, 80-211 Gdansk, Poland
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(1), 299; https://doi.org/10.3390/molecules27010299
Submission received: 18 November 2021
/
Revised: 30 December 2021
/
Accepted: 31 December 2021
/
Published: 4 January 2022
(This article belongs to the Special Issue Compounds Modulating Mitochondrial Ion Channels)
Abstract
:Mitochondrial potassium channels control potassium influx into the mitochondrial matrix and thus regulate mitochondrial membrane potential, volume, respiration, and synthesis of reactive oxygen species (ROS). It has been found that pharmacological activation of mitochondrial potassium channels during ischemia/reperfusion (I/R) injury activates cytoprotective mechanisms resulting in increased cell survival. In cancer cells, the inhibition of these channels leads to increased cell death. Therefore, mitochondrial potassium channels are intriguing targets for the development of new pharmacological strategies. In most cases, however, the substances that modulate the mitochondrial potassium channels have a few alternative targets in the cell. This may result in unexpected or unwanted effects induced by these compounds. In our review, we briefly present the various classes of mitochondrial potassium (mitoK) channels and describe the chemical compounds that modulate their activity. We also describe examples of the multidirectional activity of the activators and inhibitors of mitochondrial potassium channels.
1. Introduction
Earlier studies on isolated mitochondria have indicated that the transport of potassium ions is an important element in regulating their volume. This potassium transport process has also been observed in various cellular systems. Western blot analysis of isolated mitochondria has shown the presence of protein-binding antibodies specific to potassium channel counterparts in the plasma membrane. Finally, the presence of the mitoK channels in the inner mitochondrial membrane (IMM) was confirmed by measuring the activity of a single channel by the patch clamp technique. Mitochondrial potassium channels play an important role in mitochondrial functioning due to the flow of positive charge (in the form of K+) through the IMM. These channel proteins have been identified in the mitochondria of various tissues, including the heart, skeletal muscle, and neuronal cells. Activation of mitoK channels by passing K+ through the IMM causes changes in the mitochondrial membrane potential that increase mitochondrial oxygen consumption and influence mitochondrial ROS synthesis. Therefore, mitoK channels are an important part of cell signaling pathways and can be thought of as triggers for cell survival or cell death [1,2,3].
Several potassium channels have been identified in the IMM, such as ATP-sensitive potassium channels (mitoKATP); small (mitoSKCa), intermediate (mitoIKCa), and large conductance (mitoBKCa) calcium activated potassium channels; sodium dependent potassium (mitoSlo2) channels; and voltage-dependent (mitoKv1.3 and mitoKv7.4) potassium channels [4,5].
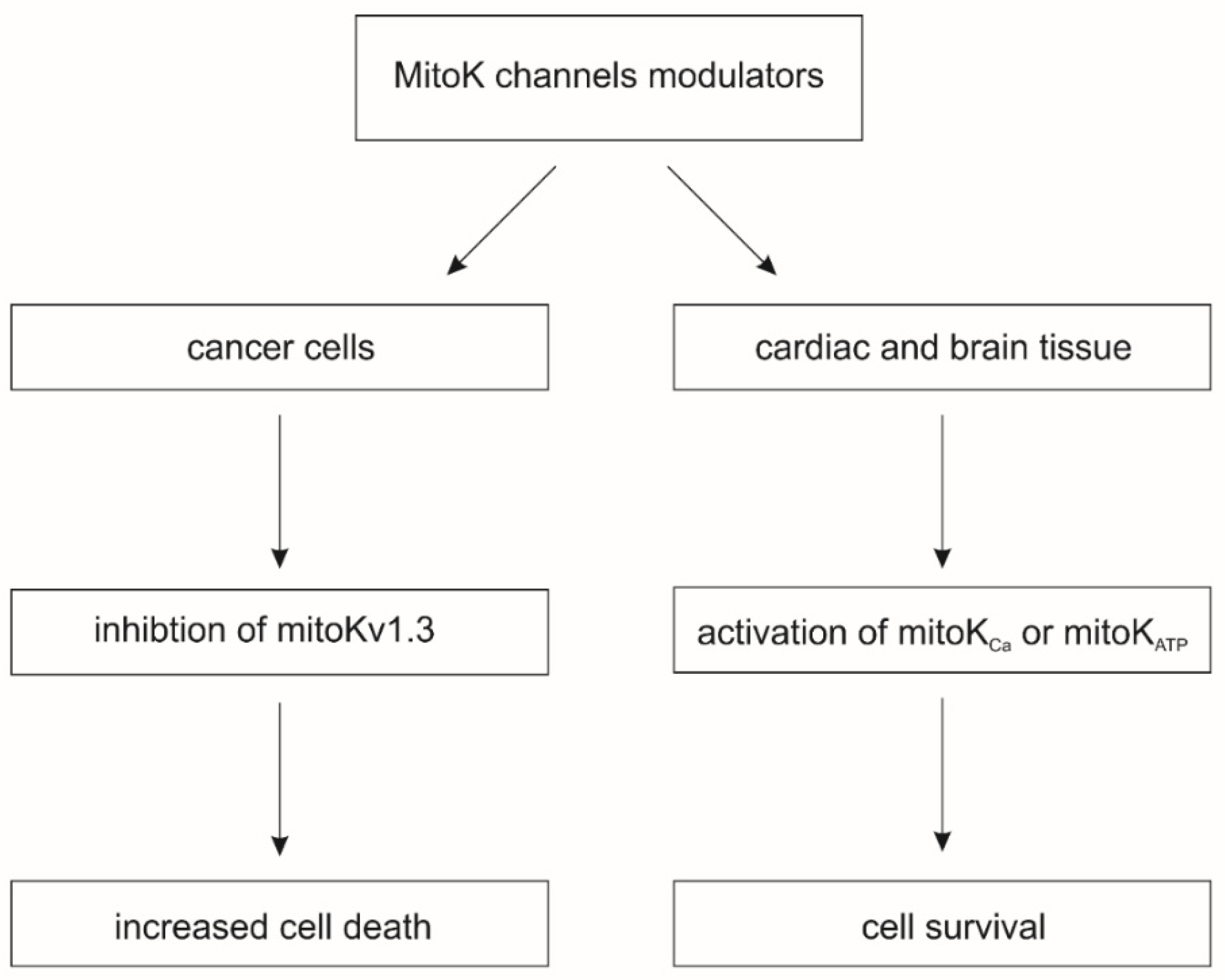
The activation of mitoK channels by means of small molecule potassium channel openers (KCOs) induces cardio- and neuroprotective effects against various injuries, including ischemia-reperfusion. On the other hand, the pharmacological inhibition of mitoKv channels induces cell death in cancer cells (Figure 1) [1]. The beneficial effects induced by modulators of mitoK channels are related to changes in mitochondrial physiology. The activity of mitoK channels regulates the synthesis of mitochondrial ROS and mitochondrial matrix Ca2+ uptake, which directly regulates the opening of the mitochondrial permeability transition pore (mPTP), a key trigger of cell death pathways [6].
For these reasons, mitoK channels are promising targets for the development of new pharmacological tools to modulate their activity. However, the use of pharmacological modulators of mitoK channels carries a high risk that these compounds will interact with other targets in the cell, which can lead to unwanted effects or make it difficult to distinguish which effects are related to mitoK channels. In this review, we will provide basic information about the key modulators of mitoK channels and their alternative sites of action.
2. Modulators of Mitochondrial ATP-Sensitive Potassium Channels
The first potassium channel identified in the IMM was the ATP-sensitive potassium channel in rat livers [7]. This channel is inhibited by ATP and the antidiabetic sulfonylurea, glibenclamide [7]. Later, the same type of channel was identified in other tissues, such as the heart [8,9,10], brain [11,12,13], skeletal muscle [14], human T-lymphocytes [15], and skin fibroblasts [16]. Electrophysiological experiments revealed that the conductance of the channel is usually close to 100 pS [11,17,18,19]; however, channels with lower conductance, close to 10 pS, have also been reported [7,8]. These differences in reported conductance may result from the various experimental conditions.
Initial findings suggested that the channel was formed by the Kir6.1 or Kir6.2 subunits, yet the molecular identity of mitoKATP remained unclear [20]. However, later studies showed that the ROMK2 isoform of the renal outer medullar potassium channel could be the structural component of the channel [9,21]. Interestingly, it was also suggested that the subunits of ATP synthase could form channels with mitoKATP pharmacological properties [22]. Recently, it was shown that the pore-forming subunit of the mitoKATP channel is a product of the CCDC51 gene [10]. Many studies have demonstrated that mitoKATP is inhibited by glibenclamide; therefore, it was speculated that the SUR subunit, which is a glibenclamide receptor, is an integral part of the channel. Indeed, CCDC51 interacts with mitoSUR, encoded by the ABCB8 gene. The channel formed by these two proteins has the canonical pharmacological properties of the mitoKATP channel [10]. Activation of the mitoKATP channel induces or mediates cardio- or neuroprotection against various insults, including I/R. This phenomenon is well documented; however, the details of the cytoprotection mechanism are still not fully understood [17,23,24].
The list of pharmacological modulators of the mitoKATP channel is relatively long. Diazoxide and BMS191095 can be treated as canonical pharmacological activators of the mitoKATP channel [25], along with nicorandil, cromakalim, pinacidil, or P1075. Inhibitors of the mitoKATP channel include glibenclamide (glyburide), 5-hydroxydecanoic acid (5-HD), tetraphenylphosphonium, and 4-aminopyridine [1,25]. 5-HD is described as a relatively selective inhibitor of mitoKATP channels, and it does not inhibit plasma membrane KATP channels [6,23]. However, all these compounds either have well-described alternative targets in the cell or induce cellular processes, which makes it difficult to reliably assess the detailed mechanism of their operation [26]. Importantly, most of these compounds were tested in the mitochondrial context.
The most notable opener of mitoKATP channels is diazoxide, which is used in the treatment of diabetes [27]. Early studies revealed that this compound shows higher specificity toward mitoKATP than to KATP channels from the plasma membrane and sarcoplasmic reticulum [28]. Therefore, it is believed that the application of diazoxide at appropriately low concentrations should result in selective activation of the mitoKATP channel. However, diazoxide inhibits the activity of respiratory chain complex II [29]. Moreover, diazoxide directly activates protein kinase c epsilon (PKC-ε) (Table 1) [30]. Interestingly, it was shown that diazoxide and glibenclamide modulate the channel formed by CCDC51 only in the presence of the mitoSUR subunit [10]. Activation of ROMK2 found in mitochondria by diazoxide has also been observed [21].
Initial studies on isolated mitochondria suggested that BMS191095 was a specific activator of mitoKATP channels [31,32] that activated the channels in the nanomolar range of concentrations [32]. Electrophysiological studies confirmed that this compound opens cardiac, neuronal, and dermal fibroblast mitoKATP channels [11,16,33]. Several studies have shown that BMS191095 induces cytoprotection by activating mitoKATP channels. For example, BMS191095 induced cytoprotection against I/R injury in mice and rat cardiac tissue [32,34] and pig skeletal muscle [35]; cytoprotection induced by this compound was reversed by 5-HD. Additionally, BMS191095 was shown to activate neuronal mitoKATP, regulate neuronal mitochondrial function [36,37], and induce neuroprotection against various insults [36,38,39]. It was also shown that BMS191095 and the less-selective BMS180448 inhibited human platelet aggregation; preincubation with both glyburide and 5-HD blocked this effect [40]. In contrast, the application of BMS191095 protected myoblast C2C12 cells against toxicity induced by the deregulation of calcium homeostasis, and these effects were not reversed by 5-HD [41]. This compound was also shown to induce neurotoxicity effects [24,32].
Nicorandil, like diazoxide, was shown to mimic ischemic preconditioning of heart tissue by the activation of mitoKATP channels [42,43]. However, this compound can also activate sarcolemmal KATP channels [44]. Other studies have reported that nicorandil can be a donor of nitric oxide (NO) [45,46]. Additionally, it can stimulate superoxide dismutase and inhibit xanthine oxidase activity, therefore acting as an antioxidant [46,47,48]. The bidirectional effects of nicorandil were recently observed in a study describing the beneficial effects of this compound on fatigue in slow skeletal muscle fibers [43]. On the one hand, nicorandil activated the channel; on the other hand, it acted as an antioxidant and NO donor [43]. Further experiments revealed that nicorandil blocks mitochondrial glutamate malate-driven respiration in skeletal muscle [49]. Nicorandil can induce cytoprotection in dystrophin-deficient cardiomyocytes, and these effects are only partially reversed by the mitoKATP blocker 5-HD [50]. Another recent example of the bidirectional activity of nicorandil can be seen in the regulation of the expression of selected genes, such as heme oxygenase-1 or interleukin-8, by nicorandil in human umbilical vein endothelial cells (HUVECs). Only some of the observed effects were reversed by 5-HD, and thus were linked to mitoKATP activity [51]. In addition to the other openers listed above, it has also been suggested that malonate and atpenin A5, which are inhibitors of complex II, can activate mitoKATP channels and induce cardioprotection [52,53].
Similar to mitoKATP openers, inhibitors of this channel can either interact with other proteins or enter various metabolic pathways. For example, 5-HD, which is a canonical mitoKATP inhibitor, was shown to inhibit sarcolemmal KATP channels [54]. In contrast, 5-HD does not inhibit mitoKATP from rat brains [11]. It was also found that 5-HD is metabolized by acyl-CoA synthetase and enters beta-oxidation, which can induce the switch from glucose to fatty acid metabolism [55,56,57]. Therefore, the direct interaction of 5-HD with mitoKATP has been questioned. Additionally, 500 μM of 5-HD was shown to stimulate the activity of mitochondrial complex II and inhibit complex III in skeletal muscle cells [49]. Another study revealed that both glibenclamide and 5-HD can interact with ADP/ATP carriers from the inner mitochondrial membrane [58]. Additionally, glibenclamide can activate the mitochondrial permeability transition pore by increasing its sensitivity to calcium ions [59]. Moreover, glibenclamide inhibits chloride channels [60,61].
To examine whether the ROMK protein acts as a possible mitoKATP channel-forming component, research has focused on the use of compounds regulating the activity of this protein. For example, it was reported that VU591 inhibits ROMK channels with high specificity [62,63,64]. However, this molecule induced mitochondrial depolarization and matrix contraction in mitochondria from both wild-type and ROMK knockout cells. [65]. Moreover, VU591 reversed matrix swelling induced by BMS191095 in the ROMK knockout cells. These observations suggest that VU591 has some alternative targets in mitochondria [65].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Examples of mitoKATP channel modulators and their off-target activity.
| Name | Function | Example of Off-Target Activity | Ref. |
|---|---|---|---|
| Diazoxide | Opener | Inhibition of mitochondrial complex II. Protonophoric properties. PKC-ε activator. ROS inducer. | [16,29,30,37,57] |
| BMS191095 | Opener | Induction of mitoKATP-independent cytoprotection. Induction of neurotoxicity. | [16,24,32,37,41] |
| Nicorandil | Opener | NO donor. Antioxidant, inhibition of xanthine oxidase activity. | [43,45,46,47] |
| 5-HD | Inhibitor | Sarcolemmal KATP channel inhibitor. Substrate for acyl-CoA synthetase. Stimulation of mitochondrial complex II. Inhibition of mitochondrial complex III. Possible interaction with mitochondrial ADP/ATP carriers. | [49,54,55,56] |
| Glibenclamide | Inhibitor | Activation of mPTP. Possible interaction with mitochondrial ADP/ATP carriers. Inhibitor of cAMP-activated chloride channels. | [58,59,60,61] |
| VU591 | Inhibitor (ROMK) | General inhibitor of ROMK channels. Mitochondria uncoupling. | [64,65] |
3. Mitochondrial Calcium-Activated Potassium Channels
In the IMM, a group of calcium-activated potassium channels have been identified, namely large conductance K+ (mitoBKCa) channels [66], intermediate conductance K+ (mitoIKCa) channels [67], and small conductance K+ (mitoSKCa) channels [68]. Their common property is regulation by calcium ions—their open probability increases in the presence of Ca2+ [17]. This common activation mechanism may indicate that these proteins are activated under the same physiological conditions. However, these channels differ in terms of other regulatory mechanisms. In addition, a different spectrum of pharmacological substances regulates their activity.
3.1. Large Conductance Calcium-Activated Potassium Channels
The mitoBKCa channel was originally identified in the mitochondria of LN229 glioma cells by the patch clamp technique. The recorded channel had a conductance of 295 pS measured in a 150 mM KCl bath and pipette solution [66]. Later, the channel was described in other tissues, including the heart [69], brain [70,71], skeletal muscle [72], endothelium [73], dermal fibroblasts [74], and pulmonary and kidney epithelial cells [75,76]. Interestingly, similar channels have been identified in the mitochondria of mammals, lower organisms, and plants [77,78,79,80].
The pore-forming α subunit of both the mitoBKCa and the plasma membrane BKCa channels is encoded by the KCNMA1 gene. The VEDEC isoform of the α subunit (the name comes from the amino acid sequence at the C-terminus of the protein) is targeted to cardiac mitochondria [81,82]. Recently, it was shown that transfection with a VEDEC-encoding plasmid results in mitoBKCa channel activity in HEK293T cells [83].
Similar to their plasma membrane counterparts, mitoBKCa channels are modulated by various endogenous modulators. It was shown that heme and hemin inhibit channel activity, and that carbon monoxide reactivates channels blocked by heme [84,85,86]. The channel is also modulated by other gasotransmitters, such as hydrogen sulfide (H2S) [87,88,89,90,91,92], redox signals, and protons [93]. The channel is also activated by 17β-estradiol, which can lead to cardioprotection [94].
Some of the first synthetic BKCa channel activators used for mitoBKCa channels were the benzimidazolone derivatives NS1619 and NS004 [69,96,97]. Interestingly, NS004 was also described as a cystic fibrosis transmembrane conductance regulator (CFTR) channel opener [98]. Application of NS1619 protects the heart tissue from I/R injury, and many studies have shown that this effect may be mediated by mitoBKCa [69,82,99]. NS1619 acts by shifting the voltage-sensor of BKCa channels toward the activated state, as the open probability is barely altered when the voltage sensing domain (VSD) is at rest [100,101]. Although NS1619 is a known mitoBKCa channel opener, this compound has many effects unrelated to the channel. For example, it can induce nonselective ion transport across the inner mitochondrial membrane [102] and inhibit complexes of the respiratory chain [102,103]. Another study showed that NS1619 has oligomycin-like properties and inhibits mitochondrial ATP synthase. Additionally, NS1619 inhibits sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) [104], and L-type calcium channels (Figure 2) [105]. NS1619 also stimulates Ca2+-gated chloride currents in smooth muscle cells of the rabbit pulmonary artery [106]. BKCa channel-independent immediate and delayed preconditioning of neuronal cells was also observed after the application of NS1619 [107,108].
NS11021, a biarylthiourea derivative, has a higher potency and selectivity as a BKCa channel opener than NS1619 [109]. Application of NS11021 shifts the voltage-activation curve of the channel to more negative potentials, and activation is independent of free Ca2+ concentration [100,110,111]. NS11021 increases the open probability of BKCa channels by altering the gating kinetics without affecting the single-channel conductance. It also protects the heart from I/R injury via mitoBKCa channel activation [110]. Interestingly, NS11021-induced activation of mitoBKCa channels prevents cold-storage induced injury of kidney epithelial cells [75].
Although NS11021 was thought to be more specific than NS1619, it was later shown to induce mitochondrial depolarization in a sucrose medium in the absence of K+ salt [112]. It was also found that NS11021 applied at higher concentrations (10–30 μM) can activate KV7.4 and have minor inhibitory effects on Kv7.2/7.3 channels. The same study showed that NS11021 is a positive modulator of α7 nicotinic acetylcholine receptors at concentrations of 10–30 µM [109].
Another set of mitoBKCa channel openers is CGS7181 and CGS7184. Mitoplast patch clamp recording has revealed that the mitoBKCa channel of glioma cells is activated by CGS7184 [113]. Application of this opener decreased ROS synthesis by brain mitochondria in a model of reversed electron flow, and this effect was reversed by mitoBKCa channel inhibitors [114]. However, in contrast to the effects of NS1619, CGS7184 and its derivative CGS7181 induced cell death, an effect unrelated to mitoBKCa channel activation [113,115]. CGS7184 can also directly activate ryanodine receptor (RyR2) calcium release [116]. Evidently, the modulation of alternative targets by these compounds may be responsible for the observed increase in cytosolic calcium ion concentration [95].
Interestingly, both CGS7184 and NS1619 show mitoBKCa channel activation in single channel patch clamp technique measurements; however, they exert opposing effects on cells. CGS7184 has a cytotoxic effect while NS1619 is cytoprotective. Therefore, the cytotoxic outcome of CGS7184 must be related to the off-target effects of this compound. Conversely, in the case of NS1619, the off-target effect is synergistic to some extent, leading to cytoprotection, especially in I/R processes. One of the mechanisms of action that distinguishes these compounds is the different manner in which calcium ions are released from the endoplasmic reticulum. CGS7184 releases Ca2+ by opening RyR2 channels, while NS1619 releases Ca2+ through the inhibition of SERCA in a pH-dependent manner [117,118]. Opening of RyR2 channels by CGS7184 naturally activates SERCA by Ca2+, which leads to a significant increase in the consumption of intracellular ATP, while NS1619 causes inhibition of SERCA, reducing ATP consumption. NS1619′s inhibition of SERCA increases as pH decreases and is reversible. Notably, a drop in pH occurs during ischemia (Figure 2).
Another interesting mitoBKCa channel modulator is chlorzoxazone, which is an FDA-approved centrally acting muscle relaxant [119]. This compound produces a left shift in the activation curve of BKCa channels, yet it does not affect the Ca2+-sensitivity of the channels during that process [119]. It was used to treat a patient with progressive cerebellar degeneration, who had a BKG354S mutation in the KCNMA1 gene [120]. Chlorzoxazone also activates SKCa and IKCa channels, and 30 μM chlorzoxazone suppresses voltage-dependent L-type Ca2+ currents [119]. Another mitoBKCa channel modulator is a synthetic inhibitor with a natural origin: diCl-DHAA (12,14-dichlorodehydroabietic acid). It was reported to reduce I/R in rat cardiac myocytes [121,122].
The mitoBKCa channel is also activated by naturally occurring openers. Similar to the plasma membrane BKCa channel, the mitoBKCa channel is activated by the sex hormone 17-estradiol (17β-estradiol, EST). 17β -estradiol has been applied to the mitoBKCa channels in vascular smooth muscle using the black lipid membrane technique or the ventricular mitoplasts of rats using patch clamp [94]. The activation of mitoBKCa channels by 17β-estradiol requires the β1 subunit [123]. 17β-estradiol has been described as an L-type channel agonist that induces a rapid increase in intracellular calcium concentration through the potentiation of the channel and the activation of cascade signaling pathways, such as Src/ERK/CREB/Bcl-2 [124]. Another example of a naturally occurring mitoBKCa channel modulator is naringenin, a plant-derived flavonoid found in a variety of fruits and herbs [125]. Naringenin has been found to induce cardioprotection by activating mitoBKCa channels [126]. Recently, it was shown that naringenin also activates mitoBKCa and mitoKATP channels in skin fibroblasts [127]. Naringenin also has a stimulatory effect on BKCa channels in the absence of the auxiliary β subunits of the channel, increasing the probability of channel opening [128]. The beneficial effects of naringenin have been described for many disorders, including cardiovascular, pulmonary, metabolic, and neurological issues [95]. When applied to NSC-34 neuronal cells, naringenin shifted the activation curve of the M-type K+ current (Kv7.2, 7.3, and 7.5) to more negative potentials [128].
Several inhibitors of mitoBKCa channels have been described. This group contains the short peptides charybdotoxin (ChTx) and iberiotoxin (IbTx), quinine, and diterpene paxilline. Charybdotoxin is a scorpion (Leiurus quinquestriatus) toxin identified in 1985 as a BKCa channel blocker [66,129,130,131]. Later, it was also shown to inhibit mitoBKCa channels [66]. In addition, charybdotoxin also inhibits the IKCa, Kv1.2, and Kv1.3 channels and, at lower potency, the Kv1.6 channels [132].
Iberiotoxin, a toxin from the red scorpion Buthus tamulus is 68% homologous to charybdotoxin; however, it does not block any other ChTX-sensitive K+ channels [133,134]. Moreover, iberiotoxin can block the BKCa channel in the presence of the auxiliary β1 subunit [135]. Interestingly, both charybdotoxin and iberiotoxin are inactive in the presence of the auxiliary β4 subunit [136,137].
Paxilline, a tremorgenic indole toxic alkaloid produced by Penicillium paxilli, has been used to block mitoBKCa channels in guinea pig ventricular cells, rat ventricular myocytes, and rat heart and liver cells [138,139]. Higher concentrations of paxilline modulate SERCA at the phosphoenzyme level [140,141]. At a concentration of 10–50 µM, paxilline decreases light scattering, slightly uncouples respiration [142], and protects HT22 cells against glutamate-induced cytotoxicity [143].
Lastly, quinine is a compound from cinchona trees that has been found to inhibit BKCa channel-caused K+ uptake into isolated mitochondria. Quinine is also known to inhibit KV2.2 and K2P18.1 channels [144]. Structural studies have shown that BKCa channels possess a conserved heme-binding sequence motif. Hemin binds to the linkage segment between the RCK1 and RCK2 domains and induces conformational changes different than those induced by Ca2+ [85]. It regulates the inactivation of the K+ channel and N-type inactivation of Kv1.4 and Kv3.4 channels [145].
3.2. Intermediate Conductance Calcium-Activated Potassium Channels
Intermediate conductance calcium-activated potassium channels from the mitochondrial inner membrane show a conductance close to 27 pS and were identified for the first time in human colon cancer cells [67]. The pore-forming subunit of IKCa channels from the plasma membrane has six transmembrane spanning regions (S1–S6), with the conducting pore located between S5 and S6. The gating is conferred upon Ca2+ binding to calmodulin (CaM), which is constitutively bound to the C-terminus of each channel subunit [146,147]. It was shown that the activity of IKCa channels in the mitochondria and plasma membrane influences oxidative phosphorylation in pancreatic ductal adenocarcinoma cells [148].
The IKCa channels from the plasma membrane and mitochondria are activated by several synthetic modulators, such as NS309 [148,149,150,151,152,153], DCEBIO [150,154,155,156], and riluzole (Table 2). Riluzole has not been tested on mitoIKCa channels per se, though it was used to measure mitochondrial membrane potential (Δψ). However, these compounds are not selective modulators of IKCa channels, as they can also activate SKCa channels [157]. Riluzole is an FDA-approved drug [154] that activates K2P10.1, TRPC5 [158], K2P2.1, K2P4.1, and Slo2 channels [159]. This compound can also inhibit Na+, Trpm4 [160,161,162,163], and Cl- channels [164], as well as GABA reuptake [165]. Moreover, riluzole increases glutamate uptake and modulates glutamate receptors [166]. Interestingly, NS309 is believed to be a more potent activator of the IKCa channel than 1-EBIO or DCEBIO [149].
IKCa channels are inhibited by clotrimazole [67,155,167,168,169], which can also inhibit BKCa channels [170]. Clotrimazole plays a role in the acute inhibition and chronic induction of human cytochrome P450-dependent enzymes, leading to liver damage. The second inhibitor of the channel, TRAM-34, is a clotrimazole-based derivative [67,148,155,171,172]. It is more selective for IKCa channels than it is for KV, BKCa, and SKCa channels [173]. TRAM-34 decreased astrogliosis and microglial activation, and attenuated memory loss in an Alzheimer’s disease mouse model [174].
Table 2.
Examples of mitoIK channel modulators and their off-target activity.
| Name | Function | Example of Off-Target Activity | Ref. |
|---|---|---|---|
| Riluzole | Opener | Activation of SKCa, K2P2.1, K2P10.1, K2P4.1, TRPC5, Slo2 channels. Inhibition of Trpm4 and chloride channels. | [161,162,164,167] |
| NS309 | Opener | Opener of SKCa channels. | [149,151] |
| DCEBIO | Opener | Opener of SKCa channels. | [153,159,160] |
| Clotrimazol | Inhibitor | Inhibition of BKCa channels. Inhibition of cytochrome P450-dependent enzymes. | [67,170] |
| TRAM-34 | Inhibitor | Inhibition of Kv, BKCa, SKCa channels when applied at higher concentration. | [150,173] |
3.3. Small Conductance Calcium-Activated Potassium Channels
In the inner mitochondrial membrane, small conductance calcium-activated potassium channels have also been identified [68]. The conductance of SKCa channels is approximately 4–14 pS. The gating of these channels is conferred by Ca2+ binding to calmodulin, which, as with in IKCa channels, is constitutively bound to the C-terminus of each channel subunit [146,147]. The plasma membrane and mitochondrial SKCa channels are activated by several compounds, such as 1-EBIO, DCEBIO, NS309, CyPPA, or riluzole [151,155,175,176,177]. As mentioned above, these compounds can also activate IKCa channels; however, not all of these compounds were used in mitoSKCa/IKCa channel studies. The activation of the mitoSKCa channel with NS309 is known to reduce neurotoxicity [151]. NS309 is a more potent activator of the recombinant SK2 channels than DCEBIO and 1-EBIO [178]. It has also been shown that 1-EBIO activates CFTR channels [179]. CyPPA-mediated activation of the mitoSK2 channel leads to neuroprotection of HT-22 cells [177,180]. CyPPA can also inhibit melanogenesis by modulating the GSK3β/β-catenin/MITF pathway, as well as reduce nitric oxide release in microglia [181].
The mitoSKCa channel is inhibited by a canonical SKCa channel peptide blocker, apamin [151,155,182]. This inhibitor blocks SKCa channels in concentrations ranging from pM to nM. Apamin is known to bind to the outer pore region of the SKCa channel; however, it is membrane impermeable and can show off-target activity at higher concentrations. Apamin regulates gene expression in various signaling pathways [183]; for example, it downregulates or inhibits the expression of TNF-α, intracellular cell adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, transforming growth factor (TGF)-β1, fibronectin, the NF-κB signaling pathway, and signal transducers and activators of transcription (STAT) in vitro, thereby inhibiting proinflammatory cytokines and type 2 helper (Th2) lymphocyte chemokines [184].
The mitoSKCa channel, similar to its plasma membrane counterpart, can be blocked by several synthetic inhibitors, such as NS8593 or UCL1684 [182,185]. UCL1684 mimics the binding residues of the structural elements of a naturally occurring inhibitory neurotoxin: apamin. This compound displaces apamin binding and is considered a pore blocker, acting at the apamin binding site [186]. SKCa channels are also blocked by chlorzoxazone [187,188,189]. This compound, which serves as a muscle-relaxing drug and a probe for human liver cytochrome P-450IIE1 (CYP2E1), suppresses voltage-dependent L-type Ca2+ current.
4. Mitochondrial Voltage-Dependent Potassium Channels
In the inner mitochondrial membrane, voltage-gated potassium channels, including mitoKv1.3, mitoKv1.5, and mitoKv7.4, have been identified [1]. These channels have also been found in the plasma membrane. Interestingly, the Kv1.3 channel is also present in the endoplasmic reticulum and the Golgi apparatus. The activity of Kv channels is regulated by changes in the cell membrane potential [190]. These channels are composed of voltage-sensing and pore-forming α-subunits and auxiliary β-subunits that enable them to perform a wide variety of physiological functions [191]. Each α-subunit is composed of six transmembrane domains (S1–S6) with the S1–S4 transmembrane domains surrounding a central pore domain built of two S5–S6 helices. The four α-subunits link together to form the functional square-pore structure of the Kv channel [192]. Previous work has shown that mitoKv1.3 channels are highly overexpressed in cancer cells and immune cells, and that modulation of these channels is effective in the treatment of cancer [193]. There are many Kv channel modulators, and we will focus only on those modulators that have been described as acting on the Kv channels found in mitochondria. Most of these modulators also modulate Kv channels present in various cellular compartments with different specificity dependent on the concentration used.
4.1. Mitochondrial Kv1.3 and Kv1.5 Channels and Their Modulators
The pore-forming subunit of the Kv1.3 channel is encoded by the KCNA3 gene. The presence of mitoKv1.3 channels was originally observed in T cells [194]; however, mitoKv1.3 channels are also present in cancer cells, such as leukemia Jurkat T cells, prostate cancer PC-3 cells, and breast cancer MCF-7 cells [195]. The Kv1.3 channel is also found in the nuclear membrane of Jurkat T cells, MCF-7 cells, A549 lung cancer cells, and SNU-484 gastric cancer cells, as well as in human brain tissue [196]. The mitoKv1.5 channel has been found in the inner mitochondrial membrane of J774 macrophages [197].
Kv1.3 channels have been identified in mitochondria by incubating isolated mitochondria with plasma membrane Kv1.3 channel-specific inhibitors, such as margatoxin (MgTx) and Stichodactyla toxin (ShK). These compounds are effective in inducing hyperpolarization of the mitochondrial membrane [194].
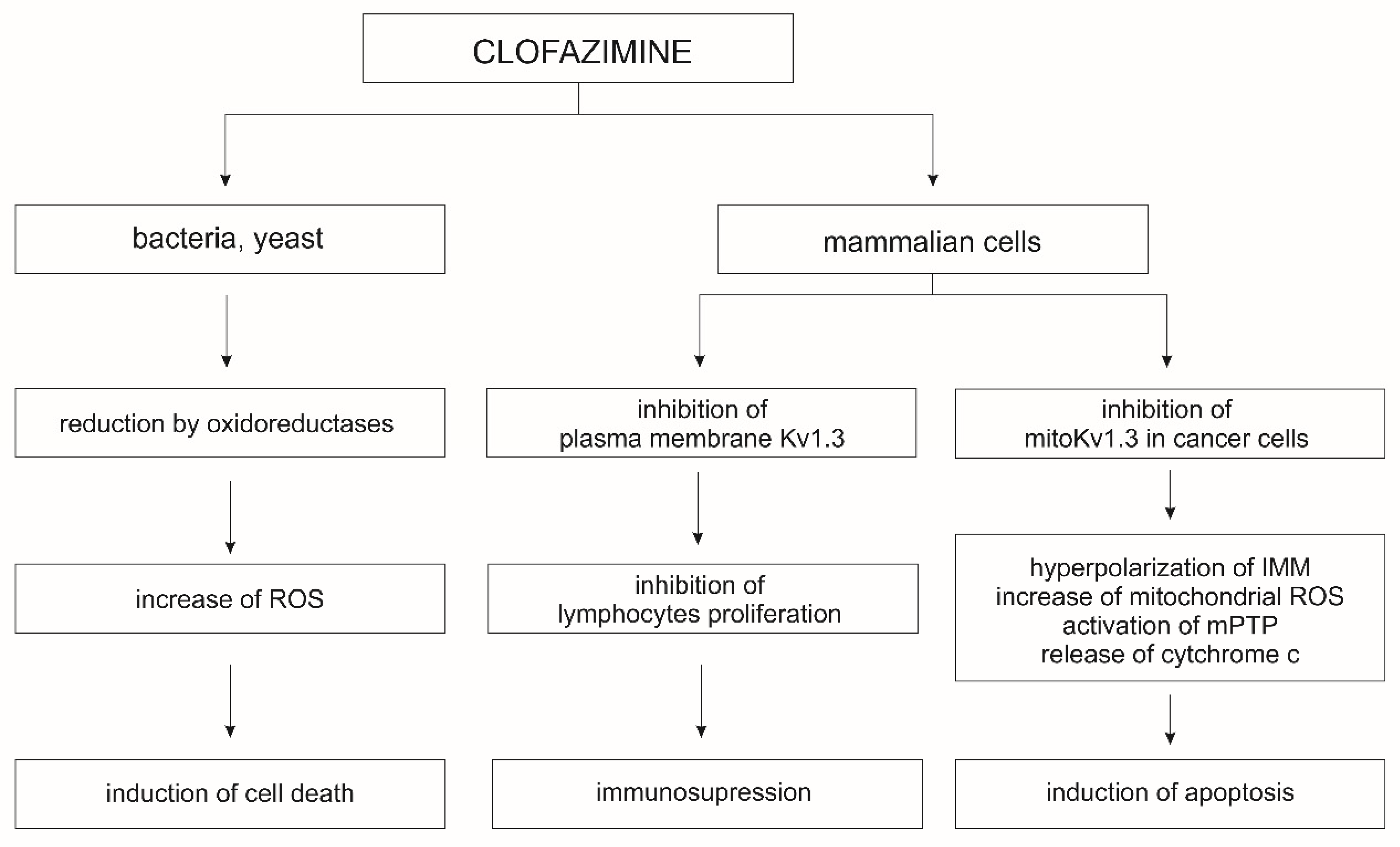
The Kv1.3 and Kv1.5 channels of the plasma membrane play a key role in the regulation of cell proliferation, migration, and differentiation; their inhibition leads to blockage of the cell cycle and cell proliferation [198]. In contrast, the inhibition of mitoKv1.3 and mitoKv1.5 channels, either by the Bax protein or by specific inhibitors, plays a key role in the activation of the apoptotic pathway [195,197]. Other known inhibitors of the mitoKv1.3 channel are clofazimine and the non-phototoxic 5-methoxy-psoralen derivatives, Psora-4 and PAP-1 [199]. Application of MgTx, ShK, or Psora-4, results in changes in membrane potential, synthesis of reactive oxygen species (ROS), and release of cytochrome c from the mitochondrial intramembrane space [200]. Both MgTx and ShK act directly on the mitoKv1.3 channel when applied to mitoplasts. However, these inhibitors are peptides, which limits their permeation of cellular membranes; therefore, they are not useful as mitoKv1.3 channel inhibitors in vivo and in whole-cell studies. The peptide nature of these blockers also limits their ability to block channels in the plasma membrane, and their long-term administration carries the risk of provoking an immune response. This is not the case with psoralen derivatives, such as Psora-4 and PAP-1, and clofazimine, an antibacterial drug used in the treatment of leprosy (Table 3). Clofazimine inhibits the mitoKv1.3 and mitoKv1.5 channels, making it highly effective for studies on cancer treatment. In Vivo studies on the murine B16-F10 orthotopic melanoma model have demonstrated that the application of clofazimine resulted in a 90% reduction in tumor mass (Figure 3) [197].
In mice transplanted with orthotopic human COLO 357 cells, intraperitoneal administration of clofazimine resulted in a 50% reduction in tumor mass [201]. The use of clofazimine also led to apoptosis of B cells in patients suffering from chronic lymphocytic leukemia [199]. Additionally, clofazimine inhibits in vitro Wnt signaling in a wide range of triple-negative breast cancer subtypes that do not respond to hormonal drugs and targeted therapies [202]. Clofazimine also has an immunosuppressive effect that results from the selective blocking of the Kv1.3 channel, which is involved in the proliferation and survival of T effector cells, and, therefore, is a good target for the treatment of autoimmune diseases. Blocking the Kv1.3 channel changes the amplitude and frequency of intracellular Ca2+ oscillation and inhibits the calcineurin/nuclear factor of activated T-cells (NFAT) signaling pathway [203]. Clofazimine interacts with and eventually blocks Kv1.3 channels in a precise and state-dependent manner. It may block open Kv1.3 channels during long depolarizations or block inactivated channels once they have been opened by brief depolarizations. Clofazimine’s mechanism of blocking plasma membrane Kv1.3 channels opens up its possibility for use in the treatment of autoimmune diseases, where the expression of these channels in immune cells is increased [204]. Clofazimine is also a promising drug for the treatment of cryptosporidiosis and nonviral diarrhea in children [205]. In addition, it was effective at preventing skin graft rejection in a mouse transplant model [203]. The use of clofazimine in the treatment of drug-resistant tuberculosis has also been described. The mechanism of action in this case is not entirely clear; however, most likely it acts on Mycobacterium tuberculosis as a pro-drug that is reduced by NADH dehydrogenase, releasing reactive oxygen species upon reoxidation. Clofazimine likely competes with menaquinone (MK-4), a key cofactor in the mycobacterial electron transfer chain, for reduction by NADH [206].
Psora-4 has been shown to bind to the central, highly-conserved pore of Kv channels, and it may also bind to the side pockets formed by the backsides of the S5 and S6 helices and the S4–S5 linker [207]. The simultaneous occupation of both binding sites by the drug results in an extremely stable nonconducting state and increases Psora-4 selectivity for mitoKv1.3 and mitoKv1.5 channels [207]. Due to the inhibition of the plasma membrane Kv1.3 channel, Psora-4 can promote the differentiation and morphological, as well as electrophysiological, maturation of neurons derived from progenitor cells; therefore, it can be used in the treatment of multiple sclerosis [208]. By inhibiting the Kv1.3 channel present on T cells and glomerular infiltrating macrophages, Psora-4 halts rapidly progressive glomerulonephritis and may also be used to treat other autoimmune diseases [209].
PAP-1-MHEG is a recently synthetized PAP-1 derivative that has a higher degree of solubility due to the presence of an oligomeric chain of ethylene glycol. At a concentration of 10 µM, PAP-1-MHEG was able to induce apoptosis more effectively than could PAP-1 in cells isolated from leukemia patients that express mitoKv1.3. PAP-1-MHEG, similar to the other derivatives, blocks the mitoKv1.3 channel through the alkyl-O-phenyl part and causes depolarization of the mitochondrial membrane, which is crucial for the induction of apoptosis. It has also been found that the coumarin ring of PAP-1 and its derivatives acts as a “ROS generator” (Table 3) [210]. However, the unique aspect of PAP-1-MHEG is its ability to block the activity of respiratory chain complex I, via its the methoxy oligoethylene glycol (MOEG) domain, which may interfere with electron transport and the ubiquinone redox cycle. This interference is likely due to the proximity of the mitoKv1.3 channel and the NDUFS1 and NDUFS3 subunits of respiratory chain complex I; however, this requires further research [210]. None of the 5-methoxy-psoralen derivatives (Psora-4, PAP-1, PAPTP, PCARBTP, and PAP-1-MHEG) show any cytotoxic effect in cells with significantly fewer mitoKv1.3 channels, and do not significantly increase the mortality of tumor cells that overexpress mitoKv1.3 channels and have elevated ROS levels [199,210,211].
4.2. Mitochondrial Kv7.4 Channel and Its Modulators
The Kv7 channel subfamily consists of five members named sequentially from Kv7.1 to Kv7.5, encoded by the KCNQ1-5 genes. Each of these channels forms homotetramers, or sometimes heterotetramers, that exhibit different tissue distributions and physiological roles [212]. The Kv7.1 channel is present in the heart, pancreas, gastrointestinal tract, thyroid gland, brain, portal vein, and inner ear. The Kv7.2 and 7.3 channels are mainly expressed in the central nervous system; Kv7.4 is the major voltage-gated potassium channel in the inner ear and bladder smooth muscle; and Kv7.5 is expressed in skeletal muscle and plays a key role in contractility [213].
The presence of mitoKv7.4 channels was demonstrated in the mitochondria of H9c2 cardiomyoblasts and in isolated adult cardiomyocytes. This channel can be activated by retigabine and flupirtine, which induce mitochondrial depolarization and reduce the uptake of calcium ions into the matrix, resulting in cardioprotection against ischemia/reperfusion. This effect was abolished by the Kv7.4 channel inhibitor XE991 [214].
Both flupirtine and retigabine are activators of virtually all types of Kv7 channels, except Kv7.1, and thus have a broad spectrum of action and several side effects (Table 3).
Flupirtine is a triaminopyridine compound that serves as a nonopioid analgesic in the treatment of acute and chronic musculoskeletal pain [215]. It also has muscle relaxant [216] and anticonvulsant properties [217]. Flupirtine activates Kv7.2 and Kv7.3 channels, thus inhibiting the firing of sustained neuronal action potentials. This inhibits the excitability of neurons and reduces the excitatory transmission of neurotransmitters [218].
Flupirtine also acts as an N-methyl-D-aspartate (NMDA) antagonist and can reverse the effects of NMDA receptors, which play a key role in learning, although flupirtine does not bind directly to them. Additionally, flupirtine normalizes the level of intracellular glutathione and increases the expression of the antiapoptotic protein Bcl-2 in neuronal cells exposed to prion proteins. This neuroprotective effect of flupirtine can be used in the treatment of prion diseases [219]. Flupirtine has also been studied in the treatment of tinnitus and overactive bladder [220]. Approximately 8–12% of bioavailable flupirtine in humans is eliminated as a derivative of mercapturic acid by conjugating glutathione with intermediate quinone diimine, which is believed to be responsible for the hepatotoxicity of flupirtine [221].
Retigabine (RTG), also known as ezogabine, is used as an anticonvulsant drug (Table 3) [222]. This potassium channel opener has been approved as an adjunct to treat drug-resistant partial seizures [223]. However, due to its low benefit-to-risk ratio, the use of retigabine was discontinued in 2017, at which time the search for safer and more effective alternatives commenced [212].
RTG binds to Kv7.2-5 channels (encoded by KCNQ2-5) in neurons near the channel gate, which stabilizes the open channel state. The amino acid residues of glycine (G) 301 and tryptophan (W) 236 are crucial for this binding. The absence of W236 in KCNQ1 (which encodes the Kv7.1 channel) explains the lack of RTG binding to that channel [224]. Retigabine and flupirtine have shown antinociceptive effects in a rat model of gout. It is believed that Kv7 channels generate low-threshold, inactivating voltage-gated potassium currents that play an important role in regulating the excitability of nociceptive neurons [225]. To date, the analgesic effect of RTG has been demonstrated in models of pain due to neuroplasticity, bone cancer [226], inflammation [227], nerve degeneration [228], and the application of capsaicin to the viscera [229]. RTG at high concentrations may also result in positive allosteric modulation of γ-aminobutyric acid (GABA) receptors, specifically GABAA, and affect GABA metabolism (at concentrations > 10 µM). At high concentrations (>100 μM) RTG may also be a weak inhibitor of sodium and calcium channels [230]. RTG and ethanol show a similar effect on GABAergic and glutamatergic neurotransmission, which suggests they may interact. RTG reduces the basal rate of dopamine release in a dose-dependent manner in the mesolimbic system, which may be important in the treatment of psychotic diseases [231]. In contrast to RTG, high doses of ethanol increase dopaminergic neurotransmission [232]. Repeated administration of RTG significantly reduces neurological changes in the hippocampus caused by long-term administration of ethanol [233]. RTG also reduces NMDA-induced cell apoptosis in organotypic hippocampal cultures and in models of hypoxia and hypoglycemia [234]. A reduction in memory impairment and a slight improvement in learning post-alcohol consumption were also observed in rats treated with RTG [235]. There are few other reports on the effects of RTG on memory; however, one study has shown that even a single administration of RTG immediately after a traumatic experience can prevent fear memory consolidation and thus prevent posttraumatic stress disorder [236]. Retigabine has shown a sedative effect in studies of mania and bipolar disorder treatment [237]. It also significantly reduces the duration of in vitro myotonia, which causes excessive muscle stiffness, by activating K+ currents during the sequences of action potentials [238]. After cerebral ischemia and reperfusion, there was a reduction in the area of cerebral infarction, tightening of tight connections between the cells of the blood vessel epithelium, reduced permeability of the blood-brain barrier, and reduced levels of MMP-2 and MMP-9 proteins in the ischemic area under the influence of RTG [239]. RTG and flupirtine, due to their oxidizing properties, reduced serum-induced ROS levels in neurons in the dentate gyrus; thus, they showed neuroprotective effects unrelated to the activity of Kv7 channels [234]. At high concentrations (>100 μM), retigabine inhibits Kv1-9 and Kv11 channels; at low concentrations (0.3–3 µM) it also inhibits the Kv2.1 channel. This inhibition is partially irreversible and may account for some of the undesirable effects of RTG application. The neuroprotective effect of RTG in neurons may result from the coordinated action of RTG on the Kv2.1 and Kv7 channels [240].
Table 3.
Examples of modulators of mitoKv channels, their off-target activity, and clinical use.
| Name | Function | Example of Off-Target Activity | Clinical Use | Ref. |
|---|---|---|---|---|
| Flupirtine | Opener | Activator of Kv7.2/Kv7.3 heterotetramer, Kv7.2, Kv7.3 and Kv7.5, NMDA antagonist, normalizes the level of glutathione, increases the expression of Bcl-2. | Approved for treatment of acute and chronic musculoskeletal pain. Anticonvulsant. | [217,218,220,222] |
| Retigabine (ezogabine) | Opener | Activator of Kv7.2/Kv7.3 heterotetramer, Kv7.2, Kv7.3 and Kv7.5, modulator of GABAA receptors, inhibitor of Kv2.1. | Potentially effective painkiller, potentially effective in the treatment of mental and neurodegenerative diseases, approved as an anticonvulsant drug in years 2011–2017. | [222,223,224,226,227,228,230,234,237,239,240] |
| Psora-4 | Inhibitor | Inhibitor of plasma membrane Kv1.3, promotes the differentiation and maturation of neurons. | Potentially effective in the treatment of cancer, autoimmune diseases, and multiple sclerosis. | [199,207,208,209] |
| PAP-1 and PAP-1-MHEG | Inhibitor | Inhibitor of the respiratory chain complex I, induces ROS. | Potentially effective in the treatment of cancer. | [210,211] |
| Clofazimine | Inhibitor | Inhibitor of the calcineurin/NFAT pathway, inhibitor of mycobacterial electron transfer chain. | Potentially effective in the treatment of cancer and autoimmune diseases, approved for treatment of leprosy and drug-resistant tuberculosis. | [202,204,206,209] |
5. Mitochondrial Sodium-Activated Potassium Channels
Sodium-activated potassium channels belonging to the Slo2 (KNa) family have been identified in the mitochondria of cardiac tissue [241,242,243]. Two genes encode Slo2 channels in mammalian cells: KCNT2, which encodes Slo2.1 (also known as Slick) channels, and KCNT1, which encodes Slo2.2 (Slack) channels [244]. The conductance of the Slack channel was found to be approximately 180 pS, while Slick was found to be approximately 140 pS [244]. Structurally, a part of the pore domain and the transmembrane S6 segment of the Slack channel share similarities with Slo1 channels [245]. Interestingly, Slo2 channels can form calcium-sensitive channels after interaction with BKCa channels [244]. The activity of both Slo2.1 and Slo2.2 channels is stimulated by sodium ions [244,246,247]. These channels are also activated by several pharmacological compounds, including bithionol, riluzole, loxapine, and niclosamide [244,247]. Slo2 channels are inhibited by compounds such as bepridil [246], clofilium [248], R56865 [246], and quinidine [244,247] (Table 4). It was found that the activation of mitochondrial Slo2 channels by bithionol induced thallium uptake into isolated mouse cardiac mitochondria as well as mitochondria of C. elegans [243]. The activity of mitoSlo2 channels is inhibited by bepridil. It has been proposed that these channels play an important role in cardioprotection, similar to mitoBKCa and mitoKATP channels [241,242,243,249]. Recently, using the black lipid membrane technique, sodium-activated potassium channels with a conductance of 150 pS were described in brain mitochondria [250]. Sodium ions have been shown to decrease complex I activity and increase complex IV activity in isolated mitochondria. These effects are accompanied by a slight decrease in mitochondrial ROS synthesis and an increase in mitochondrial membrane potential. It was suggested that these effects were related to the activity of the channel. However, no pharmacology specific to Slo2 channels was used in the study [250].
The modulators used to identify and describe mitoSlo2 channels have multiple targets in the cell. For example, bithionol inhibits several enzymes, such as mammalian mitochondrial glutamate dehydrogenase [251,252], human adenylyl cyclase [253], and N-acyl-phosphatidylethanolamine phospholipase D [254]. Bithionol also induces effects at the cellular level. For example, the application of bithionol induced apoptosis in ovarian cancer cells via cell cycle arrest, ROS generation, and inhibition of autotaxin [255,256]. Bithionol has been identified as an apoptosis-inducing photosensitizer for keratinocytes [257]. Its antiseptic and anthelminthic activities have also been studied, e.g., it inhibits the 3-oxoacyl acyl-carrier-protein reductase of Plasmodium falciparum and the large tumor antigen of polyomaviruses [258,259,260].
Another cardiac mitoSlo2 channel opener, bepridil, is a known calcium channel inhibitor (inhibiting channels such as L-type Ca2+ channels) and is used as an antiarrhythmic drug [261]. Additionally, it was shown that bepridil modulates the activity of a few potassium channels. In HEK 293 cells, a low micromolar concentration of bepridil (2–5 μM) inhibited the slow component of cardiac delayed rectifier K+ currents composed of KCNQ1/KCNE1 gene products in a concentration-dependent manner [262]. Additionally, bepridil and R56865 inhibited KATP channels in guinea pig ventricular myocytes, with an estimated IC50 value of 10.5 μM for outward KATP channel currents at a +60 mV holding potential and 6.6 μM for inward currents recorded at −60 mV [246]. Another study showed that 1–10 μM bepridil activated the mitoKATP channels of cardiac mitochondria in guinea pigs and induced cardioprotection against ischemia/reperfusion injury [263].
Clofilium is another modulator of Slo2 channels. This inhibitor can modulate a broad spectrum of channels, including Slo3, Kv1.5 [264], and TASK-2 channels [265], as well as NMDA receptors [266]. NMDA inhibition was observed after the application of 0.1 μM of clofilium [266]. Clofilium can also induce apoptosis in human promyelocytic leukemia cells [267]. Although clofilium has not been used for mitoSlo2 channel modulation, it was shown to play a role in mitochondrial DNA (mtDNA) maintenance, which was detected by an increase in mtDNA copy number in C. elegans and in yeast. A similar effect was observed in POLG-deficient cultured human fibroblasts after the application of 0.5–2.5 μM clofilium [268]. It was concluded that the observed effects were unrelated to the inhibition of potassium channels and that clofilium has a new target in yeast and human cells [268]. Interestingly, the application of clofilium reduced hypoxia-induced neuronal cell death; however, the neuroprotective mechanisms were related to the inhibition of K+ efflux [269].
Table 4.
Examples of mitochondrial sodium-activated potassium channel modulators and their off-target activity.
Table 4.
Examples of mitochondrial sodium-activated potassium channel modulators and their off-target activity.
| Name | Function | Example of Off-Target Activity | Clinical Use | Ref. |
|---|---|---|---|---|
| Bithionol | Opener | Inhibition of mammalian mitochondrial glutamate dehydrogenase, human adenylyl cyclase, and N-acyl-phosphatidylethanolamine phospholipase D. Induces apoptosis of ovarian cancer cells. Apoptosis-inducing photosensitizer for keratinocytes. | Treatment of helminthic infection, inhibits 3-oxoacyl acylcarrier-protein reductase of Plasmodium falciparum and the large tumor antigen of polyomaviruses; inhibits host caspases and also reduces the detrimental effects of anthrax lethal toxin, diphtheria toxin, cholera toxin, Pseudomonas aeruginosa exotoxin A, Botulinum neurotoxin, ricin, and Zika. | [250,251,252,253,254,255,256,257,258,259] |
| Bepridil | Inhibitor | Inhibition of calcium channel. Inhibition of KATP channels in guinea pig ventricular myocytes. | An antiarrhythmic drug. | [261,262,263] |
| Clofilium | Inhibitor | Modulation of activity of Slo3, Kv1.5, and TASK-2 channels and NMDA receptors. Influences mtDNA maintenance. | Potentially useful for treatment of POLG-related diseases. | [267,269,270] |
6. Modulators of Mitochondrial TASK Channels
To date, 15 genes encoding a group of two-pore domain K+ channels (K2P) have been identified in mammalian cells [159,271]. The synthesized proteins are distinguished by a unique subunit architecture with a molecular weight of approximately 70 kDa [270,272,273]. The group of K2P channels has been divided into 7 functional groups: TREK, TRAAK, TALK, TWIK, TASK, THIK, and TRESK. TREK (K2P 2, K2P 10) and TRAAK (K2P 4) activity is regulated by pressure, temperature, lipid environment, and the presence of volatile anesthetics. TALK (K2P 16, K2P 17) is activated in an alkaline environment or by the presence of NO, and TWIK (K2P 7, K2P 1, K2P 6) has weak rectifying properties. TASK (K2P 15, K2P 3, K2P 9) is inhibited in an acidic environment and activated by volatile anesthetics. THIK (K2P 12, K2P 13) is inhibited by halothane; lastly, TRESK (K2P 18) is activated by calcium ions. The structure of the K2P channel includes four transmembrane (4TM) domains, two re-entry pore loop (P) domains, two selectivity filters (SF), and intracellular amine and carboxyl terminals. K2P also includes a so-called cap structure, which plays a special role in its regulation [270]. Two identical K2P subunits form a single, central selective K+ pore, although some K2P subunits may also form heterodimeric complexes in vivo, e.g., TASK-1 and TASK-3 subunits [274,275]. The 4TM domains of the K2P channel family mediate the background potassium currents observed in many cells of bioelectric excitable tissues, such as the heart, brain, muscles, and sensory organs. Interestingly, K2P channels open in the physiological voltage range of the cell membrane and are simultaneously regulated by a number of neurotransmitters and biochemical mediators. It should be noted that, while single functional channels do not have two pores in their structure, each α-subunit has two P domains in its base sequence, hence the name K2P (two-pore domain and not two-porous) channels [270]. The K2P channel family includes the TASK potassium channel subfamily, composed of TASK-1, TASK-3, and TASK-5 [276,277], all of which display K+ outwardly rectifying currents that do not depend on membrane voltage. TASK-1 and TASK-3-mediated currents are highly sensitive to extracellular pH [277,278,279]. TASK-5 has no functional expression [280]. The TASK-3 channel (K2P9.1, KCNK9, 8q24) was discovered more than 20 years ago in the plasma membrane of mammalian cells. Since then, TASK-3 potassium channels have also been identified in the inner mitochondrial membrane (mitoTASK-3) of melanocytes, melanomas (WM35 and B16F10), and keratinocytes [6,281,282,283]. The mitoTASK-3 channel was detected in the human keratinocyte HaCaT cell line, with a channel conductance of 83 pS at positive voltages and 12 pS at negative voltages in symmetric 150 mM KCl, as measured by the single-channel patch clamp technique. Lidocaine and an acidic pH (<6.2) have been shown to block channel activity. The mitoTASK-3 channel, similar to its plasma membrane version, is also sensitive to an acidic pH [282,283]. Silencing TASK-3 gene expression leads to changes in mitochondrial structure and induces apoptosis in human melanoma cells [284]. It seems particularly interesting that thus far, only the TASK-3 channel from the K2P channel group has been identified in the inner mitochondrial membrane. The apparent lack of other K2P channels in the IMM to date is all the more curious because, as mentioned above, TASK-3 and TASK-1 can form a functional heterodimer of potassium ion transporting channels [274,275]. This may be due to the tissue-specific localization of the TASK-1 and TASK-3 channels. TASK-3-like channels have been located in the mitochondria of the aldosterone-producing layer of the adrenal cortex zona glomerulosa cells [285]. It was shown that knockdown of the TASK-3 gene caused changes in migration and cell survival in gastric cancer, and that the TASK-3 gene could be a potential target for gastric cancer treatment; however, it was not possible to distinguish the role of mitoTASK-3 in addition to the plasma membrane isoform [286]. The role of mitochondrial potassium channels in cancer development was discussed in a recent review [287]. Studies have also shown that overexpression of TASK-3 channels occurs in several types of cancer, such as melanoma, ovarian carcinoma, and breast cancer [281,282,288,289,290,291]. One of the major problems in distinguishing the role of mitochondrial potassium channels from those located in the plasma membrane is their molecular identity or close similarity. Nevertheless, despite the extensive structural similarity of K2P channels, there are differences in the interaction of small molecule compounds among these channels, which may also explain small structural differences as well as why only TASK-3 is located in the IMM [270]. Unfortunately, there are only a few known small-molecule compounds that modulate the activity of the TASK-3 channel [292]. To date, the verified modulators of TASK-3 channels include: the recently described plant-derived compound withaferin A (known in traditional medicine for centuries) [293], an inhibitor based on THPP (5,6,7,8-tetrahydropyrido [4,3-d]pyrimidine) [292] and its recently synthesized derivatives IN-THPP and mitoIN-THPP [294], ruthenium red (which has an IC50 of 0.7 μM) [295], and DR16 and DR16.1, which can penetrate biological membranes [276].
Recently, a study by Zúñiga et al. showed that withaferin A, the active biological component of Withania somnifera plant extract, has properties that inhibit the activity of the TASK-3 channel on the cell membrane (Table 5) [293]. In light of recent studies, this is an extremely interesting observation, as the expression of the KCNK9 gene for TASK-3 channels is increased in human breast tumors and lung tumors [295] and in 90% of ovarian tumors [288]. Despite the demonstration of a direct effect of withaferin A on the activity of the TASK-3 channel, it should be noted that it is still unclear whether it only affects the channel located in the plasma membrane or also that in the IMM. Withaferin A also acts on cellular pathways, such as cell cycle arrest [296,297], inhibits apoptosis via activated Akt-mediated inhibition of oxidative stress and ROS-dependent mitochondrial dysfunction in human colorectal cancer cells [298,299], and inhibits endoplasmic reticulum stress-associated apoptosis [300]. Low doses of withaferin A are cardioprotective in ischemia/reperfusion events via upregulation of the antiapoptotic mitochondrial pathway in an AMPK-dependent manner [301]. Another group of recently-synthetized inhibitors of TASK-3 channel activity is the 5,6,7,8-tetrahydropyrido [4,3-d]pyrimidine (THPP)-derived compounds IN-THPP and mitoIN-THPP, which differs by the addition of a triphenylphosphonium (TPP) group [294]. Because of its positive charge, the TPP group directs the mitoIN-THPP compound to the IMM. MitoIN-THPP is thus able to inhibit cancer cell migration to a higher extent than IN-THPP, which suggests that mitoTASK-3 is important in cell melanoma cell survival. It should be mentioned that after accumulation in the IMM, mitoIN-THPP is hydrolyzed to IN-THPP-COOH (the active molecule) and TPP-propyl-OH, which is itself unable to trigger apoptosis [292]. Ruthenium red and Ru360, other potential inhibitors of the TASK-3 potassium channel, are only useful for measurements in isolated TASK-3 channels systems because of their broad spectrum of interactions with different proteins in the cell, e.g., ryanodine receptors and mitochondrial calcium uniporter (MCU), respectively [302,303,304,305]. Because ruthenium red and Ru360 are known compounds that disturb intracellular calcium homeostasis, it is difficult to examine the role of TASK-3 and mitoTASK-3 in cellular processes [295,304,306,307,308]. Two novel TASK-3 channel inhibitors (DR16 and DR16.1) were found using virtual screening, and their inhibitory potency was tested using the whole cell patch clamp technique. DR16 had an IC50 of 56.8 μM, and DR16.1 had an IC50 of 14.2 μM. DR16 and DR16.1 also have inhibitory potency for the TASK-1 channel, with an IC50 of 24.7 μM and IC50 of 21.21 μM, respectively [276]. It is worth noting that these compounds are not able to distinguish between TASK-1 and TASK-3, nor between those located in the plasma membrane and IMM.
7. Pharmacology of Mitochondrial HCN Channels
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are nonselective cation channels encoded by four genes, HCN-1,-2,-3, and -4, located on chromosomes 5, 19, 1, and 15, respectively, in humans. HCN channels belong to the superfamily of channels with six transmembrane segments [311]. Functional channels result from the assembly of four subunits, which together form a centralized pore that regulates ion flow across the membrane. Each subunit has several domains: a transmembrane voltage-sensing domain, a transmembrane pore-forming domain, a cytoplasmic C-linker, and a cytoplasmic cyclic nucleotide-binding domain (CNBD), which is critical for the modulation of the channels by cyclic nucleotides [312,313]. HCN channels also have an auxiliary TRIP8b subunit (Rab8b-interacting protein), which acts as an antagonist of cAMP. In the presence of TRIP8b, HCN channels only open at more hyperpolarized potentials [314]. The cryo-EM structures of the human HCN1 channel (hHCN1) have been determined in the absence and in the presence of cAMP, a physiological ligand of HCN channels [184].
HCN channels are mainly expressed throughout the heart and the central nervous system. HCN1 is highly expressed in the neocortex, hippocampus, cerebellar cortex, brainstem, and spinal cord. HCN2 is especially abundant in thalamic and brainstem nuclei. The expression of HCN3 is relatively modest, scattered throughout the brain. HCN4 is expressed in the olfactory bulb and the thalamus. All HCN subunits are expressed in the peripheral nervous system [315]. In the heart, all four isoforms have been detected and were differentially expressed according to the cardiac region. HCN2 and HCN4 are the most abundant isoforms in most mammalian atria and ventricles [316], which also display a minor presence of HCN1. HCN3 is expressed at a low level throughout cardiac regions. There is also evidence for the expression of HCN channels in cells outside of the nervous system and heart, such as the kidneys [317], pancreas [318], and bladder [319]. All HCN isoforms, except HCN3, are present in the retina [320]. HCN channels are localized mainly in the plasma membrane, although they have also been detected in the mitochondria of rat kidneys, human HEK 293 cells [321], and human cardiac tissue [321].
HCN channels are voltage-gated, and, in contrast to most other voltage-gated Kv channels, they are activated by membrane hyperpolarization [322]. HCN channels are permeable to both Na+ and K+ ions but are weakly selective for potassium compared with other voltage-gated Kv channels [323]. Functionally, they differ from each other in terms of activation time. HCN1 channels are the fastest; HCN4 channels are the slowest; and HCN2 and HCN3 are intermediate. In the case of physiological modulators, HCN channels are activated by direct binding of cyclic nucleotides, including cyclic monophosphate nucleotides, such as cAMP, cGMP, and cCMP. There is also evidence that HCN channels are regulated by changes in intracellular pH [155], cholesterol [324], phosphatidylinositol-4,5-bisphosphate [64], and caveolin 3 [33].
Dysfunction of HCN channels has been implicated in heart arrhythmogenic diseases and nervous system diseases, such as pain disorders, epilepsy, and ataxia [325,326,327]. Many pharmacological compounds influencing the activity of HCN channels affect not only the HCN channels of the cell membrane themselves, but also the mitochondrial HCN channels (Table 6). Unfortunately, some of them also have off-targets. One example is the bradycardic drug ZD7288. Experiments on isolated mitochondria showed that ZD7288 inhibits the flow of ions through HCN channels not only in the heart but also in the kidneys [321]. Additionally, ZD7288 was shown to reduce oxygen consumption coupled to ATP synthesis and hyperpolarization of the inner mitochondrial membrane [321]. ZD7288 may also directly block T-type Ca2+ channels in mouse spermatogenic cells [328] and the sodium channel NaV1.4 in dorsal root ganglion [329], and can cause a robust depression of mossy fiber basal synaptic transmission [330]. Another HCN drug, ivabradine, also seems to be targeted to mitochondria. After ischemia/reperfusion mitochondrial stimulation, ivabradine reduced mitochondrial ROS formation and increased mitochondrial ATP production [331]. Ivabradine, a member of benzazepines, was the first clinically approved HCN channel blocker and is used to decrease heart rate. Ivabradine binds to the HCN channel pore and blocks HCN channels in pacemaker cells within the sinoatrial node; however, it is not specific to HCN isoforms. More specific to HCN channel isoforms are the modulators zatebradine and EC18 for HCN4 channels, MEL55A for HCN1 and HCN2 channels, and MEL57A for HCN1 channels. Additionally, another HCN channel modulator, gabapentin (an anesthetic, analgesic, and antiepileptic drug), reduces HCN4 channel-mediated currents in neurons [332] and inhibits mitochondrial branched-chain aminotransferase [333], opening mitochondrial ATP-dependent potassium channels and blocking voltage-gated calcium channels in a mouse model [334]. Lamotrigine, an HCN channel agonist (and anticonvulsant drug) may also block T-type Ca2+ channels [335].
8. Summary and Perspectives
Over the last dozen years, emerging research on potassium channels clearly indicates their involvement in both protective processes and processes influencing cell death. It seems that one of the targets for the regulation of both cellular protection and apoptotic and necrotic processes is the potassium channels found in the inner mitochondrial membrane. A well-documented phenomenon is a protection of heart muscle cells from I/R injury with the use of small-molecule chemical compounds and potassium channel activators of the IMM, e.g., diazoxide and NS1619. Unfortunately, due to the presence of the same types of potassium channels in both the plasma membrane and the IMM, it is extremely difficult to distinguish their participation in both protective and cytotoxic processes. Additionally, the knockdown of the genes responsible for the expression of individual isoforms does not give the desired results, because potassium channels are usually expressed by the same genes, and the difference may result from alternative splicing. Small-molecule chemical compounds modulating the activity of the potassium ion channels in the plasma membrane also modulate the activity of those in the IMM. Although diazoxide acts through an auxiliary subunit, it exhibits the concentration differences necessary to activate a channel in the plasma membrane as well as that in the IMM. In light of recent studies, it seems extremely important to find ways to regulate the expression or activity of the channel only in the IMM, which could protect heart cells in the processes of ischemia/reperfusion or to induce apoptotic processes in cancer cells. Examining the side effects of small-molecule potassium channel modulators may also reveal synergistic pathways that regulate cytoprotection and cellular cytotoxicity, in addition to modulating potassium channels in the IMM.
Author Contributions
Conceptualization, A.W., M.Ż., and B.K.; writing, A.W., S.G., A.O., M.Ż., and B.K.; supervision, A.W. and B.K.; funding acquisition, B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Polish National Science Center, grant No. 2015/18/E/NZ1/00737 to BK.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Szabo, I.; Zoratti, M.; Biasutto, L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. 2021, 42, 101846. [Google Scholar] [CrossRef]
- Rotko, D.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Signaling pathways targeting mitochondrial potassium channels. Int. J. Biochem. Cell. Biol. 2020, 125, 105792. [Google Scholar] [CrossRef]
- Kulawiak, B.; Bednarczyk, P.; Szewczyk, A. Multidimensional Regulation of Cardiac Mitochondrial Potassium Channels. Cells 2021, 10, 1554. [Google Scholar] [CrossRef]
- Szewczyk, A.; Bednarczyk, P.; Jedraszko, J.; Kampa, R.P.; Koprowski, P.; Krajewska, M.; Kucman, S.; Kulawiak, B.; Laskowski, M.; Rotko, D.; et al. Mitochondrial potassium channels—An overview. Postepy Biochem. 2018, 64, 196–212. [Google Scholar] [CrossRef]
- Urbani, A.; Prosdocimi, E.; Carrer, A.; Checchetto, V.; Szabo, I. Mitochondrial Ion Channels of the Inner Membrane and Their Regulation in Cell Death Signaling. Front. Cell. Dev. Biol. 2020, 8, 620081. [Google Scholar] [CrossRef]
- Checchetto, V.; Leanza, L.; De Stefani, D.; Rizzuto, R.; Gulbins, E.; Szabo, I. Mitochondrial K+ channels and their implications for disease mechanisms. Pharmacol. Ther. 2021, 227, 107874. [Google Scholar] [CrossRef]
- Inoue, I.; Nagase, H.; Kishi, K.; Higuti, T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991, 352, 244–247. [Google Scholar] [CrossRef]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar] [CrossRef]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Choma, K.; Bednarczyk, P.; Koszela-Piotrowska, I.; Kulawiak, B.; Kudin, A.; Kunz, W.S.; Dolowy, K.; Szewczyk, A. Single channel studies of the ATP-regulated potassium channel in brain mitochondria. J. Bioenergy Biomembr. 2009, 41, 323–334. [Google Scholar] [CrossRef]
- Debska, G.; May, R.; Kicinska, A.; Szewczyk, A.; Elger, C.E.; Kunz, W.S. Potassium channel openers depolarize hippocampal mitochondria. Brain Res. 2001, 892, 42–50. [Google Scholar] [CrossRef]
- Bajgar, R.; Seetharaman, S.; Kowaltowski, A.J.; Garlid, K.D.; Paucek, P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J. Biol. Chem. 2001, 276, 33369–33374. [Google Scholar] [CrossRef] [Green Version]
- Debska, G.; Kicinska, A.; Skalska, J.; Szewczyk, A.; May, R.; Elger, C.E.; Kunz, W.S. Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim. Biophys. Acta 2002, 1556, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Dahlem, Y.A.; Horn, T.F.; Buntinas, L.; Gonoi, T.; Wolf, G.; Siemen, D. The human mitochondrial KATP channel is modulated by calcium and nitric oxide: A patch-clamp approach. Biochim. Biophys. Acta 2004, 1656, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk, P.; Kicinska, A.; Laskowski, M.; Kulawiak, B.; Kampa, R.; Walewska, A.; Krajewska, M.; Jarmuszkiewicz, W.; Szewczyk, A. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochim. Biophys. Acta Bioenergy 2018, 1859, 309–318. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Kicinska, A.; Kominkova, V.; Ondrias, K.; Dolowy, K.; Szewczyk, A. Quinine inhibits mitochondrial ATP-regulated potassium channel from bovine heart. J. Membr. Biol. 2004, 199, 63–72. [Google Scholar] [CrossRef]
- Kravenska, Y.; Checchetto, V.; Szabo, I. Routes for Potassium Ions across Mitochondrial Membranes: A Biophysical Point of View with Special Focus on the ATP-Sensitive K+ Channel. Biomolecules 2021, 11, 1172. [Google Scholar] [CrossRef]
- Lacza, Z.; Snipes, J.A.; Miller, A.W.; Szabo, C.; Grover, G.; Busija, D.W. Heart mitochondria contain functional ATP-dependent K+ channels. J. Mol. Cell. Cardiol. 2003, 35, 1339–1347. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Bednarczyk, P.; Zochowska, M.; Kalisz, J.; O’Rourke, B.; Szewczyk, A.; Kulawiak, B. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. Int. J. Mol. Sci. 2019, 20, 5323. [Google Scholar] [CrossRef] [Green Version]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP synthase K+-and H+-flux drive ATP synthesis and enable mitochondrial K+-uniporter function. bioRxiv 2019, 355776. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Testai, L.; Rapposelli, S.; Martelli, A.; Breschi, M.C.; Calderone, V. Mitochondrial potassium channels as pharmacological target for cardioprotective drugs. Med. Res. Rev. 2015, 35, 520–553. [Google Scholar] [CrossRef]
- Pereira, O.; Kowaltowski, A.J. Mitochondrial K+ Transport: Modulation and Functional Consequences. Molecules 2021, 26, 2935. [Google Scholar] [CrossRef]
- Szewczyk, A.; Kajma, A.; Malinska, D.; Wrzosek, A.; Bednarczyk, P.; Zablocka, B.; Dolowy, K. Pharmacology of mitochondrial potassium channels: Dark side of the field. FEBS Lett. 2010, 584, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, A.; Skalska, J.; Glab, M.; Kulawiak, B.; Malinska, D.; Koszela-Piotrowska, I.; Kunz, W.S. Mitochondrial potassium channels: From pharmacology to function. Biochim. Biophys. Acta 2006, 1757, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Sun, X.; Schindler, P.A. The mitochondrial KATP channel as a receptor for potassium channel openers. J. Biol. Chem. 1996, 271, 8796–8799. [Google Scholar] [CrossRef] [Green Version]
- Busija, D.W.; Katakam, P.; Rajapakse, N.C.; Kis, B.; Grover, G.; Domoki, F.; Bari, F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res. Bull. 2005, 66, 85–90. [Google Scholar] [CrossRef]
- Kim, M.Y.; Kim, M.J.; Yoon, I.S.; Ahn, J.H.; Lee, S.H.; Baik, E.J.; Moon, C.H.; Jung, Y.S. Diazoxide acts more as a PKC-e activator, and indirectly activates the mitochondrial KATP channel conferring cardioprotection against hypoxic injury. Br. J. Pharmacol. 2006, 149, 1059–1070. [Google Scholar] [CrossRef]
- Grover, G.J.; Atwal, K.S. Pharmacologic profile of the selective mitochondrial-KATP opener BMS-191095 for treatment of acute myocardial ischemia. Cardiovasc. Drug Rev. 2002, 20, 121–136. [Google Scholar] [CrossRef]
- Grover, G.J.; D’Alonzo, A.J.; Garlid, K.D.; Bajgar, R.; Lodge, N.J.; Sleph, P.G.; Darbenzio, R.B.; Hess, T.A.; Smith, M.A.; Paucek, P.; et al. Pharmacologic characterization of BMS-191095, a mitochondrial KATP opener with no peripheral vasodilator or cardiac action potential shortening activity. J. Pharmacol. Exp. Ther. 2001, 297, 1184–1192. [Google Scholar]
- Bednarczyk, P.; Dolowy, K.; Szewczyk, A. New properties of mitochondrial ATP-regulated potassium channels. J. Bioenergy Biomembr. 2008, 40, 325–335. [Google Scholar] [CrossRef]
- Ahmad, N.; Wang, Y.; Haider, K.H.; Wang, B.; Pasha, Z.; Uzun, O.; Ashraf, M. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2402–H2408. [Google Scholar] [CrossRef] [Green Version]
- Moses, M.A.; Addison, P.D.; Neligan, P.C.; Ashrafpour, H.; Huang, N.; Zair, M.; Rassuli, A.; Forrest, C.R.; Grover, G.J.; Pang, C.Y. Mitochondrial KATP channels in hindlimb remote ischemic preconditioning of skeletal muscle against infarction. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H559–H567. [Google Scholar] [CrossRef] [Green Version]
- Kis, B.; Nagy, K.; Snipes, J.A.; Rajapakse, N.C.; Horiguchi, T.; Grover, G.J.; Busija, D.W. The mitochondrial KATP channel opener BMS-191095 induces neuronal preconditioning. Neuroreport 2004, 15, 345–349. [Google Scholar] [CrossRef]
- Katakam, P.V.; Dutta, S.; Sure, V.N.; Grovenburg, S.M.; Gordon, A.O.; Peterson, N.R.; Rutkai, I.; Busija, D.W. Depolarization of mitochondria in neurons promotes activation of nitric oxide synthase and generation of nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1097–H1106. [Google Scholar] [CrossRef] [Green Version]
- Mayanagi, K.; Gaspar, T.; Katakam, P.V.; Kis, B.; Busija, D.W. The mitochondrial KATP channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2007, 27, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, T.; Snipes, J.A.; Busija, A.R.; Kis, B.; Domoki, F.; Bari, F.; Busija, D.W. ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095. J. Cereb. Blood Flow Metab. 2008, 28, 1090–1103. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.R.; Park, J.W.; Jung, I.S.; Yi, K.Y.; Yoo, S.E.; Chung, H.J.; Yun, Y.P.; Kwon, S.H.; Shin, H.S. BMS-191095, a cardioselective mitochondrial KATP opener, inhibits human platelet aggregation by opening mitochondrial KATP channels. Arch. Pharm. Res. 2005, 28, 61–67. [Google Scholar] [CrossRef]
- Malinska, D.; Kulawiak, B.; Wrzosek, A.; Kunz, W.S.; Szewczyk, A. The cytoprotective action of the potassium channel opener BMS-191095 in C2C12 myoblasts is related to the modulation of calcium homeostasis. Cell Physiol. Biochem. 2010, 26, 235–246. [Google Scholar] [CrossRef]
- Sato, T.; Sasaki, N.; O’Rourke, B.; Marbán, E. Nicorandil, a potent cardioprotective agent, acts by opening mitochondrial ATP-dependent potassium channels. J. Am. Coll. Cardiol. 2000, 35, 514–518. [Google Scholar] [CrossRef]
- Sanchez-Duarte, E.; Trujillo, X.; Cortes-Rojo, C.; Saavedra-Molina, A.; Camargo, G.; Hernandez, L.; Huerta, M.; Montoya-Perez, R. Nicorandil improves post-fatigue tension in slow skeletal muscle fibers by modulating glutathione redox state. J. Bioenergy Biomembr. 2017, 49, 159–170. [Google Scholar] [CrossRef]
- Tsuchida, A.; Miura, T.; Tanno, M.; Sakamoto, J.; Miki, T.; Kuno, A.; Matsumoto, T.; Ohnuma, Y.; Ichikawa, Y.; Shimamoto, K. Infarct size limitation by nicorandil. J. Am. Coll. Cardiol. 2002, 40, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y. Cardiac Na+/Ca2+ exchange stimulators among cardioprotective drugs. J. Physiol. Sci. 2019, 69, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Taira, N. Nicorandil as a hybrid between nitrates and potassium channel activators. Am. J. Cardiol. 1989, 63, 18J–24J. [Google Scholar] [CrossRef]
- Mano, T.; Shinohara, R.; Nagasaka, A.; Nakagawa, H.; Uchimura, K.; Hayashi, R.; Nakano, I.; Tsugawa, T.; Watanabe, F.; Kobayashi, T.; et al. Scavenging effect of nicorandil on free radicals and lipid peroxide in streptozotocin-induced diabetic rats. Metabolism 2000, 49, 427–431. [Google Scholar] [CrossRef]
- Serizawa, K.; Yogo, K.; Aizawa, K.; Tashiro, Y.; Ishizuka, N. Nicorandil prevents endothelial dysfunction due to antioxidative effects via normalisation of NADPH oxidase and nitric oxide synthase in streptozotocin diabetic rats. Cardiovasc. Diabetol. 2011, 10, 105. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Duarte, E.; Cortes-Rojo, C.; Sanchez-Briones, L.A.; Campos-Garcia, J.; Saavedra-Molina, A.; Delgado-Enciso, I.; Lopez-Lemus, U.A.; Montoya-Perez, R. Nicorandil Affects Mitochondrial Respiratory Chain Function by Increasing Complex III Activity and ROS Production in Skeletal Muscle Mitochondria. J. Membr. Biol. 2020, 253, 309–318. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Reiter, M.; Gastonguay, C.; McGivern, J.V.; Guan, X.; Ge, Z.D.; Mack, D.L.; Childers, M.K.; Ebert, A.D.; Strande, J.L. Nicorandil, a Nitric Oxide Donor and ATP-Sensitive Potassium Channel Opener, Protects Against Dystrophin-Deficient Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.H.; Hong, H.J.; Cheng, T.H.; Shih, N.L.; Loh, S.H.; Liu, J.C.; Chen, J.J.; Sung, L.C. Nicorandil Inhibits Cyclic Strain-Induced Interleukin-8 Expression in Human Umbilical Vein Endothelial Cells. Pharmacology 2016, 98, 42–50. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Brookes, P.S. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 2009, 104, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Wojtovich, A.P.; Brookes, P.S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim. Biophys. Acta 2008, 1777, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Rapedius, M.; Baukrowitz, T.; Liu, G.X.; Srivastava, D.K.; Daut, J.; Hanley, P.J. 5-Hydroxydecanoate and coenzyme A are inhibitors of native sarcolemmal KATP channels in inside-out patches. Biochim. Biophys. Acta 2010, 1800, 385–391. [Google Scholar] [CrossRef]
- Hanley, P.J.; Gopalan, K.V.; Lareau, R.A.; Srivastava, D.K.; von Meltzer, M.; Daut, J. Beta-oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. J. Physiol. 2003, 547, 387–393. [Google Scholar] [CrossRef]
- Hanley, P.J.; Drose, S.; Brandt, U.; Lareau, R.A.; Banerjee, A.L.; Srivastava, D.K.; Banaszak, L.J.; Barycki, J.J.; Van Veldhoven, P.P.; Daut, J. 5-Hydroxydecanoate is metabolised in mitochondria and creates a rate-limiting bottleneck for beta-oxidation of fatty acids. J. Physiol. 2005, 562, 307–318. [Google Scholar] [CrossRef]
- Drose, S.; Brandt, U.; Hanley, P.J. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J. Biol. Chem. 2006, 281, 23733–23739. [Google Scholar] [CrossRef] [Green Version]
- Ziemys, A.; Toleikis, A.; Kopustinskiene, D.M. Molecular modelling of KATP channel blockers-ADP/ ATP carrier interactions. Syst. Biol. 2006, 153, 390–393. [Google Scholar] [CrossRef]
- Skalska, J.; Debska, G.; Kunz, W.S.; Szewczyk, A. Antidiabetic sulphonylureas activate mitochondrial permeability transition in rat skeletal muscle. Br. J. Pharmacol. 2005, 145, 785–791. [Google Scholar] [CrossRef]
- Tominaga, M.; Horie, M.; Sasayama, S.; Okada, Y. Glibenclamide, an ATP-sensitive K+ channel blocker, inhibits cardiac cAMP-activated Cl- conductance. Circ. Res. 1995, 77, 417–423. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Matsuura, H.; Ehara, T. Swelling-induced Cl- current in guinea-pig atrial myocytes: Inhibition by glibenclamide. J. Physiol. 1997, 505 Pt 1, 41–52. [Google Scholar] [CrossRef]
- Swale, D.R.; Sheehan, J.H.; Banerjee, S.; Husni, A.S.; Nguyen, T.T.; Meiler, J.; Denton, J.S. Computational and functional analyses of a small-molecule binding site in ROMK. Biophys. J. 2015, 108, 1094–1103. [Google Scholar] [CrossRef] [Green Version]
- Weaver, C.D.; Denton, J.S. Next-generation inward rectifier potassium channel modulators: Discovery and molecular pharmacology. Am. J. Physiol. Cell. Physiol. 2021, 320, C1125–C1140. [Google Scholar] [CrossRef]
- Bhave, G.; Chauder, B.A.; Liu, W.; Dawson, E.S.; Kadakia, R.; Nguyen, T.T.; Lewis, L.M.; Meiler, J.; Weaver, C.D.; Satlin, L.M.; et al. Development of a selective small-molecule inhibitor of Kir1.1, the renal outer medullary potassium channel. Mol. Pharmacol. 2011, 79, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Papanicolaou, K.N.; Ashok, D.; Liu, T.; Bauer, T.M.; Sun, J.; Li, Z.; da Costa, E.; D’Orleans, C.C.; Nathan, S.; Lefer, D.J.; et al. Global knockout of ROMK potassium channel worsens cardiac ischemia-reperfusion injury but cardiomyocyte-specific knockout does not: Implications for the identity of mitoKATP. J. Mol. Cell. Cardiol. 2020, 139, 176–189. [Google Scholar] [CrossRef]
- Siemen, D.; Loupatatzis, C.; Borecky, J.; Gulbins, E.; Lang, F. Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 1999, 257, 549–554. [Google Scholar] [CrossRef]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell. Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef]
- Dolga, A.M.; Netter, M.F.; Perocchi, F.; Doti, N.; Meissner, L.; Tobaben, S.; Grohm, J.; Zischka, H.; Plesnila, N.; Decher, N.; et al. Mitochondrial Small Conductance SK2 Channels Prevent Glutamate-induced Oxytosis and Mitochondrial Dysfunction*. J. Biol. Chem. 2013, 288, 10792–10804. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef]
- Douglas, R.M.; Lai, J.C.K.; Bian, S.; Cummins, L.; Moczydlowski, E.; Haddad, G.G. The calcium-sensitive large-conductance potassium channel (BK/MAXI K) is present in the inner mitochondrial membrane of rat brain. Neuroscience 2006, 139, 1249–1261. [Google Scholar] [CrossRef]
- Skalska, J.; Bednarczyk, P.; Piwońska, M.; Kulawiak, B.; Wilczynski, G.; Dołowy, K.; Kunz, W.; Kudin, A.; Szewczyk, A. Calcium Ions Regulate K+ Uptake into Brain Mitochondria: The Evidence for a Novel Potassium Channel. Int. J. Mol. Sci. 2009, 10, 1104–1120. [Google Scholar] [CrossRef] [Green Version]
- Skalska, J.; Piwońska, M.; Wyroba, E.; Surmacz, L.; Wieczorek, R.; Koszela-Piotrowska, I.; Zielińska, J.; Bednarczyk, P.; Dołowy, K.; Wilczynski, G.M.; et al. A novel potassium channel in skeletal muscle mitochondria. Biochim. Biophys. Acta Bioenergy 2008, 1777, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk, P.; Koziel, A.; Jarmuszkiewicz, W.; Szewczyk, A. Large-conductance Ca2+-activated potassium channel in mitochondria of endothelial EA.hy926 cells. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1415–H1427. [Google Scholar] [CrossRef]
- Kicinska, A.; Augustynek, B.; Kulawiak, B.; Jarmuszkiewicz, W.; Szewczyk, A.; Bednarczyk, P. A large-conductance calcium-regulated K+ channel in human dermal fibroblast mitochondria. Biochem. J. 2016, 473, 4457–4471. [Google Scholar] [CrossRef]
- Shrum, S.; Rusch, N.J.; MacMillan-Crow, L.A. Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage-Induced Mitochondrial Injury In Vitro. Biomolecules 2019, 9, 825. [Google Scholar] [CrossRef] [Green Version]
- Sek, A.; Kampa, R.P.; Kulawiak, B.; Szewczyk, A.; Bednarczyk, P. Identification of the Large-Conductance Ca2+-Regulated Potassium Channel in Mitochondria of Human Bronchial Epithelial Cells. Molecules 2021, 26, 3233. [Google Scholar] [CrossRef]
- Laskowski, M.; Kicinska, A.; Szewczyk, A.; Jarmuszkiewicz, W. Mitochondrial large-conductance potassium channel from Dictyostelium discoideum. Int. J. Biochem. Cell. Biol. 2015, 60, 167–175. [Google Scholar] [CrossRef]
- Jarmuszkiewicz, W.; Matkovic, K.; Koszela-Piotrowska, I. Potassium channels in the mitochondria of unicellular eukaryotes and plants. FEBS Lett. 2010, 584, 2057–2062. [Google Scholar] [CrossRef] [Green Version]
- Matkovic, K.; Koszela-Piotrowska, I.; Jarmuszkiewicz, W.; Szewczyk, A. Ion conductance pathways in potato tuber (Solanum tuberosum) inner mitochondrial membrane. Biochim. Biophys. Acta 2011, 1807, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sanabria, N.; Echeverria, F.; Segura, I.; Alvarado-Sanchez, R.; Latorre, R. BK in Double-Membrane Organelles: A Biophysical, Pharmacological, and Functional Survey. Front. Physiol. 2021, 12, 761474. [Google Scholar] [CrossRef]
- Sakai, Y.; Harvey, M.; Sokolowski, B. Identification and quantification of full-length BK channel variants in the developing mouse cochlea. J. Neurosci. Res. 2011, 89, 1747–1760. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Lu, R.; Bopassa, J.C.; Meredith, A.L.; Stefani, E.; Toro, L. MitoBKCa is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef] [Green Version]
- Gałecka, S.; Kulawiak, B.; Bednarczyk, P.; Singh, H.; Szewczyk, A. Single channel properties of mitochondrial large conductance potassium channel formed by BK-VEDEC splice variant. Sci. Rep. 2021, 11, 10925. [Google Scholar] [CrossRef]
- Jaggar, J.H.; Li, A.; Parfenova, H.; Liu, J.; Umstot, E.S.; Dopico, A.M.; Leffler, C.W. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 2005, 97, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.D.; Xu, R.; Reynolds, M.F.; Garcia, M.L.; Heinemann, S.H.; Hoshi, T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 2003, 425, 531–535. [Google Scholar] [CrossRef]
- Augustynek, B.; Kudin, A.P.; Bednarczyk, P.; Szewczyk, A.; Kunz, W.S. Hemin inhibits the large conductance potassium channel in brain mitochondria: A putative novel mechanism of neurodegeneration. Exp. Neurol. 2014, 257, 70–75. [Google Scholar] [CrossRef]
- Brazier, S.P.; Telezhkin, V.; Mears, R.; Muller, C.T.; Riccardi, D.; Kemp, P.J. Cysteine residues in the C-terminal tail of the human BKCaa subunit are important for channel sensitivity to carbon monoxide. Adv. Exp. Med. Biol. 2009, 648, 49–56. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G.; Weiger, T. Oxidative Stress and Maxi Calcium-Activated Potassium (BK) Channels. Biomolecules 2015, 5, 1870–1911. [Google Scholar] [CrossRef] [Green Version]
- Rotko, D.; Bednarczyk, P.; Koprowski, P.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Heme is required for carbon monoxide activation of mitochondrial BKCa channel. Eur. J. Pharmacol. 2020, 881, 173191. [Google Scholar] [CrossRef]
- Walewska, A.; Szewczyk, A.; Koprowski, P. Gas Signaling Molecules and Mitochondrial Potassium Channels. Int. J. Mol. Sci. 2018, 19, 3227. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kalogeris, T.; Wang, M.; Zuidema, M.; Wang, Q.; Dai, H.; Davis, M.J.; Hill, M.A.; Korthuis, R.J. Hydrogen sulfide preconditioning or neutrophil depletion attenuates ischemia-reperfusion-induced mitochondrial dysfunction in rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G44–G54. [Google Scholar] [CrossRef]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.; Muller, C.T.; Riccardi, D.; Kemp, P.J. Hydrogen sulfide inhibits human BKCa channels. Adv. Exp. Med. Biol. 2009, 648, 65–72. [Google Scholar] [CrossRef]
- Szteyn, K.; Singh, H. BKCa Channels as Targets for Cardioprotection. Antioxidants 2020, 9, 760. [Google Scholar] [CrossRef]
- Ohya, S.; Kuwata, Y.; Sakamoto, K.; Muraki, K.; Imaizumi, Y. Cardioprotective effects of estradiol include the activation of large-conductance Ca2+-activated K+ channels in cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1635–H1642. [Google Scholar] [CrossRef] [Green Version]
- Wrzosek, A.; Augustynek, B.; Zochowska, M.; Szewczyk, A. Mitochondrial Potassium Channels as Druggable Targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef]
- Debska, G.; Kicinska, A.; Dobrucki, J.; Dworakowska, B.; Nurowska, E.; Skalska, J.; Dolowy, K.; Szewczyk, A. Large-conductance K+ channel openers NS1619 and NS004 as inhibitors of mitochondrial function in glioma cells. Biochem. Pharmacol. 2003, 65, 1827–1834. [Google Scholar] [CrossRef]
- Olesen, S.P.; Munch, E.; Moldt, P.; Drejer, J. Selective activation of Ca2+-dependent K+ channels by novel benzimidazolone. Eur. J. Pharmacol. 1994, 251, 53–59. [Google Scholar] [CrossRef]
- Gribkoff, V.K.; Champigny, G.; Barbry, P.; Dworetzky, S.I.; Meanwell, N.A.; Lazdunski, M. The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J. Biol. Chem. 1994, 269, 10983–10986. [Google Scholar] [CrossRef]
- Stowe, D.F.; Aldakkak, M.; Camara, A.K.; Riess, M.L.; Heinen, A.; Varadarajan, S.G.; Jiang, M.T. Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H434–H440. [Google Scholar] [CrossRef] [Green Version]
- Rockman, M.E.; Vouga, A.G.; Rothberg, B.S. Molecular mechanism of BK channel activation by the smooth muscle relaxant NS11021. J. Gen. Physiol. 2020, 152, e201912506. [Google Scholar] [CrossRef] [Green Version]
- Gessner, G.; Cui, Y.M.; Otani, Y.; Ohwada, T.; Soom, M.; Hoshi, T.; Heinemann, S.H. Molecular mechanism of pharmacological activation of BK channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3552–3557. [Google Scholar] [CrossRef] [Green Version]
- Cancherini, D.V.; Queliconi, B.B.; Kowaltowski, A.J. Pharmacological and physiological stimuli do not promote Ca2+-sensitive K+channel activity in isolated heart mitochondria. Cardiovasc. Res. 2007, 73, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Kicinska, A.; Szewczyk, A. Large-Conductance Potassium Cation Channel Opener NS1619 Inhibits Cardiac Mitochondria Respiratory Chain. Toxicol. Mech. Methods 2004, 14, 59–61. [Google Scholar] [CrossRef]
- Łukasiak, A.; Skup, A.; Chlopicki, S.; Łomnicka, M.; Kaczara, P.; Proniewski, B.; Szewczyk, A.; Wrzosek, A. SERCA, complex I of the respiratory chain and ATP-synthase inhibition are involved in pleiotropic effects of NS1619 on endothelial cells. Eur. J. Pharmacol. 2016, 786, 137–147. [Google Scholar] [CrossRef]
- Park, W.S.; Kang, S.H.; Son, Y.K.; Kim, N.; Ko, J.-H.; Kim, H.K.; Ko, E.A.; Kim, C.D.; Han, J. The mitochondrial Ca2+-activated K+ channel activator, NS 1619 inhibits L-type Ca2+ channels in rat ventricular myocytes. Biochem. Biophys. Res. Commun. 2007, 362, 31–36. [Google Scholar] [CrossRef]
- Saleh, S.N.; Angermann, J.E.; Sones, W.R.; Leblanc, N.; Greenwood, I.A. Stimulation of Ca2+-gated Cl- currents by the calcium-dependent K+ channel modulators NS1619 [1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzi midazol-2-one] and isopimaric acid. J. Pharmacol. Exp. Ther. 2007, 321, 1075–1084. [Google Scholar] [CrossRef]
- Gaspar, T.; Domoki, F.; Lenti, L.; Katakam, P.V.; Snipes, J.A.; Bari, F.; Busija, D.W. Immediate neuronal preconditioning by NS1619. Brain Res. 2009, 1285, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, T.; Katakam, P.; Snipes, J.A.; Kis, B.; Domoki, F.; Bari, F.; Busija, D.W. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J. Neurochem. 2008, 105, 1115–1128. [Google Scholar] [CrossRef] [Green Version]
- Bentzen, B.H.; Nardi, A.; Calloe, K.; Madsen, L.S.; Olesen, S.P.; Grunnet, M. The small molecule NS11021 is a potent and specific activator of Ca2+-activated big-conductance K+ channels. Mol. Pharmacol. 2007, 72, 1033–1044. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Osadchii, O.; Jespersen, T.; Hansen, R.S.; Olesen, S.P.; Grunnet, M. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflugers Arch. 2009, 457, 979–988. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; Wei, A.C.; Grunnet, M.; O’Rourke, B. Energetic performance is improved by specific activation of K+ fluxes through KCa channels in heart mitochondria. Biochim. Biophys. Acta 2010, 1797, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Augustynek, B.; Koprowski, P.; Rotko, D.; Kunz, W.; Szewczyk, A.; Kulawiak, B. Mitochondrial BK Channel Openers CGS7181 and CGS7184 Exhibit Cytotoxic Properties. Int. J. Mol. Sci. 2018, 19, 353. [Google Scholar] [CrossRef] [Green Version]
- Kulawiak, B.; Kudin, A.P.; Szewczyk, A.; Kunz, W.S. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp. Neurol. 2008, 212, 543–547. [Google Scholar] [CrossRef]
- Debska-Vielhaber, G.; Godlewski, M.M.; Kicinska, A.; Skalska, J.; Kulawiak, B.; Piwonska, M.; Zablocki, K.; Kunz, W.S.; Szewczyk, A.; Motyl, T. Large-conductance K+ channel openers induce death of human glioma cells. J. Physiol. Pharmacol. 2009, 60, 27–36. [Google Scholar]
- Wrzosek, A.; Tomaskova, Z.; Ondrias, K.; Łukasiak, A.; Szewczyk, A. The potassium channel opener CGS7184 activates Ca2+ release from the endoplasmic reticulum. Eur. J. Pharmacol. 2012, 690, 60–67. [Google Scholar] [CrossRef]
- Wrzosek, A. The potassium channel opener NS1619 modulates calcium homeostasis in muscle cells by inhibiting SERCA. Cell. Calcium 2014, 56, 14–24. [Google Scholar] [CrossRef]
- Wrzosek, A.; Tomaskova, Z.; Ondrias, K.; Łukasiak, A.; Szewczyk, A. CGS7184 a potassium channel opener modulates activity of mitochondria and Ca2+ homeostasis. Biochim. Biophys. Acta Bioenergy 2012, 1817, S88–S89. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-C.; Lo, Y.-K.; Wu, S.-N. Stimulatory effects of chlorzoxazone, a centrally acting muscle relaxant, on large conductance calcium-activated potassium channels in pituitary GH3 cells. Brain Res. 2003, 959, 86–97. [Google Scholar] [CrossRef]
- Du, X.; Carvalho-de-Souza, J.L.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-function BK channel mutation causes impaired mitochondria and progressive cerebellar ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef]
- Sakamoto, K.; Nonomura, T.; Ohya, S.; Muraki, K.; Ohwada, T.; Imaizumi, Y. Molecular mechanisms for large conductance Ca2+-activated K+ channel activation by a novel opener, 12,14-dichlorodehydroabietic acid. J. Pharmacol. Exp. Ther. 2006, 316, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Ohya, S.; Muraki, K.; Imaizumi, Y. A novel opener of large-conductance Ca2+-activated K+ (BK) channel reduces ischemic injury in rat cardiac myocytes by activating mitochondrial KCa channel. J. Pharmacol. Sci. 2008, 108, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Valverde, M.A.; Rojas, P.; Amigo, J.; Cosmelli, D.; Orio, P.; Bahamonde, M.I.; Mann, G.E.; Vergara, C.; Latorre, R. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the b subunit. Science 1999, 285, 1929–1931. [Google Scholar] [CrossRef]
- Vega-Vela, N.E.; Osorio, D.; Avila-Rodriguez, M.; Gonzalez, J.; García-Segura, L.M.; Echeverria, V.; Barreto, G.E. L-Type Calcium Channels Modulation by Estradiol. Mol. Neurobiol. 2017, 54, 4996–5007. [Google Scholar] [CrossRef]
- Hernández-Aquino, E.; Muriel, P. Chapter 46—Naringenin and the Liver. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 633–651. [Google Scholar]
- Testai, L.; Da Pozzo, E.; Piano, I.; Pistelli, L.; Gargini, C.; Breschi, M.C.; Braca, A.; Martini, C.; Martelli, A.; Calderone, V. The Citrus Flavanone Naringenin Produces Cardioprotective Effects in Hearts from 1 Year Old Rat, through Activation of mitoBK Channels. Front. Pharmacol. 2017, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Kampa, R.P.; Kicinska, A.; Jarmuszkiewicz, W.; Pasikowska-Piwko, M.; Dolegowska, B.; Debowska, R.; Szewczyk, A.; Bednarczyk, P. Naringenin as an opener of mitochondrial potassium channels in dermal fibroblasts. Exp. Dermatol. 2019, 28, 543–550. [Google Scholar] [CrossRef]
- Hsu, H.-T.; Tseng, Y.-T.; Lo, Y.-C.; Wu, S.-N. Ability of naringenin, a bioflavonoid, to activate M-type potassium current in motor neuron-like cells and to increase BKCa-channel activity in HEK293T cells transfected with α-hSlo subunit. BMC Neurosci. 2014, 15, 135. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.; Moczydlowski, E.; Latorre, R.; Phillips, M. Charybdotoxin, a protein inhibitor of single Ca2+-activated K+ channels from mammalian skeletal muscle. Nature 1985, 313, 316–318. [Google Scholar] [CrossRef]
- Kang, J.; Huguenard, J.R.; Prince, D.A. Development of BK channels in neocortical pyramidal neurons. J. Neurophysiol. 1996, 76, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.Q.; Pamenter, M.E.; Siemen, D.; Sun, X.; Haddad, G.G. Mitochondrial but not plasmalemmal BK channels are hypoxia-sensitive in human glioma. Glia 2014, 62, 504–513. [Google Scholar] [CrossRef]
- Panyi, G.; Possani, L.D.; Rodríguez de la Vega, R.C.; Gáspár, R.; Varga, Z. K+ channel blockers: Novel tools to inhibit T cell activation leading to specific immunosuppression. Curr. Pharm. Des. 2006, 12, 2199–2220. [Google Scholar] [CrossRef]
- Galvez, A.; Gimenez-Gallego, G.; Reuben, J.P.; Roy-Contancin, L.; Feigenbaum, P.; Kaczorowski, G.J.; Garcia, M.L. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem. 1990, 265, 11083–11090. [Google Scholar] [CrossRef]
- Fahanik-babaei, J.; Eliassi, A.; Jafari, A.; Sauve, R.; Salari, S.; Saghiri, R. Electro-pharmacological profile of a mitochondrial inner membrane big-potassium channel from rat brain. Biochim. Biophys. Acta Biomembr. 2011, 1808, 454–460. [Google Scholar] [CrossRef] [Green Version]
- Kaczorowski, G.J.; Garcia, M.L. Developing Molecular Pharmacology of BK Channels for Therapeutic Benefit. Int. Rev. Neurobiol. 2016, 128, 439–475. [Google Scholar] [CrossRef]
- Bergeron, Z.L.; Bingham, J.P. Scorpion toxins specific for potassium K+ channels: A historical overview of peptide bioengineering. Toxins 2012, 4, 1082–1119. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yan, J. Modulation of BK Channel Function by Auxiliary Beta and Gamma Subunits. Int. Rev. Neurobiol. 2016, 128, 51–90. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef] [Green Version]
- Balderas, E.; Torres, N.S.; Rosa-Garrido, M.; Chaudhuri, D.; Toro, L.; Stefani, E.; Olcese, R. MitoBKCa channel is functionally associated with its regulatory b1 subunit in cardiac mitochondria. J. Physiol. 2019, 597, 3817–3832. [Google Scholar] [CrossRef] [Green Version]
- Bilmen, J.G.; Wootton, L.L.; Michelangeli, F. The mechanism of inhibition of the sarco/endoplasmic reticulum Ca2+ ATPase by paxilline. Arch. Biochem. Biophys. 2002, 406, 55–64. [Google Scholar] [CrossRef]
- Bilmen, J.G.; Wootton, L.L.; Godfrey, R.E.; Smart, O.S.; Michelangeli, F. Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+-binding sites. Eur. J. Biochem. 2002, 269, 3678–3687. [Google Scholar] [CrossRef]
- Bednarczyk, P.; Barker, G.D.; Halestrap, A.P. Determination of the rate of K+ movement through potassium channels in isolated rat heart and liver mitochondria. Biochim. Biophys. Acta 2008, 1777, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Kulawiak, B.; Szewczyk, A. Glutamate-induced cell death in HT22 mouse hippocampal cells is attenuated by paxilline, a BK channel inhibitor. Mitochondrion 2012, 12, 169–172. [Google Scholar] [CrossRef]
- Wulff, H.; Zhorov, B.S. K+Channel Modulators for the Treatment of Neurological Disorders and Autoimmune Diseases. Chem. Rev. 2008, 108, 1744–1773. [Google Scholar] [CrossRef] [Green Version]
- Coburger, I.; Yang, K.; Bernert, A.; Wiesel, E.; Sahoo, N.; Swain, S.M.; Hoshi, T.; Schonherr, R.; Heinemann, S.H. Impact of intracellular hemin on N-type inactivation of voltage-gated K+ channels. Pflugers Arch. 2020, 472, 551–560. [Google Scholar] [CrossRef]
- Pedarzani, P.; Stocker, M. Molecular and cellular basis of small- and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell. Mol. Life Sci. 2008, 65, 3196–3217. [Google Scholar] [CrossRef] [Green Version]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Kovalenko, I.; Glasauer, A.; Schockel, L.; Sauter, D.R.; Ehrmann, A.; Sohler, F.; Hagebarth, A.; Novak, I.; Christian, S. Identification of KCa3.1 Channel as a Novel Regulator of Oxidative Phosphorylation in a Subset of Pancreatic Carcinoma Cell Lines. PLoS ONE 2016, 11, e0160658. [Google Scholar] [CrossRef]
- Strobaek, D.; Teuber, L.; Jorgensen, T.D.; Ahring, P.K.; Kjaer, K.; Hansen, R.S.; Olesen, S.P.; Christophersen, P.; Skaaning-Jensen, B. Activation of human IK and SK Ca2+ -activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 2004, 1665, 1–5. [Google Scholar] [CrossRef] [Green Version]
- John, V.H.; Dale, T.J.; Hollands, E.C.; Chen, M.X.; Partington, L.; Downie, D.L.; Meadows, H.J.; Trezise, D.J. Novel 384-Well Population Patch Clamp Electrophysiology Assays for Ca2+-Activated K+ Channels. J. Biomol. Screen. 2007, 12, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Dolga, A.M.; De Andrade, A.; Meissner, L.; Knaus, H.G.; Höllerhage, M.; Christophersen, P.; Zischka, H.; Plesnila, N.; Höglinger, G.U.; Culmsee, C. Subcellular expression and neuroprotective effects of SK channels in human dopaminergic neurons. Cell Death Dis. 2014, 5, e999. [Google Scholar] [CrossRef]
- Coleman, N.; Brown, B.M.; Olivan-Viguera, A.; Singh, V.; Olmstead, M.M.; Valero, M.S.; Kohler, R.; Wulff, H. New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1. Mol. Pharmacol. 2014, 86, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.Y.; Terentyeva, R.; Roder, K.H.; Li, W.; Liu, M.; Greener, I.; Hamilton, S.; Polina, I.; Murphy, K.R.; Clements, R.T.; et al. SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc. Res. 2017, 113, 343–353. [Google Scholar] [CrossRef]
- Kleger, A.; Seufferlein, T.; Malan, D.; Tischendorf, M.; Storch, A.; Wolheim, A.; Latz, S.; Protze, S.; Porzner, M.; Proepper, C.; et al. Modulation of Calcium-Activated Potassium Channels Induces Cardiogenesis of Pluripotent Stem Cells and Enrichment of Pacemaker-Like Cells. Circulation 2010, 122, 1823–1836. [Google Scholar] [CrossRef] [Green Version]
- Augustynek, B.; Kunz, W.S.; Szewczyk, A. Guide to the Pharmacology of Mitochondrial Potassium Channels. Handb. Exp. Pharmacol. 2017, 240, 103–127. [Google Scholar] [CrossRef]
- Singh, S.; Syme, C.A.; Singh, A.K.; Devor, D.C.; Bridges, R.J. Benzimidazolone activators of chloride secretion: Potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2001, 296, 600–611. [Google Scholar]
- Cui, M.; Qin, G.; Yu, K.; Bowers, M.S.; Zhang, M. Targeting the Small- and Intermediate-Conductance Ca2+-Activated Potassium Channels: The Drug-Binding Pocket at the Channel/Calmodulin Interface. Neurosignals 2014, 22, 65–78. [Google Scholar] [CrossRef]
- Richter, J.M.; Schaefer, M.; Hill, K. Riluzole activates TRPC5 channels independently of PLC activity. Br. J. Pharmacol. 2014, 171, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Duprat, F.; Lesage, F.; Patel, A.J.; Fink, M.; Romey, G.; Lazdunski, M. The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol. Pharmacol. 2000, 57, 906–912. [Google Scholar]
- Stefani, A.; Spadoni, F.; Bernardi, G. Differential Inhibition by Riluzole, Lamotrigine, and Phenytoin of Sodium and Calcium Currents in Cortical Neurons: Implications for Neuroprotective Strategies. Exp. Neurol. 1997, 147, 115–122. [Google Scholar] [CrossRef]
- Yokoo, H.; Shiraishi, S.; Kobayashi, H.; Yanagita, T.; Yamamoto, R.; Wada, A. Selective inhibition by riluzole of voltage-dependent sodium channels and catecholamine secretion in adrenal chromaffin cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 357, 526–531. [Google Scholar] [CrossRef]
- Zona, C.; Siniscalchi, A.; Mercuri, N.B.; Bernardi, G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience 1998, 85, 931–938. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Ng, K.; Van Den Bos, M.; Byth, K.; Kiernan, M.C.; Vucic, S. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 580–588. [Google Scholar] [CrossRef]
- Bausch, A.R.; Roy, G. Volume-sensitive chloride channels blocked by neuroprotective drugs in human glial cells (U-138MG). Glia 1996, 18, 73–77. [Google Scholar] [CrossRef]
- Kretschmer, B.D.; Kratzer, U.; Schmidt, W.J. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 358, 181–190. [Google Scholar] [CrossRef]
- Azbill, R.D.; Mu, X.; Springer, J.E. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000, 871, 175–180. [Google Scholar] [CrossRef]
- Brugnara, C.; Armsby, C.C.; Sakamoto, M.; Rifai, N.; Alper, S.L.; Platt, O. Oral administration of clotrimazole and blockade of human erythrocyte Ca++-activated K+ channel: The imidazole ring is not required for inhibitory activity. J. Pharmacol. Exp. Ther. 1995, 273, 266–272. [Google Scholar]
- Ishii, T.M.; Maylie, J.; Adelman, J.P. Determinants of Apamin and d-Tubocurarine Block in SK Potassium Channels. J. Biol. Chem. 1997, 272, 23195–23200. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, N.J.; Kang, J.; Togo, J.A.; Christian, E.P.; Aiyar, J. A Novel Gene, hKCa4, Encodes the Calcium-activated Potassium Channel in Human T Lymphocytes. J. Biol. Chem. 1997, 272, 32723–32726. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.N.; Li, H.F.; Jan, C.R.; Shen, A.Y. Inhibition of Ca2+-activated K+ current by clotrimazole in rat anterior pituitary GH3 cells. Neuropharmacology 1999, 38, 979–989. [Google Scholar] [CrossRef]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 37137–37149. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Kolski-Andreaco, A.; Sankaranarayanan, A.; Sabatier, J.M.; Shakkottai, V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 2007, 14, 1437–1457. [Google Scholar] [CrossRef]
- Yi, M.; Yu, P.; Lu, Q.; Geller, H.M.; Yu, Z.; Chen, H. KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer’s disease. Mol. Cell. Neurosci. 2016, 76, 21–32. [Google Scholar] [CrossRef]
- Hougaard, C.; Eriksen, B.L.; Jorgensen, S.; Johansen, T.H.; Dyhring, T.; Madsen, L.S.; Strobaek, D.; Christophersen, P. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br. J. Pharmacol. 2007, 151, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Honrath, B.; Krabbendam, I.E.; Culmsee, C.; Dolga, A.M. Small conductance Ca2+-activated K+ channels in the plasma membrane, mitochondria and the ER: Pharmacology and implications in neuronal diseases. Neurochem. Int. 2017, 109, 13–23. [Google Scholar] [CrossRef]
- Richter, M.; Nickel, C.; Apel, L.; Kaas, A.; Dodel, R.; Culmsee, C.; Dolga, A.M. SK channel activation modulates mitochondrial respiration and attenuates neuronal HT-22 cell damage induced by H2O2. Neurochem. Int. 2015, 81, 63–75. [Google Scholar] [CrossRef]
- Pedarzani, P.; McCutcheon, J.E.; Rogge, G.; Jensen, B.S.; Christophersen, P.; Hougaard, C.; Strøbæk, D.; Stocker, M. Specific Enhancement of SK Channel Activity Selectively Potentiates the Afterhyperpolarizing Current IAHP and Modulates the Firing Properties of Hippocampal Pyramidal Neurons*. J. Biol. Chem. 2005, 280, 41404–41411. [Google Scholar] [CrossRef] [Green Version]
- Cuthbert, A.W. Assessment of CFTR chloride channel openers in intact normal and cystic fibrosis murine epithelia. Br. J. Pharmacol. 2001, 132, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Honrath, B.; Matschke, L.; Meyer, T.; Magerhans, L.; Perocchi, F.; Ganjam, G.K.; Zischka, H.; Krasel, C.; Gerding, A.; Bakker, B.M.; et al. SK2 channels regulate mitochondrial respiration and mitochondrial Ca2+ uptake. Cell. Death Differ. 2017, 24, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Noh, T.K.; Bang, S.H.; Lee, Y.J.; Cho, H.I.; Jung, M.Y.; Kim, I.; Leem, C.H.; Chang, S.E. The ion channel activator CyPPA inhibits melanogenesis via the GSK3β/β-catenin pathway. Chem.-Biol. Interact. 2019, 300, 1–7. [Google Scholar] [CrossRef]
- Stowe, D.F.; Gadicherla, A.K.; Zhou, Y.; Aldakkak, M.; Cheng, Q.; Kwok, W.M.; Jiang, M.T.; Heisner, J.S.; Yang, M.; Camara, A.K. Protection against cardiac injury by small Ca2+-sensitive K+ channels identified in guinea pig cardiac inner mitochondrial membrane. Biochim. Biophys. Acta 2013, 1828, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Han, S.M.; Park, K.-K. Therapeutic Effects of Apamin as a Bee Venom Component for Non-Neoplastic Disease. Toxins 2020, 12, 195. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-H.; An, H.-J.; Kim, J.-Y.; Gwon, M.-G.; Gu, H.; Lee, S.-J.; Park, J.Y.; Park, K.-D.; Han, S.-M.; Kim, M.-K.; et al. Apamin inhibits TNF-α- and IFN-γ-induced inflammatory cytokines and chemokines via suppressions of NF-κB signaling pathway and STAT in human keratinocytes. Pharmacol. Rep. 2017, 69, 1030–1035. [Google Scholar] [CrossRef]
- Strobaek, D.; Hougaard, C.; Johansen, T.H.; Sorensen, U.S.; Nielsen, E.O.; Nielsen, K.S.; Taylor, R.D.; Pedarzani, P.; Christophersen, P. Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol. Pharmacol. 2006, 70, 1771–1782. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, U.S.; Strobaek, D.; Christophersen, P.; Hougaard, C.; Jensen, M.L.; Nielsen, E.O.; Peters, D.; Teuber, L. Synthesis and structure-activity relationship studies of 2-(N-substituted)-aminobenzimidazoles as potent negative gating modulators ofsmall conductance Ca2+-activated K+ channels. J. Med. Chem. 2008, 51, 7625–7634. [Google Scholar] [CrossRef]
- Syme, C.A.; Gerlach, A.C.; Singh, A.K.; Devor, D.C. Pharmacological activation of cloned intermediate- and small-conductance Ca2+-activated K+ channels. Am. J. Physiol. Cell Physiol. 2000, 278, C570–C581. [Google Scholar] [CrossRef]
- Cao, Y.; Dreixler, J.C.; Roizen, J.D.; Roberts, M.T.; Houamed, K.M. Modulation of recombinant small-conductance Ca2+-activated K+ channels by the muscle relaxant chlorzoxazone and structurally related compounds. J. Pharmacol. Exp. Ther. 2001, 296, 683–689. [Google Scholar]
- Alvina, K.; Khodakhah, K. KCa channels as therapeutic targets in episodic ataxia type-2. J. Neurosci. 2010, 30, 7249–7257. [Google Scholar] [CrossRef] [Green Version]
- Teisseyre, A.; Palko-Labuz, A.; Sroda-Pomianek, K.; Michalak, K. Voltage-Gated Potassium Channel Kv1.3 as a Target in Therapy of Cancer. Front. Oncol. 2019, 9, 933. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [Green Version]
- Barros, F.; de la Pena, P.; Dominguez, P.; Sierra, L.M.; Pardo, L.A. The EAG Voltage-Dependent K+ Channel Subfamily: Similarities and Differences in.n Structural Organization and Gating. Front. Pharmacol. 2020, 11, 411. [Google Scholar] [CrossRef] [Green Version]
- Leanza, L.; Checchetto, V.; Biasutto, L.; Rossa, A.; Costa, R.; Bachmann, M.; Zoratti, M.; Szabo, I. Pharmacological modulation of mitochondrial ion channels. Br. J. Pharmacol. 2019, 176, 4258–4283. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Jekle, A.; Soddemann, M.; Adams, C.; Lang, F.; Zoratti, M.; Gulbins, E. A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 2005, 280, 12790–12798. [Google Scholar] [CrossRef] [Green Version]
- Gulbins, E.; Sassi, N.; Grassme, H.; Zoratti, M.; Szabo, I. Role of Kv1.3 mitochondrial potassium channel in apoptotic signalling in lymphocytes. Biochim. Biophys. Acta 2010, 1797, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.H.; Byun, J.K.; Jeon, W.I.; Choi, S.Y.; Park, J.; Lee, B.H.; Yang, J.E.; Park, J.B.; O’Grady, S.M.; Kim, D.Y.; et al. Nuclear localization and functional characteristics of voltage-gated potassium channel Kv1.3. J. Biol. Chem. 2015, 290, 12547–12557. [Google Scholar] [CrossRef] [Green Version]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef]
- Comes, N.; Bielanska, J.; Vallejo-Gracia, A.; Serrano-Albarras, A.; Marruecos, L.; Gomez, D.; Soler, C.; Condom, E.; Ramon, Y.C.S.; Hernandez-Losa, J.; et al. The voltage-dependent K+ channels Kv1.3 and Kv1.5 in human cancer. Front. Physiol. 2013, 4, 283. [Google Scholar] [CrossRef] [Green Version]
- Leanza, L.; Trentin, L.; Becker, K.A.; Frezzato, F.; Zoratti, M.; Semenzato, G.; Gulbins, E.; Szabo, I. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 2013, 27, 1782–1785. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef] [Green Version]
- Zaccagnino, A.; Manago, A.; Leanza, L.; Gontarewitz, A.; Linder, B.; Azzolini, M.; Biasutto, L.; Zoratti, M.; Peruzzo, R.; Legler, K.; et al. Tumor-reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 38276–38293. [Google Scholar] [CrossRef]
- Ahmed, K.; Koval, A.; Xu, J.; Bodmer, A.; Katanaev, V.L. Towards the first targeted therapy for triple-negative breast cancer: Repositioning of clofazimine as a chemotherapy-compatible selective Wnt pathway inhibitor. Cancer Lett. 2019, 449, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.R.; Pan, F.; Parvez, S.; Fleig, A.; Chong, C.R.; Xu, J.; Dang, Y.; Zhang, J.; Jiang, H.; Penner, R.; et al. Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PLoS ONE 2008, 3, e4009. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Starkus, J.; Penner, R. State-dependent blocking mechanism of Kv 1.3 channels by the antimycobacterial drug clofazimine. Br. J. Pharmacol. 2015, 172, 5161–5173. [Google Scholar] [CrossRef] [Green Version]
- Love, M.S.; Beasley, F.C.; Jumani, R.S.; Wright, T.M.; Chatterjee, A.K.; Huston, C.D.; Schultz, P.G.; McNamara, C.W. A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl. Trop Dis. 2017, 11, e0005373. [Google Scholar] [CrossRef] [Green Version]
- Lechartier, B.; Cole, S.T. Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463. [Google Scholar] [CrossRef] [Green Version]
- Marzian, S.; Stansfeld, P.J.; Rapedius, M.; Rinne, S.; Nematian-Ardestani, E.; Abbruzzese, J.L.; Steinmeyer, K.; Sansom, M.S.; Sanguinetti, M.C.; Baukrowitz, T.; et al. Side pockets provide the basis for a new mechanism of Kv channel-specific inhibition. Nat. Chem. Biol. 2013, 9, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Y.; Hou, G.Q.; He, S.W.; Xiao, Z.; Xu, H.J.; Qiu, Y.T.; Jiang, S.; Zheng, H.; Li, Z.Y. Psora-4, a Kv1.3 Blocker, Enhances Differentiation and Maturation in Neural Progenitor Cells. CNS Neurosci. Ther. 2015, 21, 558–567. [Google Scholar] [CrossRef]
- Hyodo, T.; Oda, T.; Kikuchi, Y.; Higashi, K.; Kushiyama, T.; Yamamoto, K.; Yamada, M.; Suzuki, S.; Hokari, R.; Kinoshita, M.; et al. Voltage-gated potassium channel Kv1.3 blocker as a potential treatment for rat anti-glomerular basement membrane glomerulonephritis. Am. J. Physiol. Renal Physiol. 2010, 299, F1258–F1269. [Google Scholar] [CrossRef] [Green Version]
- Peruzzo, R.; Mattarei, A.; Azzolini, M.; Becker-Flegler, K.A.; Romio, M.; Rigoni, G.; Carrer, A.; Biasutto, L.; Parrasia, S.; Kadow, S.; et al. Insight into the mechanism of cytotoxicity of membrane-permeant psoralenic Kv1.3 channel inhibitors by chemical dissection of a novel member of the family. Redox Biol. 2020, 37, 101705. [Google Scholar] [CrossRef]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neurosignals 2017, 25, 26–38. [Google Scholar] [CrossRef]
- Ostacolo, C.; Miceli, F.; Di Sarno, V.; Nappi, P.; Iraci, N.; Soldovieri, M.V.; Ciaglia, T.; Ambrosino, P.; Vestuto, V.; Lauritano, A.; et al. Synthesis and Pharmacological Characterization of Conformationally Restricted Retigabine Analogues as Novel Neuronal Kv7 Channel Activators. J. Med. Chem. 2020, 63, 163–185. [Google Scholar] [CrossRef]
- Brickel, N.; Gandhi, P.; VanLandingham, K.; Hammond, J.; DeRossett, S. The urinary safety profile and secondary renal effects of retigabine (ezogabine): A first-in-class antiepileptic drug that targets KCNQ (K(v)7) potassium channels. Epilepsia 2012, 53, 606–612. [Google Scholar] [CrossRef]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.A.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Devulder, J. Flupirtine in pain management: Pharmacological properties and clinical use. CNS Drugs 2010, 24, 867–881. [Google Scholar] [CrossRef]
- Ipavec, V.; Martire, M.; Barrese, V.; Taglialatela, M.; Curro, D. KV7 channels regulate muscle tone and nonadrenergic noncholinergic relaxation of the rat gastric fundus. Pharmacol. Res. 2011, 64, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Valdez, R.; White, A.M.; Raol, Y.H. Anticonvulsant effect of flupirtine in an animal model of neonatal hypoxic-ischemic encephalopathy. Neuropharmacology 2017, 123, 126–135. [Google Scholar] [CrossRef]
- Raffa, R.B.; Pergolizzi, J.V., Jr. The evolving understanding of the analgesic mechanism of action of flupirtine. J. Clin. Pharm. Ther. 2012, 37, 4–6. [Google Scholar] [CrossRef]
- Schroder, H.C.; Muller, W.E. Neuroprotective effect of flupirtine in prion disease. Drugs Today 2002, 38, 49–58. [Google Scholar] [CrossRef]
- Michel, M.C.; Radziszewski, P.; Falconer, C.; Marschall-Kehrel, D.; Blot, K. Unexpected frequent hepatotoxicity of a prescription drug, flupirtine, marketed for about 30 years. Br. J. Clin. Pharmacol. 2012, 73, 821–825. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, W.; Modess, C.; Scheuch, E.; Methling, K.; Keiser, M.; Nassif, A.; Rosskopf, D.; Bednarski, P.J.; Borlak, J.; Terhaag, B. Metabolic activation and analgesic effect of flupirtine in healthy subjects, influence of the polymorphic NAT2, UGT1A1 and GSTP1. Br. J. Clin. Pharmacol. 2015, 79, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Porter, R.J.; Nohria, V.; Rundfeldt, C. Retigabine. Neurotherapeutics 2007, 4, 149–154. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Grippon, S.; Kirkpatrick, P. Ezogabine (retigabine). Nat. Rev. Drug Discov. 2011, 10, 729–730. [Google Scholar] [CrossRef]
- Barrese, V.; Miceli, F.; Soldovieri, M.V.; Ambrosino, P.; Iannotti, F.A.; Cilio, M.R.; Taglialatela, M. Neuronal potassium channel openers in the management of epilepsy: Role and potential of retigabine. Clin. Pharmacol. 2010, 2, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Liu, S.; Jin, L.; Tang, L.; Zhao, X.; Yang, T.; Wang, Y.; Huo, B.; Liu, R.; Li, H. Antinociceptive Efficacy of Retigabine and Flupirtine for Gout Arthritis Pain. Pharmacology 2020, 105, 471–476. [Google Scholar] [CrossRef]
- Zheng, Q.; Fang, D.; Liu, M.; Cai, J.; Wan, Y.; Han, J.S.; Xing, G.G. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 2013, 154, 434–448. [Google Scholar] [CrossRef]
- Hayashi, H.; Iwata, M.; Tsuchimori, N.; Matsumoto, T. Activation of peripheral KCNQ channels attenuates inflammatory pain. Mol. Pain 2014, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, F.; Wang, X.; Sun, R.; Chen, J.; Chen, B.; Zhang, Y. Antinociceptive Efficacy of Retigabine in the Monosodium Lodoacetate Rat Model for Osteoarthritis Pain. Pharmacology 2015, 95, 251–257. [Google Scholar] [CrossRef]
- Hirano, K.; Kuratani, K.; Fujiyoshi, M.; Tashiro, N.; Hayashi, E.; Kinoshita, M. Kv7.2-7.5 voltage-gated potassium channel (KCNQ2-5) opener, retigabine, reduces capsaicin-induced visceral pain in mice. Neurosci. Lett. 2007, 413, 159–162. [Google Scholar] [CrossRef]
- Gunthorpe, M.J.; Large, C.H.; Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012, 53, 412–424. [Google Scholar] [CrossRef]
- Sotty, F.; Damgaard, T.; Montezinho, L.P.; Mork, A.; Olsen, C.K.; Bundgaard, C.; Husum, H. Antipsychotic-like effect of retigabine [N-(2-Amino-4-(fluorobenzylamino)-phenyl)carbamic acid ester], a KCNQ potassium channel opener, via modulation of mesolimbic dopaminergic neurotransmission. J. Pharmacol. Exp. Ther. 2009, 328, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Brodie, M.S.; Appel, S.B. Dopaminergic neurons in the ventral tegmental area of C57BL/6J and DBA/2J mice differ in sensitivity to ethanol excitation. Alcohol. Clin. Exp. Res. 2000, 24, 1120–1124. [Google Scholar] [CrossRef]
- Zwierzynska, E.; Krupa, A.; Pietrzak, B. A pharmaco-EEG study of the interaction between ethanol and retigabine in rabbits. Am. J. Drug Alcohol. Abuse 2015, 41, 153–160. [Google Scholar] [CrossRef]
- Boscia, F.; Annunziato, L.; Taglialatela, M. Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology 2006, 51, 283–294. [Google Scholar] [CrossRef]
- Zwierzynska, E.; Krupa-Burtnik, A.; Pietrzak, B. Beneficial effect of retigabine on memory in rats receiving ethanol. Pharmacol. Rep. 2021, 73, 480–489. [Google Scholar] [CrossRef]
- Young, M.B.; Thomas, S.A. M1-muscarinic receptors promote fear memory consolidation via phospholipase C and the M-current. J. Neurosci. 2014, 34, 1570–1578. [Google Scholar] [CrossRef]
- Dencker, D.; Husum, H. Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav. Brain Res. 2010, 207, 78–83. [Google Scholar] [CrossRef]
- Dupont, C.; Denman, K.S.; Hawash, A.A.; Voss, A.A.; Rich, M.M. Treatment of myotonia congenita with retigabine in mice. Exp. Neurol. 2019, 315, 52–59. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Nai, Y.; Li, S.Y.; Zheng, Y.H. Retigabine protects the blood-brain barrier by regulating tight junctions between cerebral vascular endothelial cells in cerebral ischemia-reperfusion rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8509–8518. [Google Scholar] [CrossRef]
- Stas, J.I.; Bocksteins, E.; Jensen, C.S.; Schmitt, N.; Snyders, D.J. The anticonvulsant retigabine suppresses neuronal KV2-mediated currents. Sci. Rep. 2016, 6, 35080. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Smith, C.O.; Urciuoli, W.R.; Wang, Y.T.; Xia, X.M.; Brookes, P.S.; Nehrke, K. Cardiac Slo2.1 Is Required for Volatile Anesthetic Stimulation of K+ Transport and Anesthetic Preconditioning. Anesthesiology 2016, 124, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.O.; Wang, Y.T.; Nadtochiy, S.M.; Miller, J.H.; Jonas, E.A.; Dirksen, R.T.; Nehrke, K.; Brookes, P.S. Cardiac metabolic effects of KNa1.2 channel deletion and evidence for its mitochondrial localization. FASEB J. 2018, 32, 6135–6149. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Sherman, T.A.; Nadtochiy, S.M.; Urciuoli, W.R.; Brookes, P.S.; Nehrke, K. SLO-2 is cytoprotective and contributes to mitochondrial potassium transport. PLoS ONE 2011, 6, e28287. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, L.K. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262. [Google Scholar] [CrossRef] [Green Version]
- Joiner, W.J.; Tang, M.D.; Wang, L.Y.; Dworetzky, S.I.; Boissard, C.G.; Gan, L.; Gribkoff, V.K.; Kaczmarek, L.K. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat. Neurosci. 1998, 1, 462–469. [Google Scholar] [CrossRef]
- Li, Y.; Sato, T.; Arita, M. Bepridil blunts the shortening of action potential duration caused by metabolic inhibition via blockade of ATP-sensitive K+ channels and Na+-activated K+ channels. J. Pharmacol. Exp. Ther. 1999, 291, 562–568. [Google Scholar]
- Yang, B.; Gribkoff, V.K.; Pan, J.; Damagnez, V.; Dworetzky, S.I.; Boissard, C.G.; Bhattacharjee, A.; Yan, Y.; Sigworth, F.J.; Kaczmarek, L.K. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology 2006, 51, 896–906. [Google Scholar] [CrossRef]
- de Los Angeles Tejada, M.; Stolpe, K.; Meinild, A.K.; Klaerke, D.A. Clofilium inhibits Slick and Slack potassium channels. Biologics 2012, 6, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.O.; Nehrke, K.; Brookes, P.S. The Slo(w) path to identifying the mitochondrial channels responsible for ischemic protection. Biochem. J. 2017, 474, 2067–2094. [Google Scholar] [CrossRef]
- Fahanik-Babaei, J.; Rezaee, B.; Nazari, M.; Torabi, N.; Saghiri, R.; Sauve, R.; Eliassi, A. A new brain mitochondrial sodium-sensitive potassium channel: Effect of sodium ions on respiratory chain activity. J. Cell. Sci. 2020. [Google Scholar] [CrossRef]
- Li, M.; Smith, C.J.; Walker, M.T.; Smith, T.J. Novel inhibitors complexed with glutamate dehydrogenase: Allosteric regulation by control of protein dynamics. J. Biol. Chem. 2009, 284, 22988–23000. [Google Scholar] [CrossRef] [Green Version]
- Whitelaw, B.S.; Robinson, M.B. Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Front. Endocrinol. 2013, 4, 123. [Google Scholar] [CrossRef] [Green Version]
- Kleinboelting, S.; Ramos-Espiritu, L.; Buck, H.; Colis, L.; van den Heuvel, J.; Glickman, J.F.; Levin, L.R.; Buck, J.; Steegborn, C. Bithionol Potently Inhibits Human Soluble Adenylyl Cyclase through Binding to the Allosteric Activator Site. J. Biol. Chem. 2016, 291, 9776–9784. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, G.; Zarrow, J.E.; Mashhadi, Z.; Flynn, C.R.; Vinson, P.; Weaver, C.D.; Davies, S.S. Symmetrically substituted dichlorophenes inhibit N-acyl-phosphatidylethanolamine phospholipase D. J. Biol. Chem. 2020, 295, 7289–7300. [Google Scholar] [CrossRef] [Green Version]
- Ayyagari, V.N.; Brard, L. Bithionol inhibits ovarian cancer cell growth in vitro—Studies on mechanism(s) of action. BMC Cancer 2014, 14, 61. [Google Scholar] [CrossRef] [Green Version]
- Ayyagari, V.N.; Hsieh, T.H.J.; Diaz-Sylvester, P.L.; Brard, L. Evaluation of the cytotoxicity of the Bithionol—Cisplatin combination in a panel of human ovarian cancer cell lines. BMC Cancer 2017, 17, 49. [Google Scholar] [CrossRef] [Green Version]
- Kurita, M.; Shimauchi, T.; Kobayashi, M.; Atarashi, K.; Mori, K.; Tokura, Y. Induction of keratinocyte apoptosis by photosensitizing chemicals plus UVA. J. Dermatol. Sci. 2007, 45, 105–112. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Inglis, K.A.; Urch, J.E.; Muller, S.; van Aalten, D.M.; Fairlamb, A.H. Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. Biochem. J. 2006, 393, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Seguin, S.P.; Ireland, A.W.; Gupta, T.; Wright, C.M.; Miyata, Y.; Wipf, P.; Pipas, J.M.; Gestwicki, J.E.; Brodsky, J.L. A screen for modulators of large T antigen’s ATPase activity uncovers novel inhibitors of Simian Virus 40 and BK virus replication. Antivir. Res. 2012, 96, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, W.; Zilbermintz, L.; Cheng, L.W.; Zozaya, J.; Tran, S.H.; Elliott, J.H.; Polukhina, K.; Manasherob, R.; Li, A.; Chi, X.; et al. Bithionol blocks pathogenicity of bacterial toxins, ricin, and Zika virus. Sci. Rep. 2016, 6, 34475. [Google Scholar] [CrossRef]
- Hollingshead, L.M.; Faulds, D.; Fitton, A. Bepridil. A review of its pharmacological properties and therapeutic use in stable angina pectoris. Drugs 1992, 44, 835–857. [Google Scholar] [CrossRef]
- Yumoto, Y.; Horie, M.; Kubota, T.; Ninomiya, T.; Kobori, A.; Takenaka, K.; Takano, M.; Niwano, S.; Izumi, T. Bepridil block of recombinant human cardiac IKs current shows a time-dependent unblock. J. Cardiovasc. Pharmacol. 2004, 43, 178–182. [Google Scholar] [CrossRef]
- Sato, T.; Costa, A.D.; Saito, T.; Ogura, T.; Ishida, H.; Garlid, K.D.; Nakaya, H. Bepridil, an antiarrhythmic drug, opens mitochondrial KATP channels, blocks sarcolemmal KATP channels, and confers cardioprotection. J. Pharmacol. Exp. Ther. 2006, 316, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Malayev, A.A.; Nelson, D.J.; Philipson, L.H. Mechanism of clofilium block of the human Kv1.5 delayed rectifier potassium channel. Mol. Pharmacol. 1995, 47, 198–205. [Google Scholar]
- Kirkegaard, S.S.; Lambert, I.H.; Gammeltoft, S.; Hoffmann, E.K. Activation of the TASK-2 channel after cell swelling is dependent on tyrosine phosphorylation. Am. J. Physiol. Cell Physiol. 2010, 299, C844–C853. [Google Scholar] [CrossRef]
- Yang, A.; Wang, X.Q.; Sun, C.S.; Wei, L.; Yu, S.P. Inhibitory effects of clofilium on membrane currents associated with Ca channels, NMDA receptor channels and Na+, K+-ATPase in cortical neurons. Pharmacology 2005, 73, 162–168. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, H.Y.; Lee, K.H.; Cho, Y.H.; Kong, G. Clofilium, a potassium channel blocker, induces apoptosis of human promyelocytic leukemia (HL-60) cells via Bcl-2-insensitive activation of caspase-3. Cancer Lett. 1999, 147, 85–93. [Google Scholar] [CrossRef]
- Pitayu, L.; Baruffini, E.; Rodier, C.; Rotig, A.; Lodi, T.; Delahodde, A. Combined use of Saccharomyces cerevisiae, Caenorhabditis elegans and patient fibroblasts leads to the identification of clofilium tosylate as a potential therapeutic chemical against POLG-related diseases. Hum. Mol. Genet. 2016, 25, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Yu, S.P.; Gottron, F.; Snider, B.J.; Zipfel, G.J.; Choi, D.W. Potassium channel blockers attenuate hypoxia- and ischemia-induced neuronal death in vitro and in vivo. Stroke 2003, 34, 1281–1286. [Google Scholar] [CrossRef]
- Natale, A.M.; Deal, P.E.; Minor, D.L. Structural Insights into the Mechanisms and Pharmacology of K2P Potassium Channels. J. Mol. Biol. 2021, 433, 166995. [Google Scholar] [CrossRef]
- Alexander, S.; Mathie, A.; Peters, J.; Veale, E.; Striessnig, J.; Kelly, E.; Armstrong, J.; Faccenda, E.; Harding, S.; Pawson, A.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Br. J. Pharmacol. 2019, 176. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.A.; Bockenhauer, D.; O’Kelly, I.; Zilberberg, N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2001, 2, 175–184. [Google Scholar] [CrossRef]
- Herrera-Pérez, S.; Campos-Ríos, A.; Rueda-Ruzafa, L.; Lamas, J.A. Contribution of K2P Potassium Channels to Cardiac Physiology and Pathophysiology. Int. J. Mol. Sci. 2021, 22, 6635. [Google Scholar] [CrossRef]
- Czirják, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, I.; Zanzouri, M.; Honoré, E.; Duprat, F.; Ehrengruber, M.U.; Lazdunski, M.; Patel, A.J. K+-dependent cerebellar granule neuron apoptosis. Role of task leak K+ channels. J. Biol. Chem. 2003, 278, 32068–32076. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, D.; Concha, G.; Arévalo, B.; Prent-Peñaloza, L.; Zúñiga, L.; Kiper, A.K.; Rinné, S.; Reyes-Parada, M.; Decher, N.; González, W.; et al. Discovery of Novel TASK-3 Channel Blockers Using a Pharmacophore-Based Virtual Screening. Int. J. Mol. Sci. 2019, 20, 4014. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, P.; Czirják, G. Molecular Background of Leak K+ Currents: Two-Pore Domain Potassium Channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef] [Green Version]
- Duprat, F.; Lesage, F.; Fink, M.; Reyes, R.; Heurteaux, C.; Lazdunski, M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997, 16, 5464–5471. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Wischmeyer, E.; Xin Liu, G.; Preisig-Müller, R.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J. Biol. Chem. 2000, 275, 16650–16657. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Gnatenco, C. TASK-5, a New Member of the Tandem-Pore K+ Channel Family. Biochem. Biophys. Res. Communications 2001, 284, 923–930. [Google Scholar] [CrossRef]
- Pocsai, K.; Kosztka, L.; Bakondi, G.; Gonczi, M.; Fodor, J.; Dienes, B.; Szentesi, P.; Kovacs, I.; Feniger-Barish, R.; Kopf, E.; et al. Melanoma cells exhibit strong intracellular TASK-3-specific immunopositivity in both tissue sections and cell culture. Cell. Mol. Life Sci. 2006, 63, 2364–2376. [Google Scholar] [CrossRef]
- Rusznák, Z.; Bakondi, G.; Kosztka, L.; Pocsai, K.; Dienes, B.; Fodor, J.; Telek, A.; Gönczi, M.; Szűcs, G.; Csernoch, L. Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Archiv 2008, 452, 415–426. [Google Scholar] [CrossRef]
- Toczylowska-Maminska, R.; Olszewska, A.; Laskowski, M.; Bednarczyk, P.; Skowronek, K.; Szewczyk, A. Potassium channel in the mitochondria of human keratinocytes. J. Investig. Dermatol. 2014, 134, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Nagy, D.; Gönczi, M.; Dienes, B.; Szöőr, Á.; Fodor, J.; Nagy, Z.; Tóth, A.; Fodor, T.; Bai, P.; Szücs, G.; et al. Silencing the KCNK9 potassium channel (TASK-3) gene disturbs mitochondrial function, causes mitochondrial depolarization, and induces apoptosis of human melanoma cells. Arch. Dermatol. Res. 2014, 306, 885–902. [Google Scholar] [CrossRef]
- Yao, J.; McHedlishvili, D.; McIntire, W.E.; Guagliardo, N.A.; Erisir, A.; Coburn, C.A.; Santarelli, V.P.; Bayliss, D.A.; Barrett, P.Q. Functional TASK-3-Like Channels in Mitochondria of Aldosterone-Producing Zona Glomerulosa Cells. Hypertension 2017, 70, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Cikutović-Molina, R.; Herrada, A.A.; González, W.; Brown, N.; Zúñiga, L. TASK-3 Gene Knockdown Dampens Invasion and Migration and Promotes Apoptosis in KATO III and MKN-45 Human Gastric Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 6077. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M.; Pontarin, G.; Szabo, I. The Contribution of Mitochondrial Ion Channels to Cancer Development and Progression. Cell. Physiol. Biochem. 2019, 53, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Innamaa, A.; Jackson, L.; Asher, V.; Van Shalkwyk, G.; Warren, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and prognostic significance of the oncogenic K2P potassium channel KCNK9 (TASK-3) in ovarian carcinoma. Anticancer Res. 2013, 33, 1401–1408. [Google Scholar]
- Patel, A.J.; Lazdunski, M. The 2P-domain K+ channels: Role in apoptosis and tumorigenesis. Pflugers Arch. 2004, 448, 261–273. [Google Scholar] [CrossRef]
- Mu, D.; Chen, L.; Zhang, X.; See, L.-H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.Q.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Zúñiga, R.; Valenzuela, C.; Concha, G.; Brown, N.; Zúñiga, L. TASK-3 Downregulation Triggers Cellular Senescence and Growth Inhibition in Breast Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 1033. [Google Scholar] [CrossRef] [Green Version]
- Coburn, C.A.; Luo, Y.; Cui, M.; Wang, J.; Soll, R.; Dong, J.; Hu, B.; Lyon, M.A.; Santarelli, V.P.; Kraus, R.L.; et al. Discovery of a pharmacologically active antagonist of the two-pore-domain potassium channel K2P9.1 (TASK-3). ChemMedChem 2012, 7, 123–133. [Google Scholar] [CrossRef]
- Zúñiga, R.; Concha, G.; Cayo, A.; Cikutović-Molina, R.; Arevalo, B.; González, W.; Catalán, M.A.; Zúñiga, L. Withaferin A suppresses breast cancer cell proliferation by inhibition of the two-pore domain potassium (K2P9) channel TASK-3. Biomed. Pharmacother. 2020, 129, 110383. [Google Scholar] [CrossRef]
- Bachmann, M.; Rossa, A.; Antoniazzi, G.; Biasutto, L.; Carrer, A.; Campagnaro, M.; Leanza, L.; Gonczi, M.; Csernoch, L.; Paradisi, C.; et al. Synthesis and cellular effects of a mitochondria-targeted inhibitor of the two-pore potassium channel TASK-3. Pharmacol. Res. 2021, 164, 105326. [Google Scholar] [CrossRef]
- Czirjak, G.; Enyedi, P. Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol. Pharmacol. 2003, 63, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Antony, M.L.; Lee, J.; Hahm, E.R.; Kim, S.H.; Marcus, A.I.; Kumari, V.; Ji, X.; Yang, Z.; Vowell, C.L.; Wipf, P.; et al. Growth arrest by the antitumor steroidal lactone withaferin A in human breast cancer cells is associated with down-regulation and covalent binding at cysteine 303 of beta-tubulin. J. Biol. Chem. 2014, 289, 1852–1865. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Tsujioka, T.; Suemori, S.-i.; Kida, J.-i.; Kondo, T.; Tohyama, Y.; Tohyama, K. Withaferin A suppresses the growth of myelodysplasia and leukemia cell lines by inhibiting cell cycle progression. Cancer Sci. 2016, 107, 1302–1314. [Google Scholar] [CrossRef]
- Yan, Z.; Guo, R.; Gan, L.; Lau, W.B.; Cao, X.; Zhao, J.; Ma, X.; Christopher, T.A.; Lopez, B.L.; Wang, Y. Withaferin A inhibits apoptosis via activated Akt-mediated inhibition of oxidative stress. Life Sci. 2018, 211, 91–101. [Google Scholar] [CrossRef]
- Xia, S.; Miao, Y.; Liu, S. Withaferin A induces apoptosis by ROS-dependent mitochondrial dysfunction in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 2363–2369. [Google Scholar] [CrossRef]
- Chen, C.-M.; Chung, Y.-P.; Liu, C.-H.; Huang, K.-T.; Guan, S.-S.; Chiang, C.-K.; Wu, C.-T.; Liu, S.-H. Withaferin A protects against endoplasmic reticulum stress-associated apoptosis, inflammation, and fibrosis in the kidney of a mouse model of unilateral ureteral obstruction. Phytomedicine 2020, 79, 153352. [Google Scholar] [CrossRef]
- Guo, R.; Gan, L.; Lau, W.B.; Yan, Z.; Xie, D.; Gao, E.; Christopher, T.A.; Lopez, B.L.; Ma, X.; Wang, Y. Withaferin A Prevents Myocardial Ischemia/Reperfusion Injury by Upregulating AMP-Activated Protein Kinase-Dependent B-Cell Lymphoma2 Signaling. Circ. J. 2019, 83, 1726–1736. [Google Scholar] [CrossRef] [Green Version]
- Kargacin, G.J.; Ali, Z.; Kargacin, M.E. Ruthenium red reduces the Ca2+ sensitivity of Ca2+ uptake into cardiac sarcoplasmic reticulum. Pflugers Arch. 1998, 436, 338–342. [Google Scholar] [CrossRef]
- Xu, L.; Tripathy, A.; Pasek, D.A.; Meissner, G. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J. Biol. Chem. 1999, 274, 32680–32691. [Google Scholar] [CrossRef] [Green Version]
- Pope, L.; Lolicato, M.; Minor, D.L., Jr. Polynuclear Ruthenium Amines Inhibit K(2P) Channels via a "Finger in the Dam" Mechanism. Cell. Chem. Biol. 2020, 27, 511–524.e514. [Google Scholar] [CrossRef]
- Kwong, J.Q. The mitochondrial calcium uniporter in the heart: Energetics and beyond. J. Physiol. 2017, 595, 3743–3751. [Google Scholar] [CrossRef]
- Dessi, F.; Ben-Ari, Y.; Charriaut-Marlangue, C. Ruthenium red protects against glutamate-induced neuronal death in cerebellar culture. Neurosci. Lett. 1995, 201, 53–56. [Google Scholar] [CrossRef]
- Groskreutz, J.L.; Bronk, S.F.; Gores, G.J. Ruthenium red delays the onset of cell death during oxidative stress of rat hepatocytes. Gastroenterology 1992, 102, 1030–1038. [Google Scholar] [CrossRef]
- Zazueta, C.; Sosa-Torres, M.E.; Correa, F.; Garza-Ortiz, A. Inhibitory properties of ruthenium amine complexes on mitochondrial calcium uptake. J. Bioenergy Biomembr. 1999, 31, 551–557. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, R.; Cheng, Y.; Wu, X.; Xi, S.; Sun, Y.; Jiang, H. Lidocaine inhibits the proliferation of lung cancer by regulating the expression of GOLT1A. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef]
- Chang, Y.C.; Liu, C.L.; Liu, T.P.; Yang, P.S.; Chen, M.J.; Cheng, S.P. Effect of Perioperative Intravenous Lidocaine Infusion on Acute and Chronic Pain after Breast Surgery: A Meta-Analysis of Randomized Controlled Trials. Pain Pract. 2017, 17, 336–343. [Google Scholar] [CrossRef]
- Biel, M.; Schneider, A.; Wahl, C. Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc. Med. 2002, 12, 206–212. [Google Scholar] [CrossRef]
- Santoro, B.; Tibbs, G.R. The HCN gene family: Molecular basis of the hyperpolarization-activated pacemaker channels. Ann. N. Y. Acad. Sci. 1999, 868, 741–764. [Google Scholar] [CrossRef]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Romanelli, M.N.; Cerbai, E. The Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels: From Biophysics to Pharmacology of a Unique Family of Ion Channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Zobeiri, M.; Chaudhary, R.; Datunashvili, M.; Heuermann, R.J.; Luttjohann, A.; Narayanan, V.; Balfanz, S.; Meuth, P.; Chetkovich, D.M.; Pape, H.C.; et al. Modulation of thalamocortical oscillations by TRIP8b, an auxiliary subunit for HCN channels. Brain Struct. Funct. 2018, 223, 1537–1564. [Google Scholar] [CrossRef] [Green Version]
- Calejo, A.I.; Reverendo, M.; Silva, V.S.; Pereira, P.M.; Santos, M.A.; Zorec, R.; Goncalves, P.P. Differences in the expression pattern of HCN isoforms among mammalian tissues: Sources and implications. Mol. Biol. Rep. 2014, 41, 297–307. [Google Scholar] [CrossRef]
- Herrmann, S.; Layh, B.; Ludwig, A. Novel insights into the distribution of cardiac HCN channels: An expression study in the mouse heart. J. Mol. Cell. Cardiol. 2011, 51, 997–1006. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, Z.; Ayala-Aguilera, C.; Martinez-Morales, F.; Galicia-Cruz, O.; Salvador-Hernandez, C.; Pedraza-Chaverri, J.; Medeiros, M.; Hernandez, A.M.; Escobar, L.I. Immunolocalization of hyperpolarization-activated cationic HCN1 and HCN3 channels in the rat nephron: Regulation of HCN3 by potassium diets. Histochem. Cell. Biol. 2016, 145, 25–40. [Google Scholar] [CrossRef]
- Herrington, J.; Zhou, Y.P.; Bugianesi, R.M.; Dulski, P.M.; Feng, Y.; Warren, V.A.; Smith, M.M.; Kohler, M.G.; Garsky, V.M.; Sanchez, M.; et al. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes 2006, 55, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Li, Y.; Han, X.; Yao, L.; Yuan, J.; Qin, W.; Liu, F.; Wang, H. Investigation of hyperpolarization-activated cyclic nucleotide-gated channels in interstitial cells of Cajal of human bladder. Urology 2012, 80, 224.e13–224.e18. [Google Scholar] [CrossRef]
- Luque, M.; Schrott-Fischer, A.; Dudas, J.; Pechriggl, E.; Brenner, E.; Rask-Andersen, H.; Liu, W.; Glueckert, R. HCN channels in the mammalian cochlea: Expression pattern, subcellular location, and age-dependent changes. J. Neurosci. Res. 2021, 99, 699–728. [Google Scholar] [CrossRef]
- León-Aparicio, D.; Salvador, C.; Aparicio-Trejo, O.E.; Briones-Herrera, A.; Pedraza-Chaverri, J.; Vaca, L.; Sampieri, A.; Padilla-Flores, T.; López-González, Z.; León-Contreras, J.C.; et al. Novel Potassium Channels in Kidney Mitochondria: The Hyperpolarization-Activated and Cyclic Nucleotide-Gated HCN Channels. Int. J. Mol. Sci. 2019, 20, 4995. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.; Saponaro, A.; Santoro, B.; Moroni, A.; Thiel, G.; Hamacher, K. Mechanical transduction of cytoplasmic-to-transmembrane-domain movements in a hyperpolarization-activated cyclic nucleotide-gated cation channel. J. Biol. Chem. 2018, 293, 12908–12918. [Google Scholar] [CrossRef] [Green Version]
- Porro, A.; Saponaro, A.; Gasparri, F.; Bauer, D.; Gross, C.; Pisoni, M.; Abbandonato, G.; Hamacher, K.; Santoro, B.; Thiel, G.; et al. The HCN domain couples voltage gating and cAMP response in hyperpolarization-activated cyclic nucleotide-gated channels. eLife 2019, 8, e49672. [Google Scholar] [CrossRef]
- Fürst, O.; D’Avanzo, N. Isoform dependent regulation of human HCN channels by cholesterol. Sci. Rep. 2015, 5, 14270. [Google Scholar] [CrossRef] [Green Version]
- Riegelhaupt, P.M.; Tibbs, G.R.; Goldstein, P.A. HCN and K2P Channels in Anesthetic Mechanisms Research. Methods Enzymol. 2018, 602, 391–416. [Google Scholar] [CrossRef]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and neuronal HCN channelopathies. Pflügers Arch.-Eur. J. Physiol. 2020, 472, 931–951. [Google Scholar] [CrossRef]
- Santoro, B.; Shah, M.M. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels as Drug Targets for Neurological Disorders. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 109–131. [Google Scholar] [CrossRef]
- Felix, R.; Sandoval, A.; Sanchez, D.; Gomora, J.C.; De la Vega-Beltran, J.L.; Trevino, C.L.; Darszon, A. ZD7288 inhibits low-threshold Ca2+ channel activity and regulates sperm function. Biochem. Biophys. Res. Commun. 2003, 311, 187–192. [Google Scholar] [CrossRef]
- Wu, X.; Liao, L.; Liu, X.; Luo, F.; Yang, T.; Li, C. Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents? Channnels 2012, 6, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Chevaleyre, V.; Castillo, P.E. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc. Natl. Acad. Sci. USA 2002, 99, 9538–9543. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Gedik, N.; Witting, P.; Freedman, B.; Klöcker, N.; Heusch, G. Pleiotropic, heart rate-independent cardioprotection by ivabradine. Br. J. Pharmacol. 2015, 172, 4380–4390. [Google Scholar] [CrossRef] [Green Version]
- Postea, O.; Biel, M. Exploring HCN channels as novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 903–914. [Google Scholar] [CrossRef]
- Goto, M.; Miyahara, I.; Hirotsu, K.; Conway, M.; Yennawar, N.; Islam, M.M.; Hutson, S.M. Structural determinants for branched-chain aminotransferase isozyme-specific inhibition by the anticonvulsant drug gabapentin. J. Biol. Chem. 2005, 280, 37246–37256. [Google Scholar] [CrossRef] [Green Version]
- Pielen, A.; Kirsch, M.; Hofmann, H.D.; Feuerstein, T.J.; Lagreze, W.A. Retinal ganglion cell survival is enhanced by gabapentin-lactam in vitro: Evidence for involvement of mitochondrial KATP channels. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 240–244. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; McNaughton, N.C.; Pereverzev, A.; Schneider, T.; Randall, A.D. Actions of sipatrigine, 202W92 and lamotrigine on R-type and T-type Ca2+ channel currents. Eur. J. Pharmacol. 2003, 467, 77–80. [Google Scholar] [CrossRef]
- Kranzler, H.R.; Feinn, R.; Morris, P.; Hartwell, E.E. A meta-analysis of the efficacy of gabapentin for treating alcohol use disorder. Addiction 2019, 114, 1547–1555. [Google Scholar] [CrossRef]
Figure 1.
Diagram of the possible beneficial effects of activation or inhibition of mitoK channels with pharmacological substances.
Figure 1.
Diagram of the possible beneficial effects of activation or inhibition of mitoK channels with pharmacological substances.

Figure 2.
Scheme of the targets of mitoBKCa openers NS1619, NS004, and CGS7184. The targets located in mitochondria, endoplasmic reticulum, and plasma membrane are shown. “+”—activation, “−“—inhibition.
Figure 2.
Scheme of the targets of mitoBKCa openers NS1619, NS004, and CGS7184. The targets located in mitochondria, endoplasmic reticulum, and plasma membrane are shown. “+”—activation, “−“—inhibition.

Figure 3.
Diagram showing the multidirectional effects of clofazimine.

Table 5.
Examples of mitochondrial TASK-3 channels modulators and their off-target activity.
| Name | Function | Example of Off-Target Activity | Clinical Use | Ref. |
|---|---|---|---|---|
| Lidocaine | Inhibitor | Inhibition of voltage-dependent Na+ and K+ channels; Inhibition of the proliferation and metastasis of breast cancer cells by inhibiting the function of TRPM7 channels; reduces the levels of the tumor markers IL-1, TNF-α, and IL-8; inhibition of lung cancer proliferation. | A local anesthetic; used to treat ventricular tachycardia; increases the sensitivity of breast cancer cells to tamoxifen; enhances the toxicity of the cancer drugs mitomycin C, pirarubicin, softening lotion, and cisplatin. | [309,310] |
| Withaferin A | Inhibitor | Cell cycle arrest, Akt-mediated inhibition; inhibit on endoplasmic reticulum stress. | Used for centuries in traditional Indian medicine; however, there is no good evidence that it is safe or effective for treating any disease. | [292,295,296,297,298,299] |
| mitoIN-THPP | Inhibitor | Decrease in cellular ATP even when glycolysis was not impaired, up to an extent similar to that triggered by oligomycin, an inhibitor of the FOF1 ATP-synthase; activates the AMP-dependent kinase; induction of massive mitochondrial fragmentation, increase in ROS level, and apoptosis. | [293] | |
| Ruthenium red and Ru360 | Inhibitor | Inhibition of ryanodine receptors and mitochondrial calcium uniporter (MCU). | [294,302,303,304,305] | |
| DR16 and DR16.1 | Inhibitor | Inhibition of TASK-1. | [276] |
Table 6.
Examples of modulators of mitoHCN channels and their off-target activity.
| Name | Function | Example of Alternative Targets | Clinical Use | Ref. |
|---|---|---|---|---|
| ZD7288 | Inhibitor | Inhibitor of T-type Ca2+ and NaV1.4 channels. Reduced oxygen consumption coupled to ATP synthesis and hyperpolarization of the IMM causes a depression of mossy fiber basal synaptic transmission. | Bradycardic drug | [320,321,328,329,330] |
| Gabapentin | Inhibitor | Inhibitor of mitochondrial branched-chain aminotransferase. | An anesthetic, analgesic, and antiepileptic drug; reduces the symptoms of alcohol withdrawal. | [331,332,336] |
| Lamotrigine | Inhibitor | Blocks T-type Ca2+ channels. | Anticonvulsant drug | [335] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wrzosek, A.; Gałecka, S.; Żochowska, M.; Olszewska, A.; Kulawiak, B. Alternative Targets for Modulators of Mitochondrial Potassium Channels. Molecules 2022, 27, 299. https://doi.org/10.3390/molecules27010299
AMA Style
Wrzosek A, Gałecka S, Żochowska M, Olszewska A, Kulawiak B. Alternative Targets for Modulators of Mitochondrial Potassium Channels. Molecules. 2022; 27(1):299. https://doi.org/10.3390/molecules27010299
Chicago/Turabian StyleWrzosek, Antoni, Shur Gałecka, Monika Żochowska, Anna Olszewska, and Bogusz Kulawiak. 2022. "Alternative Targets for Modulators of Mitochondrial Potassium Channels" Molecules 27, no. 1: 299. https://doi.org/10.3390/molecules27010299